

Abstract
Current approaches aiming to cure type 1 diabetes (T1D) have made a negligible number of patients insulin-independent. In this review, we revisit the role of stem cell (SC)-based applications in curing T1D. The optimal therapeutic approach for T1D should ideally preserve the remaining β-cells, restore β-cell function, and protect the replaced insulin-producing cells from autoimmunity. SCs possess immunological and regenerative properties that could be harnessed to improve the treatment of T1D; indeed, SCs may reestablish peripheral tolerance toward β-cells through reshaping of the immune response and inhibition of autoreactive T-cell function. Furthermore, SC-derived insulin-producing cells are capable of engrafting and reversing hyperglycemia in mice. Bone marrow mesenchymal SCs display a hypoimmunogenic phenotype as well as a broad range of immunomodulatory capabilities, they have been shown to cure newly diabetic nonobese diabetic (NOD) mice, and they are currently undergoing evaluation in two clinical trials. Cord blood SCs have been shown to facilitate the generation of regulatory T cells, thereby reverting hyperglycemia in NOD mice. T1D patients treated with cord blood SCs also did not show any adverse reaction in the absence of major effects on glycometabolic control. Although hematopoietic SCs rarely revert hyperglycemia in NOD mice, they exhibit profound immunomodulatory properties in humans; newly hyperglycemic T1D patients have been successfully reverted to normoglycemia with autologous nonmyeloablative hematopoietic SC transplantation. Finally, embryonic SCs also offer exciting prospects because they are able to generate glucose-responsive insulin-producing cells. Easy enthusiasm should be mitigated mainly because of the potential oncogenicity of SCs.
Introduction
-
Embryonic Stem Cells (ESCs)
Characteristics
Isolation methods
Immunogenicity
Effect on immune response
Cytokine and chemokine profile
Generation of insulin-producing cells
Clinical trials
Unique features: ESC cellular extract (EXT)-based reprogramming
Pros and cons
-
Cord Blood Stem Cells (CB-SCs)
Characteristics
Isolation methods
Immunogenicity
Effect on immune response
Cytokine and chemokine profile
Generation of insulin-producing cells
Clinical trials
Unique feature: naive status
Pros and cons
-
Mesenchymal Stem Cells (MSCs)
Characteristics
Isolation methods
Immunogenicity
Effect on immune response
Cytokine and chemokine profile
Generation of insulin-producing cells
Clinical trials
Unique feature: migratory ability to pancreatic islets
Pros and cons
-
Hematopoietic Stem Cells (HSCs)
Characteristics
Isolation methods
Immunogenicity
Effect on immune response
Cytokine and chemokine profile
Generation of insulin-producing cells
Clinical trials
Unique feature: promoting β-cell regeneration
Pros and cons
-
Induced Pluripotent Stem Cells (iPS)
Characteristics
Isolation methods
Immunogenicity
Effect on immune response
Cytokine and chemokine profile
Generation of insulin-producing cells
Clinical trials
Unique features
Pros and cons
-
Future Directions
Safety
Virus-free reprogramming
Autologous vs. allogeneic
The benchmark: comparing stem cell-derived insulin-producing cells with mature β-cells
Conclusions
I. Introduction
Type 1 diabetes (T1D) is an autoimmune disorder in which the body's immune system attacks and destroys pancreatic insulin-producing cells (IPCs) (1). If left untreated, T1D can rapidly progress into a number of life-threatening conditions, including diabetic ketoacidosis, hyperglycemic hyperosmolar syndrome, and shock (2). Although insulin therapy promotes patient survival, it does not cure diabetes, nor does it necessarily prevent the possibility of the disease's devastating long-term complications, which can be serious and can affect every major body organ (3). As many as 3 million Americans may have T1D, and each year, more than 15,000–40,000 children are diagnosed with T1D in the United States (http://www.jdrf.org). A European study based on the EURODIAB register, which evaluated a population from 17 countries between 1989 and 2003, confirmed 29,311 new cases of T1D and projected a doubling of new cases of T1D in European children between 2005 and 2020 (4). The Diabetes Control and Complications Trial, a 7-yr longitudinal study, demonstrated that more stringent glycemic control (which can be obtained through intensive insulin therapy and through the preservation or restoration of β-cell function) lowered the incidence of nephropathy, retinopathy, and hypoglycemia (5). Many studies on β-cell replacement (e.g., with cadaveric pancreas or islet transplantation) have shown that restoration of glycometabolic control and C-peptide secretion is capable of slowing the progression of diabetic complications (6–8). From such data, β-cell preservation, through immunological approaches, has become an important option in the management of T1D (9). Historically, approaches aiming to cure T1D have made a negligible number of patients insulin-independent (10). A successful approach should preserve the remaining β-cells, restore β-cell function, and protect the replaced IPCs from autoimmune destruction (11, 12). The use of stem cells (SCs) holds great promise for the cure of T1D due to their propitious immunological characteristics and their regenerative capabilities (13, 14).
SCs are undifferentiated cells capable of self-renewal and giving rise to virtually any tissue or organ (15). SCs have been categorized as: 1) embryonic SCs (ESCs); 2) cord blood (CB) SCs; and 3) adult SCs (ASCs) (16). ESCs are considered clonogenic and self-renewing progenitor cells capable of generating all specialized cell types (17). ESCs have the capacity to differentiate into tissues of endodermal, mesodermal, and ectodermal origin (17) and proceed from the inner cell mass (ICM) of the human blastocyst, which at 5–6 d is a source of pluripotent ESCs (17). The use of ESCs is severely restricted by law in many countries, and new guidelines have recently been released by the National Institutes of Health (18). CB-SCs are collected at birth from the umbilical CB and are represented by different subtypes of SCs, i.e., self-renewable multipotent progenitor cells capable of differentiating into various lineages such as chondrogenic, adipogenic, osteogenic, and bone marrow tissues as well as blood components (19–21). ASCs are undifferentiated multipotent cells present in virtually every type of adult tissue and organ (14, 22). The most often used ASCs in T1D studies are considered to be bone marrow-derived mesenchymal SCs (MSCs) and hematopoietic SCs (HSCs). SCs may reestablish peripheral tolerance toward β-cells through remodeling of the immune response as well as through inhibition of autoreactive T-cell function (19). SC-derived IPCs are also capable of engrafting and reversing hyperglycemia in mice (23). To date, a number of protocols have generated IPCs in vitro using pluripotent cells from different sources, including human ESCs, induced pluripotent SCs (iPS), CB-SCs, and bone marrow-derived MSCs (11–13, 21). However, multiple issues remain when considering both regenerative and immunological uses of SCs. The primary problems when using SCs to replace β-cells are: 1) generating sufficient numbers of glucose-responsive IPCs; 2) maximizing the yield of desired IPCs; and 3) the lack of evidence that long-term survival of these newly generated IPCs has been well established thus far. Other issues related to immunological properties of SCs include: 1) eliminating the risks of tumorigenesis; 2) avoiding reprogramming strategies that involve viral vectors (13); and 3) establishing a stable and long-term reshaping of the immune system in the absence of major adverse events.
II. Embryonic Stem Cells (ESCs)
ESCs are obtained by harvesting blastocysts; they typically express Oct-4, Nanog-1, and Sox2 (three transcription factors involved in self-renewal that are markers of pluripotency and are associated with the maintenance of the undifferentiated state) (24) and possess substantial telomerase activity (25). These three transcription factors comprise a primary signaling axis, which promotes pluripotency and self-renewal (26). Oct4, Nanog-1, and Sox2 are essential for the early development and maintenance/proliferation of undifferentiated ESCs in culture by forming circuitry that consists of autoregulatory and feed-forward loops (26). ESC pluripotency is dependent upon autocrine signaling as well, for instance through leukemia inhibitory factor (LIF) and fibroblast growth factor (FGF) 4 (27, 28). LIF enhances Kruppel-factor activation 4 (Klf4), whereas Oct4 primarily induces Klf2, which preserves undifferentiation (28). Recent studies have attributed an even bigger role to LIF as an all-or-nothing cell-fate decision maker for self-renewal (29). At sufficient concentrations of exogenous LIF, the state of pluripotency is maintained, whereas at low concentrations of exogenous LIF, ESCs differentiate (29), and this effect precedes the loss of Oct4 and Nanog, suggesting an early step in the hierarchical control of differentiation (29). Microarray analysis identified FGF4 as a prime candidate for autocrine signaling and for the maintenance of pluripotency, proliferation, and homeostasis of ESCs (27), whereas an FGF4 isoform (FGF4si) can antagonize FGF4, thus inducing ESC differentiation (27). Fine-tuning of WNT, FGF, and LIF signaling network may open the door for regenerative medicine (30). Although extremely promising, ESC therapy is at the moment far from being introduced into clinical trials for T1D (13).
A. Characteristics
The unlimited capacity to proliferate by a process of self-renewal and the potential to terminally differentiate into one or more cell types in vivo and in vitro are the common characteristics that define both human and murine ESCs. Morphologically, murine ESCs grow in attached rounded masses, whereas human ESCs form flat colonies with well-defined edges (31). Both human and murine ESCs possess a high nucleus-to-cytoplasm ratio and display tight gap junctions among cells (31). Stage-specific embryonic antigens (SSEA) are expressed at different stages of development; whereas SSEA-1 is expressed on the cell surface at the preimplantation stage in murine embryos and upon differentiation by a subset of human ESCs, SSEA-3 and -4 are markers expressed only by undifferentiated human ESCs (32).
B. Isolation methods
ESCs are derived from the ICM of murine or human blastocysts through three different methods: immunosurgical isolation, mechanical isolation, or isolation of the intact embryo. Immunosurgical isolation consists first of dissolution of the zona pellucida using pronase enzyme; then, in incubation with whole serum antibodies, which attach to trophoblasts; and finally, in the transfer of blastocysts into a complement-containing medium (33). The ICM remains protected from the penetration of antibodies because of cell-cell connections within the outer layer of trophoblast cells (33). After washing away of the antibody residue, the blastocyst is transferred into a complement-containing medium and incubated until cell lysis is noted (33). The intact ICM is further rinsed and cultured with mitotically inactivated murine embryonic fibroblasts (MEFs) (33). Mechanical isolation can be achieved by mechanical dissection of the trophectoderm layer under a stereoscope using a needle (34). After the removal of the zona pellucida, the trophoblast layer is separated using a needle or pulled Pasteur pipettes. The intact ICM cells are then plated on irradiated MEFs (34). Isolation of intact embryos permits isolation of the ICM when it reaches sufficient size and could be removed mechanically, but it bears the risk of ICM differentiation (35). Contrary to mechanical and immunological methods, the method of plating whole embryos requires one passage of culture (35). The entire zona-free embryo is plated with mitotically inactivated MEFs and attaches to the feeder layer, which permits the growth of the ICM surrounded by the trophoblasts as a monolayer (35). When the ICM reaches a noticeable size, it is mechanically removed and propagated (35). After their isolation, ESCs are maintained in the undifferentiated state using similar methods for both human and murine ESCs. Murine ESCs can be cultured on MEF layers on gelatin-coated Petri dishes in the presence of fetal calf serum. Human ESCs can be cultured on MEFs or in conditioned medium containing FGF on Matrigel, laminin, or fibronectin substrates. After several days to 1 wk, proliferating colonies are removed and passed into new culture dishes. Passaging of ESCs entails using both mechanical and enzymatic techniques. It is important at this point that human ESCs remain in clusters/clumps to preserve the integrity of the culture (36).
C. Immunogenicity
The potent immunological capacities of ESCs have been recently discovered (37). Undifferentiated murine ESCs are nonimmunogenic, a property of these cells confirmed by various authors (25, 38, 39) (Fig. 1). The debate on the possible hypoimmunogenicity of differentiated ESCs and on the effect of inflammation on ESC immune recognition, however, remains unresolved. In vitro, allogeneic murine ESCs fail to stimulate T cells and are resistant to natural killer cell lysis (25). In vivo, allogeneic murine ESCs populate the thymus, spleen, and liver of sublethally irradiated allogeneic host mice without being rejected (25). Wood's group at Oxford (40) extensively studied the pattern of major histocompatibility complex (MHC) class II expression (the major determinant of immunogenicity in transplantation) of murine ESCs. Wood's group also demonstrated that higher expression of MHC class I was evident in ESCs differentiated into IPCs and that differentiated ESCs up-regulate MHC class I/II more rapidly when challenged with interferon (IFN)-γ, whereas MHC class II could not be induced on undifferentiated ESCs (40). Another study reported only a minor immune-response against human ESCs in mice conditioned to carry human leukocytes (39), confirming the lack of expression of MHC class II, as well as low expression of costimulatory molecules and MHC class I (39). The effect of inflammation on ESC immune recognition has been studied by Kofidis et al. (41), who showed that undifferentiated syngeneic and allogeneic green fluorescent protein (GFP)+ ESCs administered to treat myocardial infarction elicit a significant immune response consisting of CD3+ and CD11c+ cells with an increase in MHC class I expression on ESCs. It is therefore possible that, in vivo, inflammation may up-regulate expression of MHC antigens (25). It is also possible that the generation of lymphocytes from differentiated ESCs may equally explain the increase in immune response and inflammation. Taken together, these studies suggest that the future use of ESCs will likely require the use of systemic immunosuppression.
Fig. 1.
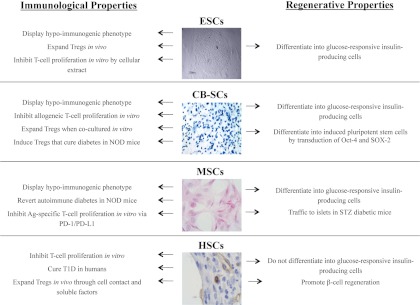
Immunological and regenerative properties of MSCs, CB-SCs, HSCs, and ESCs. Immunoregulatory and regenerative properties are described herein for each SC line, primarily focusing on T1D-related studies. Images show MSCs (hematoxylin and eosin staining of BALB/c bone marrow-derived MSCs obtained at P4); CB-SCs (chondrogenic differentiation of CB-SCs stained with toluidine blue); HSCs (murine CD34+ cells in the wound of a diabetic mouse); and ESCs [human ES cell line (H13) culture with mouse feeder cell].
D. Effect on immune response
ESCs have been shown to prevent immune activation in response to antigen-presenting cells in vitro and to have the capacity to promote allograft survival in vivo (Fig. 1) (37, 39, 42). In vitro, murine ESCs abrogate the alloimmune response, inhibit T-cell proliferation by a process that requires cell-cell contact (39, 42), and when injected in a rat cardiac allograft model, are able to protect allogeneic cardiac allografts (Fig. 1) (42). Zavazava and colleagues (37), through study of ESC-derived HSCs and their engraftment in sublethally irradiated allogeneic recipients, discovered that mixed chimerism could be established across MHC barriers, thus inducing long-term engraftment in sublethally irradiated recipients without the need for immunosuppression, the effect of which was mediated through the induction of intragraft FoxP3+ Tregs (37). In a recent study, authors were able to induce hematopoietic chimerism through rat allogeneic ESC transplantation, which allowed long-term graft acceptance of donor hearts (43). No studies on the effect of ESCs on experimental diabetes are available so far.
E. Cytokine and chemokine profile
The cytokine/chemokine profile of undifferentiated ESCs is not well-studied, particularly for human ESCs. An elegant paper by Lee et al. (44) showed that undifferentiated ESCs released few cytokines at levels significantly higher than fibroblast or MSCs. Vascular endothelial growth factor (VEGF), IGF-I, and IL-10 are considerably produced by ESCs, whereas ESCs do not release IL-1, IL-6, and TNF-α. IL-6 production by ESCs was augmented under hypoxic conditions through a reactive oxygen species mechanism involving phosphorylation of the stress-activated protein kinase family (p38/c-Jun N-terminal kinase) (44). Guo et al. (45) showed that stromal cell-derived factor-1/CXCL12 is released by murine ESCs cells, thus enhancing survival, chemotaxis, and hematopoietic differentiation of murine ESCs. Many of these cytokines are involved in diabetes onset. IL-1 (46), IL-6 (47), IL-12, IL-21, IL-27 (48), as well as chemokine axis [e.g., CXCR4/CXCL12 (49) and CXCL10] have a role in diabetes onset; thus, it is possible that some of the effects obtained with SCs may be mediated through cytokine/chemokine modulation.
F. Generation of insulin-producing cells
One of the first studies describing ESC-derived IPCs was published in 2000 by Soria et al. (50) and was seen as quite controversial. ESC-derived IPCs were selected by placing a neomycin-resistant gene under the insulin promoter's transcriptional regulation, and cells were then transplanted under the kidney capsule, leading to a transient correction of glycemia. However, surgical removal of the graft did not cause hyperglycemia, raising many doubts about the effect of these IPCs (50). The spontaneous differentiation of murine ESCs in vitro generates only a small fraction of IPCs, which can be increased by the selection of nestin-positive progenitor cells. These cells displayed some in vitro-regulated insulin release but failed to normalize high blood glucose level once transplanted in mice (51). The presence of LIF in the medium can be an important factor determining the maintenance of an undifferentiated state and the prevention of spontaneous differentiation, which in turn may represent an obstacle for a successful IPC generation (52). However, ESCs were successfully cultured in LIF-free medium (52), and the fibroblast monolayer itself may secrete LIF in LIF-free medium (52). In other studies where ESC-derived IPCs were transplanted under the kidney capsule or into the spleen, a transient correction of glycemia was observed, but treatment was associated with tumors in the kidney and the spleen (53). A novel five-step protocol for differentiation of human ESCs to IPCs has been recently established, mimicking as closely as possible the physiological multistep process of pancreatic development. First, human ESCs are transitioned from mesendoderm to definitive endoderm (DE) using high concentrations of activin A, and the human ESC-derived DE is tested for the expression of DE markers (e.g., SOX17 and CXCR4) by fluorescence-activated cell sorting analysis. The further differentiation in pancreatic-like cells is characterized by reduction of the DE marker CXCR4, the maintenance of SOX17 expression, and the appearance of the gut-tube markers HNF1B and HNF4A. The addition of retinoic acid and 3-keto-N-(aminoethyl-aminocaproyl-dihydrocinnamoyl) cyclopamine to gut-tube endoderm cells results in a rapid increase of the expression of pancreatic and duodenal homeobox factor 1 (PDX-1); and other markers that are required for pancreatic bud specification. During stages 4 and 5, PDX-1-expressing cells differentiate to pancreatic and endocrine lineages. At about 15 d of differentiation, endocrine cells are produced, so that a cell population with pancreatic identity has been generated from human ESCs (54, 55). In 2009, Lumelsky et al. (56) proposed to produce highly enriched nestin-positive cells from embryoid bodies plated in serum-free medium. Cells were then expanded in the presence of FGF and nicotinamide, followed by mitogen withdrawal to promote the cessation of cell division and differentiation (56). This novel protocol minimized the differentiation to neuronal progeny, resulting in a population of IPCs with β-cell characteristics but lacking neuronal features (57). After implantation into diabetic mice, these IPCs caused a time-dependent improvement of glucose metabolism; however, the formation of teratomas after implantation was documented in both protocols (58). In 2009, Li et al. (59) obtained ESC-derived IPCs that normalized hyperglycemia but also induced a state of hypoglycemia, which can be attributed to the IPC immature developmental state and to their “insulinoma” phenotype. Zhang et al. (60) developed a highly efficient step-wise pancreatic differentiation strategy using activin A and wortmannin to induce DE formation starting from the human ESC line H9, followed by treatment with retinoic acid to induce pancreatic specialization. After 20 d of induction, they observed approximately 25% of insulin-positive cells (as assayed by flow cytometry analysis), which released insulin/C-peptide in response to glucose stimuli in a controlled manner comparable to that of adult human islets (61). Mature β-cell-specific markers, such as NKX6-1 and PDX-1, were coexpressed by these SC-derived IPCs, thus indicating a similar gene expression pattern to adult islet β-cells in vivo (61). A recently published protocol suggested the use of Exendin-4 to increase insulin synthesis (62). Further investigations are required to better study the mechanism of pancreatic specialization and maturation in vitro, to improve differentiation efficiency, and to promote long-term function of ESC-derived IPCs after transplantation. In our hands, ESC differentiation into IPCs has always resulted in weak insulin staining (Fig. 2).
Fig. 2.

ESC-derived IPCs poorly produce insulin. ESCs were differentiated into IPCs, and cells were stained for both insulin and C-peptide. Isolated pancreatic islets were used as controls. ESC-derived IPCs displayed weak insulin staining, whereas C-peptide expression was more detectable.
G. Clinical trials
The two major limitations in the use of ESC-derived IPCs in clinical trials are tumor development and glucose sensing. Novocell scientists have developed a method to generate IPCs from human ESCs on a large-scale (54, 55) and have suggested encapsulation of these IPCs. This strategy consists of encapsulating IPCs in a durable and retrievable device, then implanting the device sc. Soft and permeable coatings have been developed to conceal cells from the immune system and to allow them to sense glucose and to subsequently secrete insulin in response (63). The Novocell team, guided by Emmanuel Baetge and Jeffrey Bluestone, were recently asked by the California Institute of Regenerative Medicine to develop a therapy for T1D using these encapsulated ESC-derived IPCs. Encapsulation appears to be a promising method to ensure prevention of tumor development and immune system attack (64).
H. Unique features: ESC cellular extract (EXT)-based reprogramming
Treatment with EXT seems to be a potential therapy, particularly given their immunomodulatory (i.e., inhibition of T-cell proliferation) and reprogramming properties (65), as well as their ability to inhibit T-cell proliferation in vitro and to reduce graft-vs.-host disease (GVHD) in vivo (66). This interesting option, while not yet explored in T1D, is based on the use of EXT to induce cellular reprogramming. These extracts are obtained through cell lysis and sonification of ESC lines. The paramount benefit of these cellular extracts, from both human and mouse ESCs, is that they retain the immunomodulatory and regenerative properties of intact cells (67). Between 12 and 24 μg of total protein obtained from ESCs, but not from fibroblasts or fibroblast extract, effectively prevented T-cell proliferation in an allogeneic mixed lymphocyte reaction (MLR). This EXT abrogates dendritic cell (DC) maturation, with reduced expression of costimulatory molecules and maturation markers (CD80, HLA-DR, and CD83) and lower secretion of IL-12(p40) (67). Accordingly, human EXT-treated DCs were hypoimmunogenic and did not stimulate allogeneic T cells (67). These surprising immunological properties coupled with the ability of EXT to dedifferentiate cells (e.g., the NIH3T3 cell line) rendered EXT an exciting option to be explored. Genome-wide expression profiling of EXT-treated NIH3T3 cells revealed the reactivation of ESC-specific transcripts (68). EXT induced epigenetic reprogramming of treated cells with demethylation of the Oct4 promoter, hyperacetylation of histones 3 and 4, and decreased lysine 9 demethylation of histone 3 (68). These EXT-reprogrammed NIH3T3 cells retain the ability to transdifferentiate into skeletal muscle, endothelial cells, and cardiomyocytes (68). Potentially, EXT-based cell reprogramming may offer a platform to generate functional multipotent stem-like cells from terminally differentiated somatic cells without the introduction of retrovirally expressed transgenes or ESC fusion (68, 69).
I. Pros and cons
A primary hurdle to utilization of ESCs is potential tumorigenesis after ESC administration. ESCs can give rise to teratomas and teratocarcinomas in humans (70). The substantial number of rounds of replication that these cells undergo before transplantation may lead to the accumulation of potentially oncogenic chromosomal abnormalities (71). Moreover, self-renewal, rapid proliferation, lack of contact inhibition and telomerase activity are some of the shared characteristics between ESCs and cancer cells (72). An efficient way to reduce the risk of tumor/teratoma development could be increasing differentiation status and commitment to the cell type of interest before transplantation into the patient. Even cell sorting using surface antigen markers for undifferentiated (negative selection) or committed (positive selection) cells can partially contribute some oncological risk. However, these approaches are unlikely to be viable for the production of large numbers of cells needed for clinical use. A “kill-gene” strategy (such as timidine kinase) or the packaging in micro/macrocapsules constitute other strategies under evaluation.
III. Cord Blood Stem Cells (CB-SCs)
CB has elevated frequencies of CD34+ and CD105+ cells, which are substantially higher than in peripheral blood (73); this substantial naiveté is confirmed by the high percentage of Oct4+ cells, which hold regenerative potential (Fig. 3) (74). CB is reported to be a source of various SCs, including HSCs, MSCs, and the very small embryonic-like SCs (VSELs), which, according to some investigators, are suitable for regenerative studies because they form in vitro colonies easily, display stable telomeres, and display almost null oncogenicity (73, 75). CB-MSCs can give rise to chondrogenic, adipogenic, and osteogenic tissues (76), whereas CB-HSCs show the ability to differentiate into bone marrow and blood components including, for instance, T cells (77). The possibility of obtaining islet-like clusters from CB-SCs has been explored, but it needs to be validated and tested more before it can become a legitimate perspective for therapy of T1D (78).
Fig. 3.

The high percentage of CD105 and Oct4 positive cells in CB compared with peripheral blood (PB) confirmed the considerable naiveté of CB-SCs and their suitability for regenerative use.
Previous papers have attempted to establish whether CB-SCs have the capacity to differentiate into insulin-expressing β-cells in humans (79). Huang et al. (79) studied pancreata obtained at autopsy from 11 individuals who had prior opposite-sex CB transplantation to reconstitute hematopoiesis. A higher percentage of XX-positive insulin-expressing islets (3.4 ± 0.3%) was detected in males who received CB from females compared with untransplanted controls (0.32 ± 0.05%) (79). CB-SCs have the capacity to differentiate into insulin-expressing cells in nondiabetic humans, but it remains to be established whether these cells possess the properties of β-cells (79). Another lesson can be learned from studies of in utero HSC transplantation (80), which suggest that pre- and peridelivery conditions are particularly favorable for SC applications, due to the naiveté of the fetal immune system, which may allow the engraftment of transplanted HSCs without the requirement for myeloablation or immunosuppression (80).
A. Characteristics
CB-SCs make up a diverse family of SCs and include undifferentiated multipotent progenitor cells capable of self-renewal for many cycles of cellular division (21, 81) with high proliferative and differentiation rates (82, 83). When CB mononuclear cells are seeded in culture, they develop SC colonies resembling MSC-like features. These CB-MSCs display a fibroblast-like shape, as revealed through microscopy analysis after 4–5 d of culture (73). CB-derived MSCs have been shown to be positive for mesenchymal markers such as desmin, nestin and vimentin (84). Over the course of postisolation culture, these cells acquire expression of CD54, CD106, SH2, SH3, and SH4 and lack expression of CD14, CD34, and CD45 (73). CB-derived HSCs can differentiate into both bone marrow tissues and blood components and are now considered an appropriate source of HSCs for hematological transplants (85). Of high scientific relevance is the presence of a population of VSELs in CB (86), which display embryonic characteristics (87). VSELs are CD34+, CD133+, and CXCR4+, and they express specific embryonic markers including SSEA-4, Oct-4, and Nanog (88). These cells are considered totipotent progenitors and can differentiate into every tissue or organ-specific cell (89). However, the totipotency of VSELs is controversial and has not been sufficiently demonstrated or reproduced by others (89).
B. Isolation methods
Using the “waterworks method,” 60–80 cc of CB can be harvested at birth from the umbilical cord, without exposing the child or the mother to any procedural risk, and then can be cryopreserved for many years (73). Evidence that CB-SC cryopreservation does not alter cellular functionality was presented in 1992 by Broxmeyer et al. (90), who demonstrated the viability of 5-yr cryopreserved CB-SCs. In 2010, Broxmeyer (91) demonstrated efficient recovery of CB-SCs stored for more than 24 yr. The U.S. National Cord Blood Program stated that more than 15,000 CB transplants have been performed worldwide through 2009.
C. Immunogenicity
CB contains various subtypes of SCs (MSCs, HSCs, vascular SCs, and VSELs), but most studies conducted thus far have focused on the mononuclear cell fraction of CB or on CB-MSCs in vivo (73). Moreover, we should remember that all studies have been performed using human CB-SCs. Undifferentiated CB-MSCs express very low levels of MHC class I, but not class II, CD40, CD40L, CD80, CD86, or costimulatory molecules like B7.1 and B7.2, and are incapable of inducing allogeneic peripheral blood mononuclear cell proliferation (92) (Fig. 1). CB-MSCs, once differentiated (for instance in hepatocytes), did not express MHC class II and were not able to induce lymphocyte proliferation in a MLR culture, contrary to undifferentiated CB-MSC (93). A study conducted on CB units stored at Massachusetts General Hospital confirmed the insubstantial immunogenicity and low levels of MHC class I/II of CB-SCs, as well as the fact that IFN-γ stimulation increases MHC class I/II (94). Immunogenicity was assessed in vivo in a fully MHC mismatched model of human CB-SC transplantation into swine using ex vivo assays including MLR, flow cytometry, and serum alloantibody detection (94). A single injection of MHC-mismatched unactivated CB did not induce a detectable immune response, yet when injected in an inflamed region, injected repeatedly in the same region, or stimulated with IFN-γ before injection, CB were found to be immunogenic (94). Therefore, regenerative strategies based on allogeneic CB-SCs should consider the use of immunosuppression.
D. Effect on immune response
In vitro, CB-MSCs dose-dependently inhibit T-cell proliferation in an MLR assay and after anti-CD3/-CD28 or phytohaemagglutinin stimulation (Fig. 1) (92, 95). Additionally, authors have found that CB-MSCs suppress the function of mature DCs (92). Using transwell systems, these studies have demonstrated an inhibitory mechanism that depends on both cell contact and soluble factors (92). Addition of prostaglandin synthesis inhibitors almost fully abrogated the immunosuppression activity of CB-MSCs, identifying prostaglandin E (2) as an important soluble mediator of CB-MSC function (95). CD14+ monocytes were found to be able to significantly enhance the immunosuppressive effect of CB-MSCs in a dose-dependent manner (95). The abundance of Tregs in CB has also been used as a basis for the use of CB as cellular therapy. Although the rarity of CD4+CD25high Tregs in peripheral blood has significantly slowed their use in clinical trials (73), these cells can be identified in CB and hold promise in the therapy of autoimmune diseases and other disorders (Fig. 1) (73). Blazar's group (96, 97) has extensively studied CB-Tregs and has found that CB was a superior source for Treg isolation and cell line generation compared with adult blood (96, 97), thus suggesting that banked CB specimens may serve as a readily available source of Treg cells for immunotherapy (96, 97). CB CD4+CD25+ cells consistently exhibit potent suppressor activity, with more than 95% suppression of allogeneic MLR, which is predominantly independent of IL-10 and TGF-β (96, 97).
Zhao et al. (93) successfully treated established autoimmune diabetes in nonobese diabetic (NOD) mice with autologous CD4+CD62L+ Tregs obtained through coculture with human CB-SCs, and investigators concluded that CB-SC-derived Tregs could reverse diabetes by promoting β-cell regeneration and reconstitution of islet architecture (Fig. 1). To increase the number of CB-SC-derived Tregs, several expansion protocols have been developed. Interestingly, Hippen et al. (98) proposed a model in which antigen-specific CB-Tregs could be expanded more than 1250-fold during an anti-CD3/-CD28 stimulation in the presence of artificial antigen-presenting cells. When used in vivo, these expanded Tregs reduced GVHD lethality with a survival rate of 75%, whereas the percentage in control mice was 0% (98). Experimental data suggested a positive effect of CB-SCs on experimental diabetes.
E. Cytokine and chemokine profile
With regard to cytokine and chemokine profile, IL-8, monocyte chemotactic protein-1, and IL-1α were consistently produced by CB mononuclear cells irrespective of culture condition (99). Liu et al. (100) reported IL-6, IL-8, tissue inhibitor of metalloproteinases 1 and 2 as the cytokines produced at highest levels by human CB-MSCs. Conversely, IL-4, IL-5, IL-7, IL-13, TGF-β1, TNF-α, and TNF-β were not produced under normal growth conditions (100). The expression of CXCR4, the IL-8 receptor (CXCL8), CXCR1, and CXCR2 was confirmed in different studies, suggesting a role for these molecules in CB-SC function (101–104). The cytokine profile also has the potential to be altered during differentiation. Penolazzi et al. (105) demonstrated the ability of CB-MSC-derived osteoblasts to produce significant amounts of soluble immune mediators different from those secreted from undifferentiated CB-MSCs (e.g., monocyte chemotactic protein-1 and VEGF).
F. Generation of insulin-producing cells
In 2004, Pessina et al. (106) identified the expression of a set of markers in CB-SCs, namely nestin, CK-8, and CK-18, as well as a group of transcription factors (Isl-1, Pax-4, and Ngn-3) all expressed by the precursors of pancreatic cells or by endocrine pancreatic cells. In view of this finding, the ability of CB-SCs to differentiate in vivo into IPCs was evaluated by Yoshida et al. (107). One to 2 months after transplant of CB-mononuclear cells into immunodeficient mice, immunofluorescence and fluorescent in situ hybridization analysis revealed the appearance of very few CB-derived IPCs (107). The evidence that human CB-SCs can be engineered to IPCs was first presented by Denner et al. (108), showing that IPCs were generated from CD34+ and CD133+ following a differentiative protocol during which nestin+ cells were first expanded [using basic FGF (bFGF)] and subsequently differentiated in a nicotinamide-supplemented medium (108). Considering that insulin was present in the culture medium, the de novo production of insulin from new IPCs was evaluated from the production of C-peptide as well as with immunostaining using anti-C-peptide IgG (108). Subsequently, Sun et al. (109) successfully generated IPCs by differentiating CB-VSELs, using an N2 medium plus nicotinamide containing DMEM/F12, progesterone, putrescine, laminin, insulin, sodium, selenite, transferrin, fibronectin, B27 media supplement, and fetal calf serum. After approximately 7 d of culture, generated IPCs and insulin/C-peptide expression were assessed by immunostaining (109). The de novo expression of C-peptide was paralleled by loss of OCT-4 expression, confirming the transition of CB-VSELs toward a differentiated cell line (109). Interestingly, Sun et al. (109) report that exposure to high-glucose medium was essential to generate IPCs. Gao et al. (110) cultured CB-MSCs in medium containing high-glucose, nicotinamide, retinoic acid, exendin-4, and epidermal growth factor (EGF) and demonstrated the appearance of IPCs expressing pancreatic markers, PDX-1, Pax- 4, Glut-2, and Ngn-3, identified by RT-PCR. Hu et al. (111) evaluated the efficiency of human CB-MSC-derived IPCs in an animal model. Insulin-positive immunostaining was evident beneath the kidney capsule (where IPCs were placed) with an increase in insulin levels in serum of transplanted mice but not in the control group, without affecting blood glucose levels (111). Recently, new approaches to improve the generation of IPCs have been tested: 1) the addition of taurine to the media used to cryopreserve CB-SCs has been shown to be capable of enhancing viability and allowing maintenance of expression of proendocrine transcription factors (78); and 2i) the inhibition of Notch signaling in CB-MSCs has demonstrated enhancement of the differentiation process of CB-MSCs into IPCs; however, these cells did not display an adequate response to glucose challenge (111).
G. Clinical trials
Haller et al. (112) have published data from a 1-yr follow-up phase II clinical trial on 15 patients (aged 1 to 18 yr) with newly diagnosed T1D who received a single iv infusion of undifferentiated autologous CB cells, previously stored in a private bank (Table 1). One year after treatment, CB-treated patients displayed no major differences compared with historical controls, despite slight improvement of insulin requirement and of glycometabolic control, without any significant adverse reaction after infusion of CB (112). Patients also showed an increase in peripheral FoxP3+ Tregs after 6 months of treatment. The 2-yr follow-up data on patients receiving umbilical CB are needed to better identify the clinical advantages (if any) offered by autologous CB cell infusion. A phase I clinical trial is ongoing in Germany (http://www.ClinicalTrials.gov), in which newly diagnosed T1D patients are treated with their own CB samples stored at the cord bank VITA34 (Table 1).
Table 1.
A brief presentation on active clinical trials evaluating SC-based therapy in T1D
| Study | Year | Identifier | Site/PI | Status/recruited pts |
|---|---|---|---|---|
| MSCs | ||||
| Phase I/II, autologous MSC infusion in new onset T1D | 2010 | NCT01068951 | Uppsala University Hospital/P. O. Carlsson | Upcoming |
| Phase II, allogeneic MSC (prochymal) infusion in new onset T1D | 2008 | NCT00690066 | Osiris Therapeutics, Inc. | Ongoing/not recruiting |
| CB-SCs | ||||
| Phase II, autologous CB-SC infusion with vitamin D and omega-3 fatty acids in recent onset T1D | 2009 | NCT00873925 | University of Florida/M. J. Haller | Follow-up with 23 pts/recruiting 15 pts |
| Phase I, autologous CB-SC infusion in new onset T1D | 2009 | NCT00989547 | Universität München/A. G. Ziegler | Recruiting (if CB stored at Vita34 bank) |
| HSCs/MSCs | ||||
| Phase I/II, autologous bone marrow transplant in T1D/T2D | 2006 | NCT00465478 | Shandong University/B. Kong | Recruiting 200 pts |
| HSCs | ||||
| Phase I/II, autologous HSC transplant in new onset T1D | 2010 | NCT01121029 | Hospital Gonzalez/F. J. Lavalle | Recruiting 15 pts |
| Phase II, autologous nonmyeloablative HSC transplant in new onset T1D | 2008 | NCT00807651 | Shanghai University/G. Ning | Recruiting 30 pts |
| Phase II, autologous bone marrow arterial infusion in long-standing T1D | 2009 | NCT00971503 | University of Moron/A. D. Mesples | Recruiting 34 pts |
| Phase I/II, autologous HSC transplant in new onset T1D | 2003 | NCT00315133 | University of Sao Paulo/J. Voltarelli | Follow-up with 23 pts/recruiting 24 pts |
Trials have been divided according to the SC line used: bone marrow-derived MSCs, CB-SCs, and HSCs. PI, Principal investigator; pts, patients.
H. Unique feature: naive status
The main and unique feature compared with the ASCs is the considerable naiveté of CB-SCs, which render them particularly suitable for regenerative purposes (19–21).
I. Pros and cons
Several advantages are attributed to the use of CB-SCs compared with other sources: 1) their safe collection; 2) their substantial naiveté (and thus plasticity), associated for instance with lower rates of GVHD; and 3) their greater regenerative potential compared with ASCs (73). Few disadvantages are correlated with the application of CB-SCs: 1) low numbers of SCs collected; and 2) the need for more extensive clinical trials as well as long-term follow-up to confirm their utility (73).
IV. Mesenchymal Stem Cells (MSCs)
MSCs are multipotent cells localized in several tissues including CB, bone marrow, and adipose tissue (113); however, we will primarily refer to the cell derived from the most widely used source: the bone marrow-derived MSCs. MSCs are multipotent progenitor cells, capable of self-renewal and differentiation into adipogenic, chondrogenic, and osteogenic lineages (114), and they can be isolated from bone marrow in a low-density cellular culture by removal of nonadherent cells (115).
A. Characteristics
Although universal agreement on a specific set of markers is lacking (19), these cells are generally negative for the HSC marker CD34, endothelial CD31 and CD117, red blood cell glycophorin A, leukocytic CD45, monocytic/macrophage CD14, and lymphocytic CD11a/LFA-1, whereas they are commonly positive for CD105 (SH2), CD73 (SH3/4), CD44, CD166 (vascular cell adhesion protein), CD54/CD102, CD49, and stromal antigen 1 (115). Interestingly, differential expression of MSC-related markers has been observed between murine MSC models in diverse strains and human MSCs. For example, a comparative study performed by Peister et al. (116), in which MSCs were characterized and compared from four inbred strains of mice (C57BL/6J, BALB/c, FVB/N, and DBA/1), revealed that murine MSCs can be either positive or negative for CD34 (but with variable expression between strains) and negative for FLK-1 (VEGF-R2) and CD90 (Thy1) (116), whereas human MSCs are always negative for CD34 and positive for FLK-1 and CD90. In addition, in the same study, investigators demonstrated higher expansion rates in murine compared with human MSCs (116). The above-mentioned studies and many others have confirmed MSC heterogeneity, which is reflected in different results obtained in the various studies, particularly when murine and human MSCs are used respectively. The primary controversial issue for MSCs, i.e., oncogenic potential, is, on the other hand, shared by murine and human MSCs. Preclinical data have shown that murine MSCs, during proliferation, develop chromosomal aberrations responsible for oncogenic transformation (117–119). Miura et al. (117) reported that, after long periods of in vitro doubling passages, murine MSCs become malignant cells and, when transplanted in immunodeficient mice, give rise to fibrosarcomas; investigators found that the transforming event was due to an increase in both c-Myc expression and telomerase activity (117). Similar results were obtained subsequently by Tolar et al. (118) and by Li et al. (119), who interestingly showed that the appearance of the tumor phenotype was also correlated to a mutation in the p53 gene. Human MSCs can be susceptible to neoplastic transformation and can also develop sarcomas as a consequence of mutations in genes responsible for cell cycle regulation or due to an abnormal activation of oncogenes (120–122). Wu et al. (123) demonstrated that aggressive fibromatosis tumors express genes and cell surface markers characteristic of MSC. In mice genetically predisposed to develop aggressive fibromatosis tumors, authors observed a proportion between the number of MSCs present and tumor formation (123) and also observed that the maintenance of an immature state by β-catenin supports tumorigenesis. Moreover, cancer/testis antigen expression was detected on MSCs isolated from different tissues (124), and MSCs may promote breast cancer metastasis through facilitation of epithelial to mesenchymal transition (125) or through the formation of mammospheres (126).
Fiorina et al. (14) showed that autologous, but not allogeneic, murine MSCs transplanted into BALB/c mice induce the formation of neoplasms, most likely because the proliferation of allogeneic MSCs is mitigated by host immunosurveillance mechanisms. On the other hand, increasingly more studies are showing the potential therapeutic opportunity offered by MSCs as a cure for cancer (127–130).
B. Isolation methods
MSCs are generally isolated from the bone marrow aspirate by density gradient fractionation (131) and subsequently separated from hematopoietic cells thanks to their ability to adhere to the plastic surface of culture dishes (132). Human MSCs represent 0.001–0.01% of the nucleated cells present in bone marrow (133); due to this low frequency and to obtain enough cells for clinical applications, several procedures have been developed to expand MSCs in vitro. Schallmoser et al. (134) developed a single-phase method for the expansion of human MSCs without the need for prior selection of mononucleated cells. Briefly, bone marrow aspirates were diluted in α-MEM containing heparin, L-glutamine, and pooled human platelet lysate. After the removal of nonadherent cells, MSCs were cultivated on a new α-MEMpHPL until confluence (reached at d 10). Subsequently, MSCs were harvested through the use of trypsin/EDTA and were cryopreserved (134). Interestingly, after this protocol, investigators showed a 4-fold increase in MSC number (134).
C. Immunogenicity
Bone marrow-derived-MSCs, obtained from bone marrow, have been shown to possess a wide range of immunomodulatory characteristics in vitro due to their hypoimmunogenic phenotype and their ability to release antiinflammatory cytokines, as well as their ability to form cell-to-cell inhibitory interactions (Fig. 1) (19). However, it is unclear whether allogeneic MSCs retain their immunoprivilege and whether after differentiation into IPCs they lose their hypoimmunogenicity. Eliopoulos et al. (135) examined the survival of murine C57BL/6 MSCs, engineered to release murine Epo, implanted sc in either syngeneic C57BL/6 mice or fully MHC-mismatched BALB/c mice (allogeneic). Although hematocrit rose rapidly from baseline levels and remained elevated for more than 200 d in syngeneic recipients, in allogeneic recipient mice, hematocrit rose only transiently and rapidly declined to baseline values, and allogeneic MSC implants were shown to be infiltrated by CD8+ natural killer T cells (135). Repeated implantations of these engineered MSCs resulted in a sensitization of mice and acquired refractoriness to the Hct response, consistent with alloimmunization to donor Epo+ MSCs (135). Another study, however, contradicted these findings. Chen et al. (136) compared engraftment of allogeneic and syngeneic MSCs with allogeneic fibroblasts in a murine model of wound healing. No decrease was observed in the number of GFP-expressing allogeneic and syngeneic MSCs, whereas allogeneic fibroblasts declined rapidly (136), and no significant infiltrate was seen when allogeneic and syngeneic MSCs were used (136). Transition from an immunoprivileged to an immunogenic state after differentiation was demonstrated in other studies and was associated with an alteration in MHC-immune antigen profile (137, 138). Recently, authors have claimed that the expression of a serine protease inhibitor (SPI6) allows MSCs to escape from the host immune response (139). Few studies examine human subjects injected with MSCs (140), and apparently these results show that a potential dichotomy may exist between human and murine MSCs and that infused human MSCs are only weakly immunogenic, which can potentially validate the clinical use of MSCs from HLA-mismatched donors (140). However, taking into account the compelling evidence from preclinical studies, it seems that MSCs (when used in an allogeneic setting) are not intrinsically immunoprivileged and cannot serve as a “universal donor” in immunocompetent MHC-mismatched recipients (135).
D. Effect on immune response
In vitro, MSCs inhibit T-cell proliferation through a PD-1/PD-L1-mediated mechanism, inhibit activity of both naive and memory antigen-specific T cells (141), and support T-cell survival and maintenance of a quiescent state, possibly through the expression of indoleamine 2,3-dioxygenase, because the addition of tryptophan significantly restores allogeneic T-cell proliferation (142, 143) (Figs. 1 and 2). MSCs inhibit both maturation and migration of monocytes/myeloid cells (144); reduce DC expression of Ia, CD11c, CD80, CD86, and CD40 while increasing CD11b expression (145); alter DC cytokine secretion profile; induce a tolerant DC phenotype; and reduce DC TNF-α secretion (Figs. 1 and 4) (146, 147). MSCs inhibit IL-2-mediated natural killer cell proliferation and IFN-γ production (148) and also inhibit B-lymphocytes proliferation and chemokine expression (149). In a murine model of multiple sclerosis, autologous MSCs administered before the onset of the disease ameliorated the disease and reduced demyelination (150), whereas allogeneic murine MSCs have been shown to reduce the severity of arthritis in murine models and to modulate the expression of inflammatory cytokines (151). Human MSCs from healthy donors reduced serum levels of anti-double stranded DNA antibodies and proteinuria in lupus-prone (MRL/lpr) mice (152). When mice deficient in Tregs were treated with human MSCs, they were protected from the onset of spontaneous autoimmune enteropathy (153). Finally, evidence of the immunomodulatory function of MSCs was shown in diabetic NOD mice, which were cured by the infusion of allogeneic and congenic MSCs (Figs. 1 and 4) and by other in vitro autoimmune relevant assays (14, 154). MSC-treated NOD mice displayed lesser islet infiltration of CD3+ and B220+ cells and a relative increase in FoxP3+ infiltrating cells (Fig. 4) (14). BALB/c-MSCs (allogeneic), but not NOD-MSCs (autologous), delayed the onset of hyperglycemia in 10-wk-old NOD mice and reversed diabetes in seven of eight newly hyperglycemic NOD mice (Fig. 4) (14). NOD mice treated with autologous, but not allogeneic, MSCs developed aggressive tumors, with transcriptome analysis revealing increased expression of genes promoting cell cycle and proliferation (14). The detected tumors formed nodular masses (0.1–0.3 cm in diameter) in the legs, tails, lungs, and livers of animals (14). Muscle, nerve, skin, bone, peritoneum, and colon were invaded by the tumors as well (14). In the lung, the tumor formed nodular masses with alveolar wall infiltration, containing a homogeneous population of malignant spindle cells in sheets and fascicles, which lacked malignant or benign-looking epithelium. A detailed electron microscopy analysis revealed the presence of compact intertwined processes covered by basal lamina, consistent with Schwann cell differentiation, suggesting a malignant peripheral nerve sheath tumor (14). Human MSCs, administered to NOD/severely compromised immunodeficient (SCID) mice rendered hyperglycemic with streptozotocin (STZ) (155) displayed lower blood glucose levels and increased pancreatic islet numbers (155). Madec et al. (156) confirmed this positive effect of allogeneic MSCs in NOD mice, with a decrease in the incidence of diabetes from 100 to 59% at 36 wk of age. Experimental data strongly suggested an effect of MSCs on experimental diabetes.
Fig. 4.
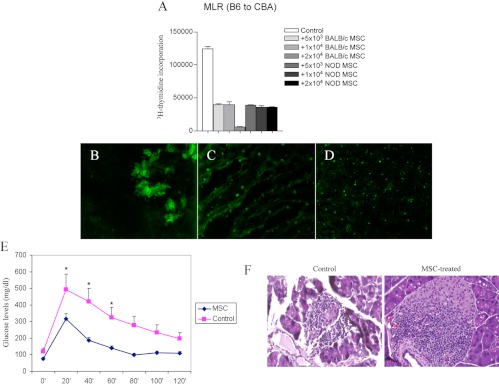
Bone marrow-derived MSCs from different third-party strains (NOD or BALB/c) inhibited the MLR (A). Once injected iv into NOD mice, MSCs obtained from BALB/c mice were detectable in the pancreas at d 1 (B), in the pancreatic lymph nodes after 7 d (C), and in the spleen up to 30 d after injection (D). NOD mice injected with BALB/c MSCs showed a near-normal glucose profile upon peritoneal glucose testing as well as reduced insulitis (14 d after injection) (E and F).
E. Cytokine and chemokine profile
The profile of soluble immunosuppressive factors is complex, and MSCs have been shown to produce (among others) nitric oxide and indoleamine 2,3-dioxygenase (157, 158). MSCs express different chemokine receptors, including CXCR4, which has been shown to be expressed at substantial levels (101). Although it will be important to provide an explanation with regard to what these profiles may represent in terms of actual pathophysiological events, the task is not a simple one due to the lack of literature and particularly due to the heterogeneity of MSCs.
F. Generation of insulin-producing cells
The ability of human and murine MSCs to differentiate into IPCs has previously been evaluated to identify novel therapeutic strategies to regenerate pancreatic islets (Table 2) (155, 159–161, 257–263). Li et al. obtained IPCs by transfecting human MSCs with an adenovirus containing integrated human PDX-1 cDNA (a transcription factor pivotal in the control of pancreatic development and in expression of the insulin gene) (261). After confirming the expression of the PDX-1 protein, human PDX+ MSCs were cultured in serum-free medium containing glucagon like peptide-1, and the resulting cells displayed expression of a set of pancreatic genes including PDX-1, brain4, Ngn3, insulin, glucagon, Glut-2, and glucokinase, suggesting potential glucose sensitivity of these human PDX+-MSC-derived IPCs (261). These IPCs demonstrated the ability to produce insulin, as revealed by immunofluorescent staining using anti-insulin and anti-C-peptide antibodies (261). When transplanted under the renal capsule in STZ-induced diabetic BALB/c mice, these human PDX+-MSC-derived IPCs normalized glucose levels 7–14 d after transplant, whereas nephrectomy induced recurrence of hyperglycemia (261). Sun et al. (262) were able to differentiate human MSCs, collected from diabetic patients, into IPCs following a three-step protocol without genetic manipulation, in which cells were: 1) cultured in serum-free high-glucose DMEM supplemented with β-mercaptoethanol; 2) seeded in medium containing B27, bFGF, EGF, L-glutamine, and nonessential amino acids for 8 d; and 3) transferred to medium supplemented with nicotinamide, β-cellulin, and activin-A for another 8 d. Although the generation of iPS as shown in this study does not represent a major technical advance compared with other studies, authors demonstrated here for the first time that persistence of long-term hyperglycemia in patients with T1D does not constitute a factor against the generation of iPS. After differentiation, these IPCs displayed high expression of PDX-1, generated structures similar to pancreatic islets, were positive for dithizone staining, and released insulin in vitro upon glucose challenge (262).
Table 2.
Summary of the studies that describe the generation of IPCs from bone marrow-derived MSCs
| Study | Protocol | Results |
|---|---|---|
| Murine MSCs | ||
| Chandra, 2009 (159) (adipose-derived SCs) | Three steps: 1) 2-d DMEM/F12, BSA 1%, ITS, activin-A, sodium butyrate, β-mercaptoethanol; 2) 2-d DMEM/F12, BSA 1%, ITS, taurine 0.3 mm; 3) 6-d DMEM/F12, BSA 1.5%, ITS, taurine 3 mm, GLP-1, nicotinamide, NEAA | In vitro, release of C-peptide in glucose-dependent manner. In vivo, restoration of normoglycemia in STZ-diabetic mice within 2 wk. |
| Hisanaga, 2008 (160) (bone marrow-MSCs) | 6-d culture in RPMI 1640, FCS 10%, 25 mm glucose, activin-A, betacellulin (BTC), conophylline, BTC-δ 4 | In vitro, glucose-stimulated insulin production. In vivo, reduction of hyperglycemia in STZ-diabetic mice for 4 wk. |
| Rat MSCs | ||
| Chen, 2004 (161) | Two steps: 1) 34-h l-DMEM, nicotinamide, β-mercaptoethanol; 2) 24-h serum-free HG-DMEM, nicotinamide, β-mercaptoethanol | In vitro, expression of insulin and nestin. In vivo, slight reduction of glucose levels. |
| Wu, 2007 (258) | Three steps: 1) 14-d FBS HG-DMEM; 2) 7-d FBS HG-DMEM, nicotinamide; 3) 7-d FBS HG-DMEM, nicotinamide, extendin-4 | In vitro, expression of insulin. In vivo, reduction of glucose levels for 2 wk. |
| Oh, 2004 (259) | Three steps: 1) 3-d serum-free HG-DMEM, 1% DMSO; 2) 7-d FBS HG-DMEM; 3) cells plated with type I collagen | In vitro, expression of insulin. In vivo, reduction of glucose levels for 3 months. |
| Human MSCs | ||
| Moriscot, 2005 (260) | Adenovirus infection with IPF1, HLXB9, FOXA2 transcription factors | In vitro, expression of insulin, NeuroD1, and Islet1. |
| Li, 2007 (261) | Adenovirus infection with PDX-1 transcription factor in serum-free DMEM with B27 and GLP-1 | In vitro, expression of insulin and C-peptide. In vivo, normoglycemia for 42 d. |
| Lee, 2006 (155) | Undifferentiated | In vivo, normoglycemia for 1 month, improvement of renal lesions. |
| Sun, 2007 (262) | Three steps: 1) 2-d serum-free HG-DMEM, β-mercaptoethanol; 2) 8-d NEAA, EGF, βFGF, B27, glutamine; 3) 8-d β-cellulin, activin-A, B27, nicotinamide | In vitro, insulin production in response to different glucose concentrations. |
| Karnieli, 2007 (263) | Retrovirus infection with rat PDX-1 under control of viral LTR | In vitro, insulin expression. In vivo, normoglycemia and expression of genes not expressed in vitro. |
| Xie, 2009 (264) | Three steps: 1) 3-d FBS HG-DMEM, bFGF, DMSO, glucose; 2) 7-d serum-free DMEM/F12, glucose, nicotinamide, EGF, bFGF, exendin-5, B27; 3) 5-d RPMI 1640, glucose, nicotinamide, HEPES, activin-A, exendin-4 | In vitro, insulin production in response to different glucose concentrations. In vivo, improved glycemia for over 1 month. |
FBS, Fetal bovine serum; FCS, fetal calf serum; HG-DMEM, high-glucose DMEM; DMSO, dimethylsulfoxide; ITS, insulin–transferrin–selenium; NEAA, non-essential amino acids; LTR, long-terminal repeats.
G. Clinical trials
Few trials in which MSCs are being administered to patients with T1D are ongoing (Table 1). The first is a U.S.-based trial on the use of allogeneic MSCs (Prochymal, a commercial formulation produced by Osiris, composed of human MSCs harvested from volunteer healthy donors) to determine safety and efficacy in patients affected by T1D (www.ClinicalTrials.gov; identifier, NCT00690066). In China, a second clinical trial is recruiting participants to evaluate the efficacy of autologous transplantation of MSCs in patients with T1D (www.ClinicalTrials.gov; identifier, NCT01157403). Finally, another clinical trial based on autologous administration of MSCs is currently planned in Europe (www.ClinicalTrials.gov; identifier, NCT01068951).
H. Unique feature: migratory ability to pancreatic islets
Sordi et al. (162) detected that the release of several chemokines (including high quantities of CXCL12) by pancreatic islets was capable of attracting human MSCs, which express several chemokine receptors including CXCR4, CCR7, CCR1, CXCR6, CX3CR1, CXCR5, CCR10, and CCR4 (162–164). Once transplanted, these human MSCs traffic to pancreatic islets where they can be detected up to 13 wk after transplantation (162). Lee et al. (155) confirmed the migration of human MSCs to pancreas into STZ diabetic mice. Interestingly, Fiorina et al. (14) observed a preferential migration of murine MSCs to pancreatic lymph nodes of NOD mice, suggesting the possibility that this homing mechanism is crucial for the in vivo modulation of the immune response due to autoreactive T cells.
I. Pros and cons
The use of MSCs in scientific research offers numerous advantages: MSCs are 1) easy to grow (165) and 2) easy to manipulate (166). On the other hand, several disadvantages are associated with the use of MSCs: 1) their oncogenicity (117–122); 2) the resulting formation of ectopic unwanted tissue (19, 167); and 3) unwanted release of cytokines.
V. Hematopoietic Stem Cells (HSCs)
HSCs are multipotent SCs found in the bone marrow, CB, peripheral blood, and fetal tissues (168). HSCs display the ability to differentiate into myeloid lineages (macrophages, erythrocyte, and granulocytes) and lymphoid lineages (natural killer, T, and B cells) (169–171). In 2003 for the first time, an HSC transplant was performed in a patient affected by T1D at the University of São Paulo, Brazil (172, 173). Given the initial and preliminary promising results, HSC treatment is being considered as a potential treatment for T1D (174); however, several issues remain to be clarified, particularly with regard to the potential contribution of concomitant immunosuppression (174). To better understand the role of HSCs and concomitant immunosuppression in pancreatic β-cell regeneration, we should recall the work by Hasegawa et al. (175), who performed bone marrow transplantation from GFP mice into chemically induced hyperglycemic mice. Bone marrow transplantation, associated with irradiation, nearly restored normoglycemia and pancreatic islet number and size, and the effect was abrogated by the avoidance of irradiation (175). However, most of the GFP+ cells (i.e., bone marrow-derived) were detected around islets and were not insulin-positive (175). It is therefore possible that a combination of bone marrow conditioning and SCs is required to restore normoglycemia, and this could be the case in humans as well.
A. Characteristics
HSCs are generally characterized by the expression of CD34 and Thy1 and the lack of expression of CD38, CD33, and HLA-DR, but many differences among mice strains and between humans and mice are evident (176, 177). Murine HSCs (or KLS cells) are identified by the signature Lin−c-Kit+Sca1+flk2−CD34− Slamf1+ phenotype, whereas other markers may be informative in some but not all mouse strains (178). Human HSCs can be identified through expression of the surface markers CD34 and CD90 together with the lack of expression for Lin, CD38, and mature markers including B220, Mac1, Gr-1, IL-7R, Ter119, CD3, CD4, and CD8 (178). The most prominent difference between human and murine HSCs is the absence of CD34 on murine HSCs. Although extensive research has been conducted, the function of this sialomucin still remains elusive, with proposed roles ranging from inhibiting differentiation to boosting proliferation or acting as an L-selectin ligand in humans (179). Murine and human HSCs are also characterized by differential behavior when used as treatment for T1D. Particularly, human HSCs are much more effective in reverting the hyperglycemic state, even if transplanted at the time of full-blown disease, whereas murine HSCs have been shown to be fully efficacious only for diabetes prevention and not reversal (173, 180).
B. Isolation methods
HSCs can be harvested relatively easily from donors by directly sampling bone marrow or from peripheral blood after granulocyte-colony stimulating factor (G-CSF) administration followed by blood apheresis (181, 182). A small number of cells are normally harvested, and it is thus essential to set up ex vivo expansion strategies to overcome this limitation. First, cell preselection is necessary because cell sorting may enrich the HSC population in the sample (183, 184). CD34 and CD133 are the optimal surface markers used for HSC enrichment; CD34 is the “gold standard” marker for HSC, whereas CD133 helps in identifying less mature cells (185). Because ex vivo culturing of HSCs is not an easy task since a cellular network supports HSC growth and survival in vivo, coculturing HSCs with a mesenchymal stromal feeder cell layer provides a physiological-like microenvironment ideal to support cell proliferation (186). To facilitate proliferation, cytokines including IL-3, IL-6, SC factor, thrombopoietin, and Flt3L are frequently added to the expansion medium, thereby promoting cell maintenance and expansion, reducing apoptosis, and protecting the self-renewal of naive SCs by preventing telomere degradation (186).
C. Immunogenicity
Recent reports of clinical benefits after the use of autologous HSCs in T1D have sparked much interest in the utility and value of HSC therapy as a tool to induce tolerance (173). The discovery of a CXCR4 antagonist capable of mobilizing HSCs has further increased interest in HSCs (187). HSCs are probably among the most immunogenic SCs, in that they can generate lymphoid and myeloid immune cells and can be promptly rejected by the recipient immune system without prior immune ablation via radiation or chemicals (188). Myeloablative allogeneic HSC transplantation can be curative for many diseases, but the procedure is unduly toxic (188) as a result of the burden of graft rejection and GVHD, which represent two major barriers to the success of HSC transplantation (188). A possible improvement in the HSC transplantation protocol could be the depletion of T cells from hematopoietic grafts, to avoid the potential lethal complication of GVHD and to reduce immunogenicity, resulting in transplantation of a pure population of HSCs. It should be noted that we believe that allogeneic HSC transplantation is not an option for T1D due to the necessity of myeloablation and the potential subsequent onset of GHVD.
D. Effect on immune response
Growing evidence suggests that autologous HSCs can induce central and peripheral immunological tolerance per se (189). Preclinical studies have demonstrated that T-cell-depleted bone marrow-resident CD34+ SCs overcome MHC barriers in sublethally irradiated mice (190) and that murine HSCs may delete effector cells through the Fas/FasL interaction or via the TNF-α pathways, which are both present on HSCs (Fig. 1) (191). This effect can be reverted by the addition of a caspase inhibitor, suggesting a deletion-based mechanism (192). Kared et al. (193) have recently demonstrated that murine HSCs may stimulate peripheral FoxP3+ cell (i.e., Treg) expansion through cell-to-cell contact activation of Notch signaling and through soluble factors such as granulocyte-macrophage colony-stimulating factor (Fig. 1) (193). With respect to human HSCs, Rachamim et al. (194) have shown that cells within the human CD34+ population are endowed with potent veto activity, referring to the ability of HSCs to neutralize precursors of cytotoxic T lymphocytes in an HLA-restricted and cell contact-dependent fashion (194). Experimental data did not clarify the effect of HSCs on experimental diabetes.
E. Cytokine and chemokine profile
The cytokine/chemokine profile of HSCs is elusive and difficult to address, primarily due to the ability of HSCs to generate lymphoid and myeloid cells. Too many uncertainties still surround HSCs' function and chemokine/cytokine production; HSC half-life is not fully clarified (195), and most of the human studies have been conducted in the setting of allogeneic HSC transplantation, with ongoing GHVD often present (196). The HSC chemokine profile has been more extensively studied, and various papers have confirmed that the interaction between CXCR4 (expressed by HSC) and its ligand CXCL12 governs HSC homing (197). The discovery of this interaction has resulted in multiple studies using CXCR4 antagonists and agonists to mobilize HSC in different diseases (187, 198, 199).
F. Generation of insulin-producing cells
β-Cell regeneration has not been observed in either human (200) or animal models (180) during HSC transplantation, and thus HSCs are not considered a possible source of IPCs; however, it is possible that HSCs may promote or facilitate β-cell regeneration (201). The question of whether bone marrow cells and HSCs can contribute to the generation of endocrine components of the pancreas has been studied by various investigators. Bone marrow cells from male GFP mice were injected into neonatal NOD/SCID mice 24 h after birth (202). Forty percent of ducts contained epithelial cells derived from donor bone marrow, with rare cells coexpressing GFP and insulin, whereas in adults transplanted with GFP+ bone marrow-derived cells, this effect was not observed (202). Bone marrow pluripotent SCs can therefore migrate to the pancreas and differentiate into complex organ-specific structures during the neonatal period (202). Nelson et al. (203) detected maternal microchimerism in peripheral blood in T1D patients by studying the presence of female islet β-cells in male pancreata from autopsies. Female islet β-cells (presumed maternal) formed 0.39–0.96% of the total β-cell mass, thus supporting the idea of circulating maternal progenitors localizing in the pancreata of children (203).
G. Clinical trials
HSCs are, to date, among the most often used SCs in the clinic for the therapy of autoimmune diseases (174). In particular, T1D studies and trials have been carried out, beginning with the trial of Voltarelli et al. (173) (Table 1). In 2003, Voltarelli et al. initiated a phase I/II study in T1D, with the goal of evaluating the safety and capacity of autologous HSC mobilization using a combined regimen of cyclophosphamide plus G-CSF as well as retransplantation after high-dose cyclophosphamide plus rabbit polyclonal anti-T-cell antibody [antithymocyte globulin (ATG)] (173). The latest analysis reported 23 treated patients with a mean follow-up of 30 months, 20 of which were insulin-free for a period of time (204). Among these, 12 were insulin-free after 31 months, whereas eight relapsed and resumed insulin therapy at lower doses compared with the pretransplant period (Fig. 5) (204). The three nonresponder patients either suffered from previous diabetic ketoacidosis or received corticosteroids to prevent ATG reactions (204). Recently, Snarski et al. (205) treated eight newly diagnosed T1D patients with autologous HSC transplantation using a very similar protocol to that previously reported, achieving insulin discontinuation in all patients, except one who resumed low-dosage insulin treatment 7 months after transplantation. In a trial conducted at the University of Nanjing in China, autologous HSC infusion induced an insulin-free state in four patients treated less than 3 months from diagnosis, but not in 11 patients treated 3–12 months after diagnosis (J. Ouyang, unpublished observations). The major issue with this study is that ATG and cyclophosphamide administration may account for some of the results obtained.
Fig. 5.

Daily insulin doses in individual T1D patients before (gray bars) and after (black bars) autologous HSC transplantation. Metabolic data from different groups of patients are presented: A, patients insulin-free since transplantation (because they are continously insulin-free, only gray bars are shown); B, patients who were never insulin-free; and C, patients who were transiently insulin-free. Updated results of 25 patients (December, 2010).
H. Unique feature: promoting β-cell regeneration
HSCs are extremely plastic and easily procured, and thus much excitement and expectation has been generated regarding their potential. Indeed, multiple works have described the ability of HSCs to normalize hyperglycemia, yet the ability of HSCs to differentiate into β-cells is still disputed (201). Hess et al. (206) in 2003 opened up the field of HSC with regard to promotion of β-cell regeneration. Authors showed that bone marrow-derived c-kit+ cells reduce hyperglycemia, once adoptively transferred into chemically induced hyperglycemic mice (206). A low frequency of donor-derived insulin-positive cells was evident (206), and the majority of the transplanted cells were localized to ductal and islet structures, with stimulation of insulin production. Tang et al. (207) isolated BALB/c HSCs and attempted to differentiate them into IPCs under high-glucose culture conditions and addition of β-cell-stimulating factors such as nicotinamide and exendin 4. When these cells were injected into mice rendered diabetic by treatment with STZ, reversal of hyperglycemia, restoration of insulin, and C-peptide secretion were observed (207). These data, however, were not highly convincing, and others confirmed a minimal or absent endogenous insulin production from these HSC-derived IPCs (208). Another proposed possibility for HSC action in T1D models was that HSCs could act by promoting regeneration of β-cells rather than by directly differentiating or through their immunological properties. Hasegawa et al. (175) observed islet regeneration after HSC mobilization and demonstrated an absence of GFP-positive bone marrow cells in regenerated islets. Kang et al. (180) transplanted allogeneic HSCs from β-gal transgenic donors into NOD mice. HSC transplantation was able to successfully prevent the onset of T1D in all treated mice, but reversal was obtained in only one mouse out of 50, despite full hematopoietic engraftment (180).
I. Pros and cons
An HSC-based therapeutic approach seems to be effective in limited groups of newly diagnosed patients (173). Some flaws are evident in the cited study, such as the absence of a randomized control group as well as a missing HSC transplantation-only cohort (without immunosuppression), which are necessary to clearly define the efficacy of HSCs per se (173). Moreover, some treated patients experienced side effects including pneumonia, oligospermia, and endocrine disorders (173). Despite these effects, we must remember that autologous nonmyeloablative HSC transplantation is one of the few treatments (together with anti-CD3 and cyclosporine) that has been proven to be capable of reversing human T1D. Thus, it is important to spend the effort attempting to optimize this procedure by carefully evaluating clinical symptoms, C-peptide levels, and genotype (HLA), and thereby determining which patients are likely to benefit from the treatment (209). Among the advantages of using HSCs in patients with T1D, we must consider: 1) the availability of HSCs; 2) the absence of ectopic tissue or tumor formation; and 3) the rapid clinical response. Some disadvantages should also be considered: 1) the occurrence of relapses in most patients treated with HSC transplantation; 2) the requirement of a conditioning regimen that comes with risks associated with transient myeloablation; and 3) potential infertility. Presently, the cohort of T1D patients treated with HSC transplantation is being followed for occurrence of long-term complications.
VI. Induced Pluripotent Stem Cells (iPS)
iPS are a hot topic of regenerative medicine, because they can be generated in an autologous setting (therefore avoid any immunological issues) and because of illimited differentiation potentials (210, 211).
A. Characteristics
iPS are reprogrammed somatic cells in which pluripotency is restored by an induced expression of transcription factors including Oct-4, Sox2, Nanog, c-Myc, LIN28, and Klf4 (210, 211). iPS are similar to ESCs with regard to morphology, self-renewal activity, and differentiative potential, and like ESCs, iPS display the ability to give rise to both embryonic bodies and teratomas and to differentiate into endodermal, mesodermal, and ectodermal cellular lineages (212).
B. Isolation methods
Demonstration of the capability of inducing pluripotency in differentiated cell lines was first given by Takahashi and Yamanaka in 2006 (213). Investigators were able to obtain iPS from embryonic, adult, and dermal fibroblasts after retroviral transduction of a set of transcription factors including Oct4, Sox2, c-Myc, and Klf4 (213, 214). These iPS gave rise in vitro to embryonic bodies and displayed the ability to initiate the differentiative process into endodermic, mesodermic, and ectodermic lineages (213). In addition, these iPS originated teratomas composed of cells from all three germ layers when transplanted sc into the dorsal flank of nude mice (213). Subsequently, Yu et al. (215) demonstrated the capability of obtaining iPS through transduction of a panel of factors composed of Oct4, Sox2, Nanog, and LIN28. Resultant iPS did not display karyotype abnormalities, exhibited telomerase activity, and expressed a set of surface antigens typical of embryonic cells including Tra-1-60, Tra-1-81, SSEA-3, and SSEA-4 (215). Recently, Huangfu et al. (216) were able to originate iPS from human fibroblasts transduced with a retroviral vector bearing cDNA of only Oct4 and Sox2 and treated with valproic acid (a histone deacetylase inhibitor). Investigators observed that these iPS were responsible for formation of embryonic bodies when cultured in vitro and of teratomas when transplanted in vivo into NOD/SCID mice (216). Because the use of viral vectors can result in the integration of the virus genome with that of somatic cells, new protocols that avoid the use of viral vectors have recently been developed. Stadtfeld et al. (217) reported obtaining iPS from tail fibroblasts and fetal liver cells of mice through the use of replication-incompetent adenoviral vectors that transiently expressed Oct4, Sox2, c-Myc, and Klf4. Shortly afterward, Yu et al. (218) generated iPS through the use of a nonintegrating episomal vector. Various nonviral techniques have been employed to generate iPS: 1) plasmid transfection; 2) micro-RNA therapy; 3) transposon insertion; and 4) treatment with recombinant proteins. 1) The successful generation of murine iPS through plasmid transfection, without viral vectors, was achieved in 2008 by Okita et al. (219). iPS were generated from MEFs after transfection with two plasmids (one containing the cDNAs of Oct3/4, Sox2, and Klf4, and the other containing c-Myc cDNA). These iPS, once transplanted into immunodeficient mice, produced teratomas (73); however, this method remained largely inefficient in the generation of iPS. 2) Lin et al. (220) first showed that micro-RNA can be used to generate iPS. Authors used mir-302 to reprogram human skin cancer cells into iPS, although it remained unclear whether iPS could be generated from human somatic cells using micro-RNA, such as fibroblasts, instead of cancer cells. 3) Woltjen et al. (221) used piggyBac transposition to insert Oct4, Sox2, c-Myc, and Klf4 into MEFs to generate iPS cells (221). Transposon technology still carries the risk of transgene transfer, yet a single piggyBac insertion can be removed from established iPS to prevent oncogene overexpression. 4) iPS can be generated using recombinant proteins as well. In 2009, Kim et al. (222) were able to reprogram human fibroblast to obtain iPS by directly delivering four reprogramming proteins (Oct4, Sox2, Klf4, and c-Myc) fused with a cell-penetrating peptide. Unfortunately, macromolecules do not cross cellular membranes, which may limit their ability to generate large numbers of iPS, resulting in poor efficiency (approximately 0.001 vs. 0.01% of initially plated cells) (222). The DNA-free iPS cells generated by Kim et al.(222) displayed features (morphology, proliferation, surface antigens, gene expression, telomerase activity, and the epigenetic status) of iPS.
Recently, another nonintegrating method to induce pluripotency into somatic cells has been reported by Rossi and colleagues (223). Investigators produced a synthetic mRNA using the open reading frame as a template for in vitro transcription of Klf4, c-Myc, Oct4, Sox2, and LIN28 cDNA (amplified from plasmids bearing them) (223). Because the possibility of inducing pluripotency in differentiated cells has been demonstrated (213), several cell lineages have been used as a source of iPS. iPS have been obtained from: 1) human somatic cells including keratinocytes (224, 225), dermal skin fibroblasts (214, 225–228), lung fibroblasts (228), astrocytes (229), and T cells (230, 231); 2) human ASCs including bone marrow MSCs (228), CD34+ bone marrow SCs (232), fetal neural SCs (233), and adipose stromal cells (234, 235); and 3) CD133+ (236) and CD34+ (232) cells obtained from CB. The possibility of obtaining and generating iPS from such a variety of sources may enhance the potential for iPS generation and access for diseased patients.
C. Immunogenicity
Few papers have studied iPS expression of: 1) MHC-I- and MHC-II-related molecules; 2) antigen-processing machinery components; or 3) NK2GD ligands (237). iPS displayed lower expression of molecules associated with MHC-I, such as HLA, -B, -C, -E, when compared with the IMR90 cells from which they were derived (237). In addition, similarly to human ESCs, iPS express neither HLA-G nor molecules associated with MHC-II (237). Real-time PCR expression studies revealed that after reprogramming, a reduction in the levels of mRNA for genes involved in antigen processing (AP-1, TPN, LMP2, and RFX5) was evident concomitant with an increase in expression of both the CNX and ERp57 chaperones and of the immunoproteasome component LMP7 (237). These results may suggest broad hypoimmunogenicity that is far from being demonstrated in iPS. A recent paper showed that, in contrast to B6 ESCs, teratomas formed by B6 iPS by retroviral and episomal approaches were mostly immune-rejected by B6 recipients with T-cell infiltration, tissue damage, and regression (238). Global gene expression analysis revealed several gene products that contribute directly to the immunogenicity of iPS in syngeneic recipients (238). Therefore, the immunogenicity of patient-specific iPS should be evaluated before any clinic application of these autologous cells into the patients (238).
D. Effect on immune response
The effect of iPS on the immune system has been scarcely studied (239). Senju et al. (239) were able to differentiate iPS into DCs and macrophages following a three-step protocol. These DC-like cells expressed CD11c but not CD80, CD86, and MHC-II-related molecules, thus displaying a partially immature state. Challenge of these iPS-derived DCs with IL-4, TNF-α, and anti-CD40 monoclonal antibody resulted in the generation of mature DCs (239). These mature DCs stimulate the mixed leukocyte response and correctly present antigen (239). Finally, macrophages derived from iPS exhibited phagocytic activity and expression of markers such as CD11b and F4/80 in a similar manner compared with macrophages derived from bone marrow cells (239). Experimental data on the effect of iPS on experimental diabetes are not available.
E. Cytokine and chemokine profile
The cytokine and chemokine profile of iPS have not been studied.
F. Generation of insulin-producing cells
A recent publication by Maehr et al. (240) reported the possibility of obtaining iPS from dermal fibroblasts of patients suffering from T1D. After skin biopsy, fibroblasts were subsequently transduced with retroviruses expressing Oct4, Sox2, and Klf4 (241). Although the method is not novel per se, the paper demonstrated for the very first time that iPS can be obtained from patients with T1D. After 4 wk following viral transduction, fibroblast-derived iPS colonies were harvested, and immunostaining and semiquantitative PCR analysis revealed that iPS were generated (240). Interestingly, these iPS showed a normal karyotype and, thanks to DNA microarrays, researchers found a higher similarity of iPS to human ESCs than to parental fibroblasts (240). When cultured in vitro, iPS gave rise to embryonic bodies composed of cells positive for endodermal, mesodermal, and ectodermal markers (SOX17 and FOXA2, smooth muscle-actin, and TUJ1, respectively) (240). In addition, when transplanted into immunocompromised mice, iPS originated teratomas in which cells of all three germ layers were recognizable (240). Researchers demonstrated that iPS-derived β-cells were C-peptide-positive and able to release insulin after in vitro stimulation with high and low levels of glucose (241). However, the full comparison of these iPS-derived IPCs with mature β-cells is far from being achieved.
G. Clinical trials
There are no clinical trials currently ongoing using iPS.
H. Unique features
1. Genetic and epigenetic abnormalities of iPS
Because ESCs may acquire genetic changes, it is reasonable to posit that this could occur in iPS as well (242). For instance, Mayshar et al. (243), in analyzing 66 lines of iPS obtained from 18 independent researchers, showed that one fifth of these iPS lines contained chromosomal aberrations (caused by deletions or duplications) (242, 243). Furthermore, it appears that iPS may display epigenetic abnormalities maintaining some memory of original cells (244). Authors carried out a systematic comparison of human iPS generated from different sources (hepatocytes, skin fibroblasts, and melanocytes), showing that iPS retained a transcriptional memory of the original cells, partially explained by incomplete promoter DNA methylation (244).
2. The potency of iPS compared with ESC
As previously reported, iPS are similar to ESCs and express genes typically associated with pluripotency; however, in some cases, differences in iPS characteristics have been found (245). Tokumoto et al. (245) reported that only 2.3% of iPS-derived cells differentiated into oligodendrocytes, whereas more than 24% of ESC-derived cells differentiated into oligodendrocytes. However, in the paper of Tokumoto et al. (245), it is unclear whether the differences observed are related to intrinsic variations between iPS cells and ESCs or are due to the fact that a different line was used (245). Hu et al. (246) induced and compared neuroepithelial and neuronal differentiation of ESCs and iPS-derived cells, with iPS demonstrating lower efficiency in the differentiative process (246). Feng et al. (247), in comparing hemangioblasts/blast cells derived from human iPS and ESCs, noted that iPS-derived blast cells displayed high rates of apoptosis with a concomitant reduced ability to grow and to expand.
I. Pros and cons
The use of iPS is advantageous because this method allows the use of pluripotent cells without manipulating embryos (235) and offers the possibility of generating cells specific to the patient (248). A few disadvantages still exist for iPS applications: 1) tumorigenicity (72); 2) appearance of chromosomal aberrations (249); and 3) viral contaminations during induction of pluripotency (250, 251). Finally, we should bear in mind that patients with T1D have severe microangiopathy, which may render the recovery of viable fibroblasts potentially problematic (252).
VII. Future Directions
A. Safety
The primary goal before introducing SCs to the clinic for T1D should be to resolve safety issues and potential oncogenicity associated with this therapy. Most importantly, we need to be absolutely certain that we are not injecting cancer precursors. This issue could represent the undoing of the SC field for the treatment of human disease. The lessons from preclinical models and anecdotal case reports have taught us that ESCs carry substantial potential risk (253), whereas the use of unmanipulated CB-SCs and adult HSCs appears very safe (73). More caution is required with MSC therapy, which holds oncogenic potential and is associated with robust immunological and proliferative abilities (73). The introduction of undifferentiated ESCs to the clinic, to make use of their immunomodulatory capacity, is prohibitive due to ESC teratogenicity in vivo (253). The use of CB-SCs or CB-mononuclear cells appears promising, whereas the use of MSCs is complicated by the same issue. Two studies by Tolar et al. (118) and by Fiorina et al. (14) confirmed that MSCs may bear marked oncogenic potential, which must be addressed before moving forward with any MSC-based therapy in humans. Infusion of MSC-derived sarcoma cells resulted in malignant lesions in secondary recipients (118). It is important to determine whether oncogenicity is linked to a specific lineage or sublineage as well as which cells are more likely to generate tumors. Documentation of the homing and engraftment of SCs as well as quantification of the dose, frequency, and duration of infusions are relevant issues but will not be milestones in the field (14, 19).
B. Virus-free reprogramming
The secondary goal should be to move in the direction of virus-free reprogramming of adult somatic cells (223) or of nontotipotent SCs (254, 255). Although a virus-based approach could be considered for severe diseases (256), it is difficult to justify the risk of viral spreading or genomic integration in patients with T1D who can be treated with insulin therapy. Therefore, we should welcome any effort in this direction. The Harvard Stem Cell Institute is currently breaking ground on developing virus-free cell reprogramming (223).
C. Autologous vs. allogeneic
The third goal should be to come to an agreement on the autologous vs. allogeneic use of SCs. Most key-opinion leaders agree that only autologous SCs are likely to be accepted as a general therapy for T1D, whereas allogeneic SCs will be considered for some specific cases (e.g., when associated with islet transplantation, in severe forms of diabetes onset, and so on). At the moment, CB-SC-based therapy appears promising but needs to be confirmed on a larger scale (112). Autologous HSC transplantation associated with nonmyeloablative conditioning and immunosuppression has been shown to reverse T1D in humans (257), whereas the safe clinical use of ESCs and MSCs awaits novel protocols focusing on various aspects such as the generation of stable ESC lines expressing key transcriptional factors involved in the development of pancreatic β-cells, as well as the necessity of avoiding any harmful procedures to patients with T1D.
D. The benchmark: comparing stem cell-derived insulin-producing cells with mature β-cells
The fourth goal, which is the most relevant for the generation of SC-derived IPCs, will be to compare them with mature β-cells and to establish whether these IPCs sense glucose levels and produce/process/release insulin in a controlled manner.
VIII. Conclusions
Evidence thus far suggests the requirement for further in vitro and in vivo studies on SC properties to achieve safety in SC-based clinical trials. Furthermore, the immunological and regenerative mechanisms of SCs need to be fully elucidated to better understand the mechanisms behind the SC effect. In the future, treatment of T1D will be, most likely, primarily based on SCs or their derivatives, in particular due to the chronic shortage of cadaveric organ donors. Thus, greater efforts are required to improve our ability to take advantage of SCs in the treatment of this devastating autoimmune disease.
Acknowledgments
P.F. acknowledges support from a pilot and feasibility award from the Boston Area Diabetes Endocrinology Research Center (5P30DK57521) and a Ministero Universita' e Ricerca Scientifica Grant (“Staminali” RF-FSR-2008-1213704). We thank Mollie Jurewicz for editing.
P.F. is the recipient of a Juvenile Diabetes Research Foundation-Career Development Award and an American Society of Nephrology Career Development Award. N.Z. was supported by Veterans Affairs Merit Grants 1I01BX001125 and NIH/NHLBI 5R01HL073015.
Disclosure Summary: N.Z. and J.V. have nothing to declare. P.F. serves in the advisory board of Sorgente srl, Italian biotech.
Footnotes
- ASC
- Adult SC
- ATG
- antithymocyte globulin
- bFGF
- basic FGF
- CB
- cord blood
- DC
- dendritic cell
- DE
- definitive endoderm
- EGF
- epidermal growth factor
- ESC
- embryonic SC
- EXT
- embryonic SC cellular extract
- FGF
- fibroblast growth factor
- G-CSF
- granulocyte-colony stimulating factor
- GFP
- green fluorescent protein
- GVHD
- graft-vs.-host disease
- HLA-DR
- human leukocyte antigen-DR
- HSC
- hematopoietic SC
- ICM
- inner cell mass
- IFN
- interferon
- IPC
- insulin-producing cell
- iPS
- induced pluripotent SCs
- Klf
- Kruppel-factor activation
- LIF
- leukemia inhibitory factor
- MEF
- murine embryonic fibroblast
- MHC
- major histocompatibility complex
- MLR
- mixed lymphocyte reaction
- MSC
- mesenchymal SC
- NOD
- nonobese diabetic
- PDX-1
- pancreatic and duodenal homeobox factor 1
- SC
- stem cell
- SCID
- severely compromised immunodeficient
- SSEA
- stage-specific embryonic antigen
- STZ
- streptozotocin
- T1D
- type 1 diabetes
- VEGF
- vascular endothelial growth factor
- VSEL
- very small embryonic-like SC.
References
- 1. Colucci F, Cilio CM. 2010. Taming killer cells may halt diabetes progression. Nat Immunol 11:111–112 [DOI] [PubMed] [Google Scholar]
- 2. Karges B, Kapellen T, Neu A, Hofer SE, Rohrer T, Rosenbauer J, Wolf J, Holl RW. 2010. Long-acting insulin analogs and the risk of diabetic ketoacidosis in children and adolescents with type 1 diabetes. Diabetes Care 33:1031–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B. 2005. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patterson CC, Dahlquist GG, Gyürüs E, Green A, Soltész G. 2009. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet 373:2027–2033 [DOI] [PubMed] [Google Scholar]
- 5. 2000 Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med [Erratum (2000) 342:1376] 342:381–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. 1998. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 339:69–75 [DOI] [PubMed] [Google Scholar]
- 7. Kennedy WR, Navarro X, Goetz FC, Sutherland DE, Najarian JS. 1990. Effects of pancreatic transplantation on diabetic neuropathy. N Engl J Med 322:1031–1037 [DOI] [PubMed] [Google Scholar]
- 8. Fiorina P, La Rocca E, Venturini M, Minicucci F, Fermo I, Paroni R, D'Angelo A, Sblendido M, Di Carlo V, Cristallo M, Del Maschio A, Pozza G, Secchi A. 2001. Effects of kidney-pancreas transplantation on atherosclerotic risk factors and endothelial function in patients with uremia and type 1 diabetes. Diabetes 50:496–501 [DOI] [PubMed] [Google Scholar]
- 9. Couri CE, Voltarelli JC. 2009. Stem cell therapy for type 1 diabetes mellitus: a review of recent clinical trials. Diabetol Metab Syndr 1:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bluestone JA, Herold K, Eisenbarth G. 2010. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 464:1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nir T, Melton DA, Dor Y. 2007. Recovery from diabetes in mice by β cell regeneration. J Clin Invest 117:2553–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zaret KS, Grompe M. 2008. Generation and regeneration of cells of the liver and pancreas. Science 322:1490–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aguayo-Mazzucato C, Bonner-Weir S. 2010. Stem cell therapy for type 1 diabetes mellitus. Nat Rev Endocrinol 6:139–148 [DOI] [PubMed] [Google Scholar]
- 14. Fiorina P, Jurewicz M, Augello A, Vergani A, Dada S, La Rosa S, Selig M, Godwin J, Law K, Placidi C, Smith RN, Capella C, Rodig S, Adra CN, Atkinson M, Sayegh MH, Abdi R. 2009. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol 183:993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. 1998. Embryonic stem cell lines derived from human blastocysts. Science 282:1145–1147 [DOI] [PubMed] [Google Scholar]
- 16. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147 [DOI] [PubMed] [Google Scholar]
- 17. Anderson DJ, Gage FH, Weissman IL. 2001. Can stem cells cross lineage boundaries? Nat Med 7:393–395 [DOI] [PubMed] [Google Scholar]
- 18. Williams DA. 2009. National Institutes of Health releases new guidelines on human stem cell research. Mol Ther 17:1485–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abdi R, Fiorina P, Adra CN, Atkinson M, Sayegh MH. 2008. Immunomodulation by mesenchymal stem cells. Diabetes 57:1759–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tyndall A, Walker UA, Cope A, Dazzi F, De Bari C, Fibbe W, Guiducci S, Jones S, Jorgensen C, Le Blanc K, Luyten F, McGonagle D, Martin I, Bocelli-Tyndall C, Pennesi G, Pistoia V, Pitzalis C, Uccelli A, Wulffraat N, Feldmann M. 2007. Immunomodulatory properties of mesenchymal stem cells: a review based on an interdisciplinary meeting held at the Kennedy Institute of Rheumatology Division, London, UK, 31 October 2005. Arthritis Res Ther 9:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Francese R, Fiorina P. 2010. Immunological and regenerative properties of cord blood stem cells. Clin Immunol 136:309–322 [DOI] [PubMed] [Google Scholar]
- 22. Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, Morando L, Busca A, Falda M, Bussolati B, Tetta C, Camussi G. 2009. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol 20:1053–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zalzman M, Gupta S, Giri RK, Berkovich I, Sappal BS, Karnieli O, Zern MA, Fleischer N, Efrat S. 2003. Reversal of hyperglycemia in mice by using human expandable insulin-producing cells differentiated from fetal liver progenitor cells. Proc Natl Acad Sci USA 100:7253–7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Friel R, van der Sar S, Mee PJ. 2005. Embryonic stem cells: understanding their history, cell biology and signalling. Adv Drug Deliv Rev 57:1894–1903 [DOI] [PubMed] [Google Scholar]
- 25. Bonde S, Zavazava N. 2006. Immunogenicity and engraftment of mouse embryonic stem cells in allogeneic recipients. Stem Cells 24:2192–2201 [DOI] [PubMed] [Google Scholar]
- 26. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. 2005. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mayshar Y, Rom E, Chumakov I, Kronman A, Yayon A, Benvenisty N. 2008. Fibroblast growth factor 4 and its novel splice isoform have opposing effects on the maintenance of human embryonic stem cell self-renewal. Stem Cells 26:767–774 [DOI] [PubMed] [Google Scholar]
- 28. Hall J, Guo G, Wray J, Eyres I, Nichols J, Grotewold L, Morfopoulou S, Humphreys P, Mansfield W, Walker R, Tomlinson S, Smith A. 2009. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 5:597–609 [DOI] [PubMed] [Google Scholar]
- 29. Davey RE, Onishi K, Mahdavi A, Zandstra PW. 2007. LIF-mediated control of embryonic stem cell self-renewal emerges due to an autoregulatory loop. FASEB J 21:2020–2032 [DOI] [PubMed] [Google Scholar]
- 30. Katoh M. 2011. Network of WNT and other regulatory signaling cascades in pluripotent stem cells and cancer stem cells. Curr Pharm Biotechnol 12:160–170 [DOI] [PubMed] [Google Scholar]
- 31. Ginis I, Luo Y, Miura T, Thies S, Brandenberger R, Gerecht-Nir S, Amit M, Hoke A, Carpenter MK, Itskovitz-Eldor J, Rao MS. 2004. Differences between human and mouse embryonic stem cells. Dev Biol 269:360–380 [DOI] [PubMed] [Google Scholar]
- 32. Hoffman LM, Carpenter MK. 2005. Characterization and culture of human embryonic stem cells. Nat Biotechnol 23:699–708 [DOI] [PubMed] [Google Scholar]
- 33. Koestenbauer S, Zech NH, Juch H, Vanderzwalmen P, Schoonjans L, Dohr G. 2006. Embryonic stem cells: similarities and differences between human and murine embryonic stem cells. Am J Reprod Immunol 55:169–180 [DOI] [PubMed] [Google Scholar]
- 34. Ström S, Inzunza J, Grinnemo KH, Holmberg K, Matilainen E, Strömberg AM, Blennow E, Hovatta O. 2007. Mechanical isolation of the inner cell mass is effective in derivation of new human embryonic stem cell lines. Hum Reprod 22:3051–3058 [DOI] [PubMed] [Google Scholar]
- 35. Suss-Toby E, Gerecht-Nir S, Amit M, Manor D, Itskovitz-Eldor J. 2004. Derivation of a diploid human embryonic stem cell line from a mononuclear zygote. Hum Reprod 19:670–675 [DOI] [PubMed] [Google Scholar]
- 36. Conley BJ, Young JC, Trounson AO, Mollard R. 2004. Derivation, propagation and differentiation of human embryonic stem cells. Int J Biochem Cell Biol 36:555–567 [DOI] [PubMed] [Google Scholar]
- 37. Bonde S, Chan KM, Zavazava N. 2008. ES-Cell derived hematopoietic cells induce transplantation tolerance. PLoS ONE 3:e3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boyd AS, Higashi Y, Wood KJ. 2005. Transplanting stem cells: potential targets for immune attack. Modulating the immune response against embryonic stem cell transplantation. Adv Drug Deliv Rev 57:1944–1969 [DOI] [PubMed] [Google Scholar]
- 39. Drukker M, Katchman H, Katz G, Even-Tov Friedman S, Shezen E, Hornstein E, Mandelboim O, Reisner Y, Benvenisty N. 2006. Human embryonic stem cells and their differentiated derivatives are less susceptible to immune rejection than adult cells. Stem Cells 24:221–229 [DOI] [PubMed] [Google Scholar]
- 40. Boyd AS, Wood KJ. 2010. Characteristics of the early immune response following transplantation of mouse ES cell derived insulin-producing cell clusters. PLoS One 5:e10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kofidis T, deBruin JL, Tanaka M, Zwierzchoniewska M, Weissman I, Fedoseyeva E, Haverich A, Robbins RC. 2005. They are not stealthy in the heart: embryonic stem cells trigger cell infiltration, humoral and T-lymphocyte-based host immune response. Eur J Cardiothorac Surg 28:461–466 [DOI] [PubMed] [Google Scholar]
- 42. Fändrich F, Lin X, Chai GX, Schulze M, Ganten D, Bader M, Holle J, Huang DS, Parwaresch R, Zavazava N, Binas B. 2002. Preimplantation-stage stem cells induce long-term allogeneic graft acceptance without supplementary host conditioning. Nat Med 8:171–178 [DOI] [PubMed] [Google Scholar]
- 43. Baertschiger RM, Gonelle-Gispert C, Morel P, Sgroi A, Serre-Beinier V, Stouffs M, Jaconi ME, Bühler LH. 2010. Transplantation of mouse embryonic stem cells induces hematopoietic and tissue chimerism in rats. Xenotransplantation 17:362–369 [DOI] [PubMed] [Google Scholar]
- 44. Lee JE, Kang MS, Park MH, Shim SH, Yoon TK, Chung HM, Lee DR. 2010. Evaluation of 28 human embryonic stem cell lines for use as unrelated donors in stem cell therapy: implications of HLA and ABO genotypes. Cell Transplant 19:1383–1395 [DOI] [PubMed] [Google Scholar]
- 45. Guo Y, Hangoc G, Bian H, Pelus LM, Broxmeyer HE. 2005. SDF-1/CXCL12 enhances survival and chemotaxis of murine embryonic stem cells and production of primitive and definitive hematopoietic progenitor cells. Stem Cells 23:1324–1332 [DOI] [PubMed] [Google Scholar]
- 46. Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, Kay TW. 2004. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes 53:113–121 [DOI] [PubMed] [Google Scholar]
- 47. Jurewicz M, Yang S, Augello A, Godwin JG, Moore RF, Azzi J, Fiorina P, Atkinson M, Sayegh MH, Abdi R. 2010. Congenic mesenchymal stem cell therapy reverses hyperglycemia in experimental type 1 diabetes. Diabetes 59:3139–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang R, Han G, Wang J, Chen G, Xu R, Wang L, Li X, Shen B, Li Y. 2008. The pathogenic role of interleukin-27 in autoimmune diabetes. Cell Mol Life Sci 65:3851–3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leng Q, Nie Y, Zou Y, Chen J. 2008. Elevated CXCL12 expression in the bone marrow of NOD mice is associated with altered T cell and stem cell trafficking and diabetes development. BMC Immunol 9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soria B, Roche E, Berná G, León-Quinto T, Reig JA, Martín F. 2000. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 49:157–162 [DOI] [PubMed] [Google Scholar]
- 51. Humphrey RK, Bucay N, Beattie GM, Lopez A, Messam CA, Cirulli V, Hayek A. 2003. Characterization and isolation of promoter-defined nestin-positive cells from the human fetal pancreas. Diabetes 52:2519–2525 [DOI] [PubMed] [Google Scholar]
- 52. Lee JH, Lee EJ, Lee CH, Park JH, Han JY, Lim JM. 2009. Requirement of leukemia inhibitory factor for establishing and maintaining embryonic stem cells in mice. Fertil Steril 92:1133–1140 [DOI] [PubMed] [Google Scholar]
- 53. Blyszczuk P, Czyz J, Kania G, Wagner M, Roll U, St-Onge L, Wobus AM. 2003. Expression of Pax4 in embryonic stem cells promotes differentiation of nestin-positive progenitor and insulin-producing cells. Proc Natl Acad Sci USA 100:998–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, Agulnick AD, D'Amour KA, Carpenter MK, Baetge EE. 2008. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 26:443–452 [DOI] [PubMed] [Google Scholar]
- 55. D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. 2006. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 24:1392–1401 [DOI] [PubMed] [Google Scholar]
- 56. Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. 2001. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 292:1389–1394 [DOI] [PubMed] [Google Scholar]
- 57. Naujok O, Francini F, Picton S, Jörns A, Bailey CJ, Lenzen S. 2008. A new experimental protocol for preferential differentiation of mouse embryonic stem cells into insulin-producing cells. Cell Transplant 17:1231–1242 [DOI] [PubMed] [Google Scholar]
- 58. Naujok O, Francini F, Picton S, Bailey CJ, Lenzen S, Jörns A. 2009. Changes in gene expression and morphology of mouse embryonic stem cells on differentiation into insulin-producing cells in vitro and in vivo. Diabetes Metab Res Rev 25:464–476 [DOI] [PubMed] [Google Scholar]
- 59. Li G, Luo R, Zhang J, Yeo KS, Lian Q, Xie F, Tan EK, Caille D, Kon OL, Salto-Tellez M, Meda P, Lim SK. 2009. Generating mESC-derived insulin-producing cell lines through an intermediate lineage-restricted progenitor line. Stem Cell Res 2:41–55 [DOI] [PubMed] [Google Scholar]
- 60. Zhang D, Jiang W, Shi Y, Deng H. 2009. Generation of pancreatic islet cells from human embryonic stem cells. Sci China C Life Sci 52:615–621 [DOI] [PubMed] [Google Scholar]
- 61. Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S, Shi Y, Deng H. 2009. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res 19:429–438 [DOI] [PubMed] [Google Scholar]
- 62. Li H, Lam A, Xu AM, Lam KS, Chung SK. 2010. High dosage of Exendin-4 increased early insulin secretion in differentiated β cells from mouse embryonic stem cells. Acta Pharmacol Sin 31:570–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee SH, Hao E, Savinov AY, Geron I, Strongin AY, Itkin-Ansari P. 2009. Human β-cell precursors mature into functional insulin-producing cells in an immunoisolation device: implications for diabetes cell therapies. Transplantation 87:983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shao S, Gao Y, Xie B, Xie F, Lim SK, Li G. 2011. Correction of hyperglycemia in type 1 diabetic models by transplantation of encapsulated insulin-producing cells derived from mouse embryo progenitor. J Endocrinol 208:245–255 [DOI] [PubMed] [Google Scholar]
- 65. Xu YN, Guan N, Wang ZD, Shan ZY, Shen JL, Zhang QH, Jin LH, Lei L. 2009. ES cell extract-induced expression of pluripotent factors in somatic cells. Anat Rec (Hoboken) 292:1229–1234 [DOI] [PubMed] [Google Scholar]
- 66. Burt RK, Verda L, Kim DA, Oyama Y, Luo K, Link C. 2004. Embryonic stem cells as an alternate marrow donor source: engraftment without graft-versus-host disease. J Exp Med 199:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mohib K, Allan D, Wang L. 2010. Human embryonic stem cell-extracts inhibit the differentiation and function of monocyte-derived dendritic cells. Stem Cell Rev 6:611–621 [DOI] [PubMed] [Google Scholar]
- 68. Rajasingh J, Lambers E, Hamada H, Bord E, Thorne T, Goukassian I, Krishnamurthy P, Rosen KM, Ahluwalia D, Zhu Y, Qin G, Losordo DW, Kishore R. 2008. Cell-free embryonic stem cell extract-mediated derivation of multipotent stem cells from NIH3T3 fibroblasts for functional and anatomical ischemic tissue repair. Circ Res 102:e107–e117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rippon HJ, Lane S, Qin M, Ismail NS, Wilson MR, Takata M, Bishop AE. 2008. Embryonic stem cells as a source of pulmonary epithelium in vitro and in vivo. Proc Am Thorac Soc 5:717–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Blum B, Benvenisty N. 2009. The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle 8:3822–3830 [DOI] [PubMed] [Google Scholar]
- 71. Knoepfler PS. 2009. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells 27:1050–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kooreman NG, Wu JC. 2010. Tumorigenicity of pluripotent stem cells: biological insights from molecular imaging. J R Soc Interface 7(Suppl 6):S753–S763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oh W, Kim DS, Yang YS, Lee JK. 2008. Immunological properties of umbilical cord blood-derived mesenchymal stromal cells. Cell Immunol 251:116–123 [DOI] [PubMed] [Google Scholar]
- 74. Verneris MR, Miller JS. 2009. The phenotypic and functional characteristics of umbilical cord blood and peripheral blood natural killer cells. Br J Haematol 147:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McGuckin CP, Forraz N. 2008. Potential for access to embryonic-like cells from human umbilical cord blood. Cell Prolif 41:31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kode JA, Mukherjee S, Joglekar MV, Hardikar AA. 2009. Mesenchymal stem cells: immunobiology and role in immunomodulation and tissue regeneration. Cytotherapy 11:377–391 [DOI] [PubMed] [Google Scholar]
- 77. La Motte-Mohs RN, Herer E, Zúñiga-Pflücker JC. 2005. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood 105:1431–1439 [DOI] [PubMed] [Google Scholar]
- 78. Parekh VS, Joglekar MV, Hardikar AA. 2009. Differentiation of human umbilical cord blood-derived mononuclear cells to endocrine pancreatic lineage. Differentiation 78:232–240 [DOI] [PubMed] [Google Scholar]
- 79. Huang CJ, Butler AE, Moran A, Rao PN, Wagner JE, Blazar BR, Rizza RA, Manivel JC, Butler PC. 2011. A low frequency of pancreatic islet insulin-expressing cells derived from cord blood stem cell allografts in humans. Diabetologia 54:1066–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Flake AW, Roncarolo MG, Puck JM, Almeida-Porada G, Evans MI, Johnson MP, Abella EM, Harrison DD, Zanjani ED. 1996. Treatment of X-linked severe combined immunodeficiency by in utero transplantation of paternal bone marrow. N Engl J Med 335:1806–1810 [DOI] [PubMed] [Google Scholar]
- 81. Rogers I, Casper RF. 2004. Umbilical cord blood stem cells. Best Pract Res Clin Obstet Gynaecol 18:893–908 [DOI] [PubMed] [Google Scholar]
- 82. Lazzari L, Lucchi S, Montemurro T, Porretti L, Lopa R, Rebulla P, Sirchia G. 2001. Evaluation of the effect of cryopreservation on ex vivo expansion of hematopoietic progenitors from cord blood. Bone Marrow Transplant 28:693–698 [DOI] [PubMed] [Google Scholar]
- 83. Domanska-Janik K, Buzanska L, Lukomska B. 2008. A novel, neural potential of non-hematopoietic human umbilical cord blood stem cells. Int J Dev Biol 52:237–248 [DOI] [PubMed] [Google Scholar]
- 84. Kita K, Gauglitz GG, Phan TT, Herndon DN, Jeschke MG. 2010. Isolation and characterization of mesenchymal stem cells from the sub-amniotic human umbilical cord lining membrane. Stem Cells Dev 19:491–502 [DOI] [PubMed] [Google Scholar]
- 85. Ljungman P, Bregni M, Brune M, Cornelissen J, de Witte T, Dini G, Einsele H, Gaspar HB, Gratwohl A, Passweg J, Peters C, Rocha V, Saccardi R, Schouten H, Sureda A, Tichelli A, Velardi A, Niederwieser D. 2010. Allogeneic and autologous transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe 2009. Bone Marrow Transplant 45:219–234 [DOI] [PubMed] [Google Scholar]
- 86. Zuba-Surma EK, Ratajczak MZ. 2010. Overview of very small embryonic-like stem cells (VSELs) and methodology of their identification and isolation by flow cytometric methods. Curr Protoc Cytom Unit 9.29 [DOI] [PubMed] [Google Scholar]
- 87. Zuba-Surma EK, Wu W, Ratajczak J, Kucia M, Ratajczak MZ. 2009. Very small embryonic-like stem cells in adult tissues—potential implications for aging. Mech Ageing Dev 130:58–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kucia M, Halasa M, Wysoczynski M, Baskiewicz-Masiuk M, Moldenhawer S, Zuba-Surma E, Czajka R, Wojakowski W, Machalinski B, Ratajczak MZ. 2007. Morphological and molecular characterization of novel population of CXCR4+ SSEA-4+ Oct-4+ very small embryonic-like cells purified from human cord blood—preliminary report. Leukemia 21:297–303 [DOI] [PubMed] [Google Scholar]
- 89. Martins AA, Paiva A, Morgado JM, Gomes A, Pais ML. 2009. Quantification and immunophenotypic characterization of bone marrow and umbilical cord blood mesenchymal stem cells by multicolor flow cytometry. Transplant Proc 41:943–946 [DOI] [PubMed] [Google Scholar]
- 90. Broxmeyer HE, Hangoc G, Cooper S, Ribeiro RC, Graves V, Yoder M, Wagner J, Vadhan-Raj S, Benninger L, Rubinstein P, Broun ER. 1992. Growth characteristics and expansion of human umbilical cord blood and estimation of its potential for transplantation in adults. Proc Natl Acad Sci USA 89:4109–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Broxmeyer HE. 2010. Cord blood hematopoietic stem cell transplantation. StemBook. Boston: Harvard Stem Cell Institute, Harvard University; [PubMed] [Google Scholar]
- 92. Wang M, Yang Y, Yang D, Luo F, Liang W, Guo S, Xu J. 2009. The immunomodulatory activity of human umbilical cord blood-derived mesenchymal stem cells in vitro. Immunology 126:220–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhao Y, Lin B, Darflinger R, Zhang Y, Holterman MJ, Skidgel RA. 2009. Human cord blood stem cell-modulated regulatory T lymphocytes reverse the autoimmune-caused type 1 diabetes in nonobese diabetic (NOD) mice. PLoS ONE 4:e4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cho PS, Messina DJ, Hirsh EL, Chi N, Goldman SN, Lo DP, Harris IR, Popma SH, Sachs DH, Huang CA. 2008. Immunogenicity of umbilical cord tissue derived cells. Blood 111:430–438 [DOI] [PubMed] [Google Scholar]
- 95. Wang D, Chen K, Du WT, Han ZB, Ren H, Chi Y, Yang SG, Bayard F, Zhu D, Han ZC. 2010. CD14+ monocytes promote the immunosuppressive effect of human umbilical cord matrix stem cells. Exp Cell Res 316:2414–2423 [DOI] [PubMed] [Google Scholar]
- 96. Li L, Godfrey WR, Porter SB, Ge Y, June CH, Blazar BR, Boussiotis VA. 2005. CD4+CD25+ regulatory T-cell lines from human cord blood have functional and molecular properties of T-cell anergy. Blood 106:3068–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Godfrey WR, Spoden DJ, Ge YG, Baker SR, Liu B, Levine BL, June CH, Blazar BR, Porter SB. 2005. Cord blood CD4(+)CD25(+)-derived T regulatory cell lines express FoxP3 protein and manifest potent suppressor function. Blood 105:750–758 [DOI] [PubMed] [Google Scholar]
- 98. Hippen KL, Harker-Murray P, Porter SB, Merkel SC, Londer A, Taylor DK, Bina M, Panoskaltsis-Mortari A, Rubinstein P, Van Rooijen N, Golovina TN, Suhoski MM, Miller JS, Wagner JE, June CH, Riley JL, Blazar BR. 2008. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4-1BB expressed on artificial antigen-presenting cells. Blood 112:2847–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Newman MB, Willing AE, Manresa JJ, Sanberg CD, Sanberg PR. 2006. Cytokines produced by cultured human umbilical cord blood (HUCB) cells: implications for brain repair. Exp Neurol 199:201–208 [DOI] [PubMed] [Google Scholar]
- 100. Liu Y, Liu T, Ma X, Fan X, Bao C, Cui Z. 2009. Effects of encapsulated rabbit mesenchymal stem cells on ex vivo expansion of human umbilical cord blood hematopoietic stem/progenitor cells. J Microencapsul 26:130–142 [DOI] [PubMed] [Google Scholar]
- 101. Son BR, Marquez-Curtis LA, Kucia M, Wysoczynski M, Turner AR, Ratajczak J, Ratajczak MZ, Janowska-Wieczorek A. 2006. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells 24:1254–1264 [DOI] [PubMed] [Google Scholar]
- 102. Castellani ML, Perrella A, Kempuraj DJ, Boucher W, Tagen M, Salini V, Vecchiet J, Tetè S, Frydas S, Theoharides TC, Conti P. 2007. Immunological activation of human umbilical cord blood mast cells induces tryptase secretion and interleukin-6, and histidine decarboxilase mRNA gene expression. Pharmacol Res 55:57–63 [DOI] [PubMed] [Google Scholar]
- 103. Newman MB, Willing AE, Manresa JJ, Davis-Sanberg C, Sanberg PR. 2005. Stroke-induced migration of human umbilical cord blood cells: time course and cytokines. Stem Cells Dev 14:576–586 [DOI] [PubMed] [Google Scholar]
- 104. Kim YJ, Broxmeyer HE. 2011. Immune regulatory cells in umbilical cord blood and their potential roles in transplantation tolerance. Crit Rev Oncol Hematol 79:112–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Penolazzi L, Lambertini E, Tavanti E, Torreggiani E, Vesce F, Gambari R, Piva R. 2008. Evaluation of chemokine and cytokine profiles in osteoblast progenitors from umbilical cord blood stem cells by BIO-PLEX technology. Cell Biol Int 32:320–325 [DOI] [PubMed] [Google Scholar]
- 106. Pessina A, Eletti B, Croera C, Savalli N, Diodovich C, Gribaldo L. 2004. Pancreas developing markers expressed on human mononucleated umbilical cord blood cells. Biochem Biophys Res Commun 323:315–322 [DOI] [PubMed] [Google Scholar]
- 107. Yoshida S, Ishikawa F, Kawano N, Shimoda K, Nagafuchi S, Shimoda S, Yasukawa M, Kanemaru T, Ishibashi H, Shultz LD, Harada M. 2005. Human cord blood-derived cells generate insulin-producing cells in vivo. Stem Cells 23:1409–1416 [DOI] [PubMed] [Google Scholar]
- 108. Denner L, Bodenburg Y, Zhao JG, Howe M, Cappo J, Tilton RG, Copland JA, Forraz N, McGuckin C, Urban R. 2007. Directed engineering of umbilical cord blood stem cells to produce C-peptide and insulin. Cell Prolif 40:367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sun B, Roh KH, Lee SR, Lee YS, Kang KS. 2007. Induction of human umbilical cord blood-derived stem cells with embryonic stem cell phenotypes into insulin producing islet-like structure. Biochem Biophys Res Commun 354:919–923 [DOI] [PubMed] [Google Scholar]
- 110. Gao F, Wu DQ, Hu YH, Jin GX, Li GD, Sun TW, Li FJ. 2008. In vitro cultivation of islet-like cell clusters from human umbilical cord blood-derived mesenchymal stem cells. Transl Res 151:293–302 [DOI] [PubMed] [Google Scholar]
- 111. Hu YH, Wu DQ, Gao F, Li GD, Zhang XC. 2010. Notch signaling: a novel regulating differentiation mechanism of human umbilical cord blood-derived mesenchymal stem cells into insulin-producing cells in vitro. Chin Med J (Engl) 123:606–614 [PubMed] [Google Scholar]
- 112. Haller MJ, Wasserfall CH, McGrail KM, Cintron M, Brusko TM, Wingard JR, Kelly SS, Shuster JJ, Atkinson MA, Schatz DA. 2009. Autologous umbilical cord blood transfusion in very young children with type 1 diabetes. Diabetes Care 32:2041–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kern S, Eichler H, Stoeve J, Klüter H, Bieback K. 2006. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 24:1294–1301 [DOI] [PubMed] [Google Scholar]
- 114. Oswald J, Boxberger S, Jørgensen B, Feldmann S, Ehninger G, Bornhäuser M, Werner C. 2004. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells 22:377–384 [DOI] [PubMed] [Google Scholar]
- 115. Pittenger MF, Martin BJ. 2004. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res 95:9–20 [DOI] [PubMed] [Google Scholar]
- 116. Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, Prockop DJ. 2004. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood 103:1662–1668 [DOI] [PubMed] [Google Scholar]
- 117. Miura M, Miura Y, Padilla-Nash HM, Molinolo AA, Fu B, Patel V, Seo BM, Sonoyama W, Zheng JJ, Baker CC, Chen W, Ried T, Shi S. 2006. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells 24:1095–1103 [DOI] [PubMed] [Google Scholar]
- 118. Tolar J, Nauta AJ, Osborn MJ, Panoskaltsis Mortari A, McElmurry RT, Bell S, Xia L, Zhou N, Riddle M, Schroeder TM, Westendorf JJ, McIvor RS, Hogendoorn PC, Szuhai K, Oseth L, Hirsch B, Yant SR, Kay MA, Peister A, Prockop DJ, Fibbe WE, Blazar BR. 2007. Sarcoma derived from cultured mesenchymal stem cells. Stem Cells 25:371–379 [DOI] [PubMed] [Google Scholar]
- 119. Li H, Fan X, Kovi RC, Jo Y, Moquin B, Konz R, Stoicov C, Kurt-Jones E, Grossman SR, Lyle S, Rogers AB, Montrose M, Houghton J. 2007. Spontaneous expression of embryonic factors and p53 point mutations in aged mesenchymal stem cells: a model of age-related tumorigenesis in mice. Cancer Res 67:10889–10898 [DOI] [PubMed] [Google Scholar]
- 120. Serakinci N, Guldberg P, Burns JS, Abdallah B, Schrødder H, Jensen T, Kassem M. 2004. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene 23:5095–5098 [DOI] [PubMed] [Google Scholar]
- 121. Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. 2007. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 11:421–429 [DOI] [PubMed] [Google Scholar]
- 122. Li N, Yang R, Zhang W, Dorfman H, Rao P, Gorlick R. 2009. Genetically transforming human mesenchymal stem cells to sarcomas: changes in cellular phenotype and multilineage differentiation potential. Cancer 115:4795–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wu C, Nik-Amini S, Nadesan P, Stanford WL, Alman BA. 2010. Aggressive fibromatosis (desmoid tumor) is derived from mesenchymal progenitor cells. Cancer Res 70:7690–7698 [DOI] [PubMed] [Google Scholar]
- 124. Saldanha-Araujo F, Haddad R, Zanette DL, De Araujo AG, Orellana MD, Covas DT, Zago MA, Panepucci RA. 2010. Cancer/testis antigen expression on mesenchymal stem cells isolated from different tissues. Anticancer Res 30:5023–5027 [PubMed] [Google Scholar]
- 125. Martin FT, Dwyer RM, Kelly J, Khan S, Murphy JM, Curran C, Miller N, Hennessy E, Dockery P, Barry FP, O'Brien T, Kerin MJ. 2010. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res Treat 124:317–326 [DOI] [PubMed] [Google Scholar]
- 126. Klopp AH, Lacerda L, Gupta A, Debeb BG, Solley T, Li L, Spaeth E, Xu W, Zhang X, Lewis MT, Reuben JM, Krishnamurthy S, Ferrari M, Gaspar R, Buchholz TA, Cristofanilli M, Marini F, Andreeff M, Woodward WA. 2010. Mesenchymal stem cells promote mammosphere formation and decrease E-cadherin in normal and malignant breast cells. PLoS One 5:e12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Secchiero P, Zorzet S, Tripodo C, Corallini F, Melloni E, Caruso L, Bosco R, Ingrao S, Zavan B, Zauli G. 2010. Human bone marrow mesenchymal stem cells display anti-cancer activity in SCID mice bearing disseminated non-Hodgkin's lymphoma xenografts. PLoS One 5:e11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. 2010. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: role of mesenchymal stem cell-derived TGF-β. J Immunol 184:5885–5894 [DOI] [PubMed] [Google Scholar]
- 129. Ling X, Marini F, Konopleva M, Schober W, Shi Y, Burks J, Clise-Dwyer K, Wang RY, Zhang W, Yuan X, Lu H, Caldwell L, Andreeff M. 2010. Mesenchymal stem cells overexpressing IFN-β inhibit breast cancer growth and metastases through Stat3 signaling in a syngeneic tumor model. Cancer Microenviron 3:83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Li GC, Ye QH, Xue YH, Sun HJ, Zhou HJ, Ren N, Jia HL, Shi J, Wu JC, Dai C, Dong QZ, Qin LX. 2010. Human mesenchymal stem cells inhibit metastasis of a hepatocellular carcinoma model using the MHCC97-H cell line. Cancer Sci 101:2546–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Caterson EJ, Nesti LJ, Danielson KG, Tuan RS. 2002. Human marrow-derived mesenchymal progenitor cells: isolation, culture expansion, and analysis of differentiation. Mol Biotechnol 20:245–256 [DOI] [PubMed] [Google Scholar]
- 132. Gnecchi M, Melo LG. 2009. Bone marrow-derived mesenchymal stem cells: isolation, expansion, characterization, viral transduction, and production of conditioned medium. Methods Mol Biol 482:281–294 [DOI] [PubMed] [Google Scholar]
- 133. Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM. 2001. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood 98:2396–2402 [DOI] [PubMed] [Google Scholar]
- 134. Schallmoser K, Rohde E, Reinisch A, Bartmann C, Thaler D, Drexler C, Obenauf AC, Lanzer G, Linkesch W, Strunk D. 2008. Rapid large-scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Eng Part C Methods 14:185–196 [DOI] [PubMed] [Google Scholar]
- 135. Eliopoulos N, Stagg J, Lejeune L, Pommey S, Galipeau J. 2005. Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood 106:4057–4065 [DOI] [PubMed] [Google Scholar]
- 136. Chen L, Tredget EE, Liu C, Wu Y. 2009. Analysis of allogenicity of mesenchymal stem cells in engraftment and wound healing in mice. PLoS One 4:e7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Schuleri KH, Feigenbaum GS, Centola M, Weiss ES, Zimmet JM, Turney J, Kellner J, Zviman MM, Hatzistergos KE, Detrick B, Conte JV, McNiece I, Steenbergen C, Lardo AC, Hare JM. 2009. Autologous mesenchymal stem cells produce reverse remodelling in chronic ischaemic cardiomyopathy. Eur Heart J 30:2722–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Huang XP, Sun Z, Miyagi Y, McDonald Kinkaid H, Zhang L, Weisel RD, Li RK. 2010. Differentiation of allogeneic mesenchymal stem cells induces immunogenicity and limits their long-term benefits for myocardial repair. Circulation 122:2419–2429 [DOI] [PubMed] [Google Scholar]
- 139. El Haddad N, Heathcote D, Moore R, Yang S, Azzi J, Mfarrej B, Atkinson M, Sayegh MH, Lee JS, Ashton-Rickardt PG, Abdi R. 2011. Mesenchymal stem cells express serine protease inhibitor to evade host immune response. Blood 117:1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sundin M, Barrett AJ, Ringdén O, Uzunel M, Lönnies H, Dackland AL, Christensson B, Blanc KL. 2009. HSCT recipients have specific tolerance to MSC but not to the MSC donor. J Immunother 32:755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R, Pennesi G. 2005. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol 35:1482–1490 [DOI] [PubMed] [Google Scholar]
- 142. Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. 2004. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 103:4619–4621 [DOI] [PubMed] [Google Scholar]
- 143. Benvenuto F, Ferrari S, Gerdoni E, Gualandi F, Frassoni F, Pistoia V, Mancardi G, Uccelli A. 2007. Human mesenchymal stem cells promote survival of T cells in a quiescent state. Stem Cells 25:1753–1760 [DOI] [PubMed] [Google Scholar]
- 144. Nauta AJ, Kruisselbrink AB, Lurvink E, Willemze R, Fibbe WE. 2006. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J Immunol 177:2080–2087 [DOI] [PubMed] [Google Scholar]
- 145. Zhang Q, Shi S, Liu Y, Uyanne J, Shi Y, Shi S, Le AD. 2009. Mesenchymal stem cells derived from human gingiva are capable of immunomodulatory functions and ameliorate inflammation-related tissue destruction in experimental colitis. J Immunol 183:7787–7798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, Mao N. 2005. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood 105:4120–4126 [DOI] [PubMed] [Google Scholar]
- 147. Spaggiari GM, Abdelrazik H, Becchetti F, Moretta L. 2009. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: central role of MSC-derived prostaglandin E2. Blood 113:6576–6583 [DOI] [PubMed] [Google Scholar]
- 148. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. 2008. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 111:1327–1333 [DOI] [PubMed] [Google Scholar]
- 149. Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, Risso M, Gualandi F, Mancardi GL, Pistoia V, Uccelli A. 2006. Human mesenchymal stem cells modulate B-cell functions. Blood 107:367–372 [DOI] [PubMed] [Google Scholar]
- 150. Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, Giunti D, Ceravolo A, Cazzanti F, Frassoni F, Mancardi G, Uccelli A. 2005. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 106:1755–1761 [DOI] [PubMed] [Google Scholar]
- 151. Augello A, Tasso R, Negrini SM, Cancedda R, Pennesi G. 2007. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum 56:1175–1186 [DOI] [PubMed] [Google Scholar]
- 152. Zhou K, Zhang H, Jin O, Feng X, Yao G, Hou Y, Sun L. 2008. Transplantation of human bone marrow mesenchymal stem cell ameliorates the autoimmune pathogenesis in MRL/lpr mice. Cell Mol Immunol 5:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Parekkadan B, Tilles AW, Yarmush ML. 2008. Bone marrow-derived mesenchymal stem cells ameliorate autoimmune enteropathy independently of regulatory T cells. Stem Cells 26:1913–1919 [DOI] [PubMed] [Google Scholar]
- 154. Zanone MM, Favaro E, Miceli I, Grassi G, Camussi E, Caorsi C, Amoroso A, Giovarelli M, Perin PC, Camussi G. 2010. Human mesenchymal stem cells modulate cellular immune response to islet antigen glutamic acid decarboxylase in type 1 diabetes. J Clin Endocrinol Metab 95:3788–3797 [DOI] [PubMed] [Google Scholar]
- 155. Lee RH, Seo MJ, Reger RL, Spees JL, Pulin AA, Olson SD, Prockop DJ. 2006. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci USA 103:17438–17443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Madec AM, Mallone R, Afonso G, Abou Mrad E, Mesnier A, Eljaafari A, Thivolet C. 2009. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia 52:1391–1399 [DOI] [PubMed] [Google Scholar]
- 157. Uccelli A, Moretta L, Pistoia V. 2008. Mesenchymal stem cells in health and disease. Nat Rev Immunol 8:726–736 [DOI] [PubMed] [Google Scholar]
- 158. Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, Santarlasci V, Mazzinghi B, Pizzolo G, Vinante F, Romagnani P, Maggi E, Romagnani S, Annunziato F. 2006. Role for interferon-γ in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 24:386–398 [DOI] [PubMed] [Google Scholar]
- 159. Chandra V, G S, Phadnis S, Nair PD, Bhonde RR. 2009. Generation of pancreatic hormone-expressing islet-like cell aggregates from murine adipose tissue-derived stem cells. Stem Cells 27:1941–1953 [DOI] [PubMed] [Google Scholar]
- 160. Hisanaga E, Park KY, Yamada S, Hashimoto H, Takeuchi T, Mori M, Seno M, Umezawa K, Takei I, Kojima I. 2008. A simple method to induce differentiation of murine bone marrow mesenchymal cells to insulin-producing cells using conophylline and betacellulin-Δ4. Endocr J 55:535–543 [DOI] [PubMed] [Google Scholar]
- 161. Chen LB, Jiang XB, Yang L. 2004. Differentiation of rat marrow mesenchymal stem cells into pancreatic islet β-cells. World J Gastroenterol 10:3016–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Sordi V, Malosio ML, Marchesi F, Mercalli A, Melzi R, Giordano T, Belmonte N, Ferrari G, Leone BE, Bertuzzi F, Zerbini G, Allavena P, Bonifacio E, Piemonti L. 2005. Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood 106:419–427 [DOI] [PubMed] [Google Scholar]
- 163. Wynn RF, Hart CA, Corradi-Perini C, O'Neill L, Evans CA, Wraith JE, Fairbairn LJ, Bellantuono I. 2004. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood 104:2643–2645 [DOI] [PubMed] [Google Scholar]
- 164. Von Lüttichau I, Notohamiprodjo M, Wechselberger A, Peters C, Henger A, Seliger C, Djafarzadeh R, Huss R, Nelson PJ. 2005. Human adult CD34-progenitor cells functionally express the chemokine receptors CCR1, CCR4, CCR7, CXCR5, and CCR10 but not CXCR4. Stem Cells Dev 14:329–336 [DOI] [PubMed] [Google Scholar]
- 165. Le Blanc K, Ringdén O. 2005. Immunobiology of human mesenchymal stem cells and future use in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 11:321–334 [DOI] [PubMed] [Google Scholar]
- 166. Gafni Y, Turgeman G, Liebergal M, Pelled G, Gazit Z, Gazit D. 2004. Stem cells as vehicles for orthopedic gene therapy. Gene Ther 11:417–426 [DOI] [PubMed] [Google Scholar]
- 167. Breitbach M, Bostani T, Roell W, Xia Y, Dewald O, Nygren JM, Fries JW, Tiemann K, Bohlen H, Hescheler J, Welz A, Bloch W, Jacobsen SE, Fleischmann BK. 2007. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood 110:1362–1369 [DOI] [PubMed] [Google Scholar]
- 168. Huang S, Law P, Young D, Ho AD. 1998. Candidate hematopoietic stem cells from fetal tissues, umbilical cord blood vs. adult bone marrow and mobilized peripheral blood. Exp Hematol 26:1162–1171 [PubMed] [Google Scholar]
- 169. Herzog EL, Chai L, Krause DS. 2003. Plasticity of marrow-derived stem cells. Blood 102:3483–3493 [DOI] [PubMed] [Google Scholar]
- 170. Wojakowski W, Tendera M, Kucia M, Zuba-Surma E, Paczkowska E, Ciosek J, Hałasa M, Król M, Kazmierski M, Buszman P, Ochała A, Ratajczak J, Machaliñski B, Ratajczak MZ. 2009. Mobilization of bone marrow-derived Oct-4+ SSEA-4+ very small embryonic-like stem cells in patients with acute myocardial infarction. J Am Coll Cardiol 53:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Sigvardsson M. 2009. New light on the biology and developmental potential of haematopoietic stem cells and progenitor cells. J Intern Med 266:311–324 [DOI] [PubMed] [Google Scholar]
- 172. Tyndall A, Black C, Finke J, Winkler J, Mertlesmann R, Peter HH, Gratwohl A. 1997. Treatment of systemic sclerosis with autologous haemopoietic stem cell transplantation. Lancet 349:254. [DOI] [PubMed] [Google Scholar]
- 173. Voltarelli JC, Couri CE, Stracieri AB, Oliveira MC, Moraes DA, Pieroni F, Coutinho M, Malmegrim KC, Foss-Freitas MC, Simões BP, Foss MC, Squiers E, Burt RK. 2007. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 297:1568–1576 [DOI] [PubMed] [Google Scholar]
- 174. Farge D, Labopin M, Tyndall A, Fassas A, Mancardi GL, Van Laar J, Ouyang J, Kozak T, Moore J, Kötter I, Chesnel V, Marmont A, Gratwohl A, Saccardi R. 2010. Autologous hematopoietic stem cell transplantation for autoimmune diseases: an observational study on 12 years' experience from the European Group for Blood and Marrow Transplantation Working Party on Autoimmune Diseases. Haematologica 95:284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Hasegawa Y, Ogihara T, Yamada T, Ishigaki Y, Imai J, Uno K, Gao J, Kaneko K, Ishihara H, Sasano H, Nakauchi H, Oka Y, Katagiri H. 2007. Bone marrow (BM) transplantation promotes β-cell regeneration after acute injury through BM cell mobilization. Endocrinology 148:2006–2015 [DOI] [PubMed] [Google Scholar]
- 176. Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. 1992. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci USA 89:2804–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Sutherland HJ, Eaves CJ, Eaves AC, Dragowska W, Lansdorp PM. 1989. Characterization and partial purification of human marrow cells capable of initiating long-term hematopoiesis in vitro. Blood 74:1563–1570 [PubMed] [Google Scholar]
- 178. Bryder D, Rossi DJ, Weissman IL. 2006. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol 169:338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Nielsen JS, McNagny KM. 2009. CD34 is a key regulator of hematopoietic stem cell trafficking to bone marrow and mast cell progenitor trafficking in the periphery. Microcirculation 16:487–496 [DOI] [PubMed] [Google Scholar]
- 180. Kang EM, Zickler PP, Burns S, Langemeijer SM, Brenner S, Phang OA, Patterson N, Harlan D, Tisdale JF. 2005. Hematopoietic stem cell transplantation prevents diabetes in NOD mice but does not contribute to significant islet cell regeneration once disease is established. Exp Hematol 33:699–705 [DOI] [PubMed] [Google Scholar]
- 181. Hai-Jiang W, Xin-Na D, Hui-Jun D. 2008. Expansion of hematopoietic stem/progenitor cells. Am J Hematol 83:922–926 [DOI] [PubMed] [Google Scholar]
- 182. Peters C, Matthes-Martin S, Fritsch G, Holter W, Lion T, Witt V, Höcker P, Fischer G, Dieckmann K, Handgretinger R, Klingebiel T, Gadner H. 1999. Transplantation of highly purified peripheral blood CD34+ cells from HLA-mismatched parental donors in 14 children: evaluation of early monitoring of engraftment. Leukemia 13:2070–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Petzer AL, Zandstra PW, Piret JM, Eaves CJ. 1996. Differential cytokine effects on primitive (CD34+CD38-) human hematopoietic cells: novel responses to Flt3-ligand and thrombopoietin. J Exp Med 183:2551–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Kwok YK, Tang MH, Law HK, Ngai CS, Lau YL, Lau ET. 2007. Maternal plasma or human serum albumin in wash buffer enhances enrichment and ex vivo expansion of human umbilical cord blood CD34+ cells. Br J Haematol 137:468–474 [DOI] [PubMed] [Google Scholar]
- 185. Hofmeister CC, Zhang J, Knight KL, Le P, Stiff PJ. 2007. Ex vivo expansion of umbilical cord blood stem cells for transplantation: growing knowledge from the hematopoietic niche. Bone Marrow Transplant 39:11–23 [DOI] [PubMed] [Google Scholar]
- 186. Koestenbauer S, Zisch A, Dohr G, Zech NH. 2009. Protocols for hematopoietic stem cell expansion from umbilical cord blood. Cell Transplant 18:1059–1068 [DOI] [PubMed] [Google Scholar]
- 187. Fiorina P, Jurewicz M, Vergani A, Petrelli A, Carvello M, D'Addio F, Godwin JG, Law K, Wu E, Tian Z, Thoma G, Kovarik J, La Rosa S, Capella C, Rodig S, Zerwes HG, Sayegh MH, Abdi R. 2011. Targeting the CXCR4-CXCL12 axis mobilizes autologous hematopoietic stem cells and prolongs islet allograft survival via programmed death ligand 1. J Immunol 186:121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Hsieh MM, Kang EM, Fitzhugh CD, Link MB, Bolan CD, Kurlander R, Childs RW, Rodgers GP, Powell JD, Tisdale JF. 2009. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 361:2309–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Steptoe RJ, Ritchie JM, Jones LK, Harrison LC. 2005. Autoimmune diabetes is suppressed by transfer of proinsulin-encoding Gr-1+ myeloid progenitor cells that differentiate in vivo into resting dendritic cells. Diabetes 54:434–442 [DOI] [PubMed] [Google Scholar]
- 190. Bachar-Lustig E, Rachamim N, Li HW, Lan F, Reisner Y. 1995. Megadose of T cell-depleted bone marrow overcomes MHC barriers in sublethally irradiated mice. Nat Med 1:1268–1273 [DOI] [PubMed] [Google Scholar]
- 191. Gur H, Krauthgamer R, Berrebi A, Klein T, Nagler A, Tabilio A, Martelli MF, Reisner Y. 2002. Tolerance induction by megadose hematopoietic progenitor cells: expansion of veto cells by short-term culture of purified human CD34(+) cells. Blood 99:4174–4181 [DOI] [PubMed] [Google Scholar]
- 192. Gur H, Krauthgamer R, Bachar-Lustig E, Katchman H, Arbel-Goren R, Berrebi A, Klein T, Nagler A, Tabilio A, Martelli MF, Reisner Y. 2005. Immune regulatory activity of CD34+ progenitor cells: evidence for a deletion-based mechanism mediated by TNF-α. Blood 105:2585–2593 [DOI] [PubMed] [Google Scholar]
- 193. Kared H, Leforban B, Montandon R, Renand A, Layseca Espinosa E, Chatenoud L, Rosenstein Y, Schneider E, Dy M, Zavala F. 2008. Role of GM-CSF in tolerance induction by mobilized hematopoietic progenitors. Blood 112:2575–2578 [DOI] [PubMed] [Google Scholar]
- 194. Rachamim N, Gan J, Segall H, Krauthgamer R, Marcus H, Berrebi A, Martelli M, Reisner Y. 1998. Tolerance induction by “megadose” hematopoietic transplants: donor-type human CD34 stem cells induce potent specific reduction of host anti-donor cytotoxic T lymphocyte precursors in mixed lymphocyte culture. Transplantation 65:1386–1393 [DOI] [PubMed] [Google Scholar]
- 195. Lord BI, Gurney H, Chang J, Thatcher N, Crowther D, Dexter TM. 1992. Haemopoietic cell kinetics in humans treated with rGM-CSF. Int J Cancer 50:26–31 [DOI] [PubMed] [Google Scholar]
- 196. Imamura M. 2002. Immunological reconstitution and immunoregulatory cells in hematopoietic stem cell transplantation. Int J Hematol 76(Suppl 1):191–194 [DOI] [PubMed] [Google Scholar]
- 197. Watt SM, Forde SP. 2008. The central role of the chemokine receptor, CXCR4, in haemopoietic stem cell transplantation: will CXCR4 antagonists contribute to the treatment of blood disorders? Vox Sang 94:18–32 [DOI] [PubMed] [Google Scholar]
- 198. Broxmeyer HE, Hangoc G, Cooper S, Campbell T, Ito S, Mantel C. 2007. AMD3100 and CD26 modulate mobilization, engraftment, and survival of hematopoietic stem and progenitor cells mediated by the SDF-1/CXCL12-CXCR4 axis. Ann NY Acad Sci 1106:1–19 [DOI] [PubMed] [Google Scholar]
- 199. Tchernychev B, Ren Y, Sachdev P, Janz JM, Haggis L, O'Shea A, McBride E, Looby R, Deng Q, McMurry T, Kazmi MA, Sakmar TP, Hunt S, 3rd, Carlson KE. 2010. Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc Natl Acad Sci USA 107:22255–22259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Butler AE, Huang A, Rao PN, Bhushan A, Hogan WJ, Rizza RA, Butler PC. 2007. Hematopoietic stem cells derived from adult donors are not a source of pancreatic β-cells in adult nondiabetic humans. Diabetes 56:1810–1816 [DOI] [PubMed] [Google Scholar]
- 201. Ianus A, Holz GG, Theise ND, Hussain MA. 2003. In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J Clin Invest 111:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wang X, Ge S, Gonzalez I, McNamara G, Rountree CB, Xi KK, Huang G, Bhushan A, Crooks GM. 2006. Formation of pancreatic duct epithelium from bone marrow during neonatal development. Stem Cells 24:307–314 [DOI] [PubMed] [Google Scholar]
- 203. Nelson JL, Gillespie KM, Lambert NC, Stevens AM, Loubiere LS, Rutledge JC, Leisenring WM, Erickson TD, Yan Z, Mullarkey ME, Boespflug ND, Bingley PJ, Gale EA. 2007. Maternal microchimerism in peripheral blood in type 1 diabetes and pancreatic islet β cell microchimerism. Proc Natl Acad Sci USA 104:1637–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Couri CE, Oliveira MC, Stracieri AB, Moraes DA, Pieroni F, Barros GM, Madeira MI, Malmegrim KC, Foss-Freitas MC, Simões BP, Martinez EZ, Foss MC, Burt RK, Voltarelli JC. 2009. C-Peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 301:1573–1579 [DOI] [PubMed] [Google Scholar]
- 205. Snarski E, Milczarczyk A, Torosian T, Paluszewska M, Urbanowska E, Król M, Boguradzki P, Jedynasty K, Franek E, Wiktor-Jedrzejczak W. 2011. Independence of exogenous insulin following immunoablation and stem cell reconstitution in newly diagnosed diabetes type I. Bone Marrow Transplant 46:562–566 [DOI] [PubMed] [Google Scholar]
- 206. Hess D, Li L, Martin M, Sakano S, Hill D, Strutt B, Thyssen S, Gray DA, Bhatia M. 2003. Bone marrow-derived stem cells initiate pancreatic regeneration. Nat Biotechnol 21:763–770 [DOI] [PubMed] [Google Scholar]
- 207. Tang DQ, Cao LZ, Burkhardt BR, Xia CQ, Litherland SA, Atkinson MA, Yang LJ. 2004. In vivo and in vitro characterization of insulin-producing cells obtained from murine bone marrow. Diabetes 53:1721–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Mathews V, Hanson PT, Ford E, Fujita J, Polonsky KS, Graubert TA. 2004. Recruitment of bone marrow-derived endothelial cells to sites of pancreatic β-cell injury. Diabetes 53:91–98 [DOI] [PubMed] [Google Scholar]
- 209. Ross LF, Philipson LH. 2007. Ethics of hematopoietic stem cell transplantation in type 1 diabetes mellitus. JAMA 298:285; author reply 285–286 [DOI] [PubMed] [Google Scholar]
- 210. Kim EY, Jeon K, Park HY, Han YJ, Yang BC, Park SB, Chung HM, Park SP. 2010. Differences between cellular and molecular profiles of induced pluripotent stem cells generated from mouse embryonic fibroblasts. Cell Reprogram 12:627–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Stefanovic S, Abboud N, Désilets S, Nury D, Cowan C, Pucéat M. 2009. Interplay of Oct4 with Sox2 and Sox17: a molecular switch from stem cell pluripotency to specifying a cardiac fate. J Cell Biol 186:665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Marchetto MC, Yeo GW, Kainohana O, Marsala M, Gage FH, Muotri AR. 2009. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PLoS One 4:e7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676 [DOI] [PubMed] [Google Scholar]
- 214. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872 [DOI] [PubMed] [Google Scholar]
- 215. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920 [DOI] [PubMed] [Google Scholar]
- 216. Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA. 2008. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 26:1269–1275 [DOI] [PubMed] [Google Scholar]
- 217. Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. 2008. Induced pluripotent stem cells generated without viral integration. Science 322:945–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. 2009. Human induced pluripotent stem cells free of vector and transgene sequences. Science 324:797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. 2008. Generation of mouse induced pluripotent stem cells without viral vectors. Science 322:949–953 [DOI] [PubMed] [Google Scholar]
- 220. Lin SL, Chang DC, Lin CH, Ying SY, Leu D, Wu DT. 2011. Regulation of somatic cell reprogramming through inducible mir-302 expression. Nucleic Acids Res 39:1054–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R, Cowling R, Wang W, Liu P, Gertsenstein M, Kaji K, Sung HK, Nagy A. 2009. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458:766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Kim DS, Lee JS, Leem JW, Huh YJ, Kim JY, Kim HS, Park IH, Daley GQ, Hwang DY, Kim DW. 2010. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev 6:270–281 [DOI] [PubMed] [Google Scholar]
- 223. Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, Daley GQ, Brack AS, Collins JJ, Cowan C, Schlaeger TM, Rossi DJ. 2010. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7:618–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Biliæ J, Pekarik V, Tiscornia G, Edel M, Boué S, Izpisúa Belmonte JC. 2008. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 26:1276–1284 [DOI] [PubMed] [Google Scholar]
- 225. Tolar J, Xia L, Riddle MJ, Lees CJ, Eide CR, McElmurry RT, Titeux M, Osborn MJ, Lund TC, Hovnanian A, Wagner JE, Blazar BR. 2011. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol 131:848–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Takayama N, Nishimura S, Nakamura S, Shimizu T, Ohnishi R, Endo H, Yamaguchi T, Otsu M, Nishimura K, Nakanishi M, Sawaguchi A, Nagai R, Takahashi K, Yamanaka S, Nakauchi H, Eto K. 2010. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med 207:2817–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Pessach IM, Ordovas-Montanes J, Zhang SY, Casanova JL, Giliani S, Gennery AR, Al-Herz W, Manos PD, Schlaeger TM, Park IH, Rucci F, Agarwal S, Mostoslavsky G, Daley GQ, Notarangelo LD. 2011. Induced pluripotent stem cells: a novel frontier in the study of human primary immunodeficiencies. J Allergy Clin Immunol 127:1400–1407.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. 2008. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451:141–146 [DOI] [PubMed] [Google Scholar]
- 229. Ruiz S, Brennand K, Panopoulos AD, Herrerías A, Gage FH, Izpisua-Belmonte JC. 2010. High-efficient generation of induced pluripotent stem cells from human astrocytes. PLoS One 5:e15526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Seki T, Yuasa S, Oda M, Egashira T, Yae K, Kusumoto D, Nakata H, Tohyama S, Hashimoto H, Kodaira M, Okada Y, Seimiya H, Fusaki N, Hasegawa M, Fukuda K. 2010. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 7:11–14 [DOI] [PubMed] [Google Scholar]
- 231. Brown ME, Rondon E, Rajesh D, Mack A, Lewis R, Feng X, Zitur LJ, Learish RD, Nuwaysir EF. 2010. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLoS One 5:e11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, Dang CV, Spivak JL, Moliterno AR, Cheng L. 2009. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 114:5473–5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Medvedev SP, Grigor'eva EV, Shevchenko AI, Malakhova AA, Dementyeva EV, Shilov AA, Pokushalov EA, Zaidman AM, Aleksandrova MA, Plotnikov EY, Sukhikh GT, Zakian SM. 2011. Human induced pluripotent stem cells derived from fetal neural stem cells successfully undergo directed differentiation into cartilage. Stem Cells Dev 20:1099–1112 [DOI] [PubMed] [Google Scholar]
- 234. Narsinh KH, Jia F, Robbins RC, Kay MA, Longaker MT, Wu JC. 2011. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat Protoc 6:78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Shao L, Feng W, Sun Y, Bai H, Liu J, Currie C, Kim J, Gama R, Wang Z, Qian Z, Liaw L, Wu WS. 2009. Generation of iPS cells using defined factors linked via the self-cleaving 2A sequences in a single open reading frame. Cell Res 19:296–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Giorgetti A, Montserrat N, Aasen T, Gonzalez F, Rodríguez-Pizà I, Vassena R, Raya A, Boué S, Barrero MJ, Corbella BA, Torrabadella M, Veiga A, Izpisua Belmonte JC. 2009. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell 5:353–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Suárez-Alvarez B, Rodriguez RM, Calvanese V, Blanco-Gelaz MA, Suhr ST, Ortega F, Otero J, Cibelli JB, Moore H, Fraga MF, López-Larrea C. 2010. Epigenetic mechanisms regulate MHC and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS One 5:e10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Zhao T, Zhang ZN, Rong Z, Xu Y. 2011. Immunogenicity of induced pluripotent stem cells. Nature 474:212–215 [DOI] [PubMed] [Google Scholar]
- 239. Senju S, Haruta M, Matsunaga Y, Fukushima S, Ikeda T, Takahashi K, Okita K, Yamanaka S, Nishimura Y. 2009. Characterization of dendritic cells and macrophages generated by directed differentiation from mouse induced pluripotent stem cells. Stem Cells 27:1021–1031 [DOI] [PubMed] [Google Scholar]
- 240. Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA. 2009. Generation of pluripotent stem cells from patients with type 1 diabetes. Proc Natl Acad Sci USA 106:15768–15773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Maehr R. 2011. iPS cells in type 1 diabetes research and treatment. Clin Pharmacol Ther 89:750–753 [DOI] [PubMed] [Google Scholar]
- 242. de Souza N. 2010. iPS cell aberrations. Nat Methods 7:948–949 [DOI] [PubMed] [Google Scholar]
- 243. Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, Plath K, Lowry WE, Benvenisty N. 2010. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 7:521–531 [DOI] [PubMed] [Google Scholar]
- 244. Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, Qi Z, Downey SL, Manos PD, Rossi DJ, Yu J, Hebrok M, Hochedlinger K, Costello JF, Song JS, Ramalho-Santos M. 2011. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol 13:541–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Tokumoto Y, Ogawa S, Nagamune T, Miyake J. 2010. Comparison of efficiency of terminal differentiation of oligodendrocytes from induced pluripotent stem cells versus embryonic stem cells in vitro. J Biosci Bioeng 109:622–628 [DOI] [PubMed] [Google Scholar]
- 246. Hu Q, Friedrich AM, Johnson LV, Clegg DO. 2010. Memory in induced pluripotent stem cells: reprogrammed human retinal-pigmented epithelial cells show tendency for spontaneous redifferentiation. Stem Cells 28:1981–1991 [DOI] [PubMed] [Google Scholar]
- 247. Feng Q, Lu SJ, Klimanskaya I, Gomes I, Kim D, Chung Y, Honig GR, Kim KS, Lanza R. 2010. Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells 28:704–712 [DOI] [PubMed] [Google Scholar]
- 248. Taylor-Fishwick DA, Pittenger GL. 2010. Harnessing the pancreatic stem cell. Endocrinol Metab Clin North Am 39:763–776 [DOI] [PubMed] [Google Scholar]
- 249. Gunaseeli I, Doss MX, Antzelevitch C, Hescheler J, Sachinidis A. 2010. Induced pluripotent stem cells as a model for accelerated patient- and disease-specific drug discovery. Curr Med Chem 17:759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Maherali N, Hochedlinger K. 2008. Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell 3:595–605 [DOI] [PubMed] [Google Scholar]
- 251. Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. 2008. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321:1218–1221 [DOI] [PubMed] [Google Scholar]
- 252. Fiorina P, Folli F, Bertuzzi F, Maffi P, Finzi G, Venturini M, Socci C, Davalli A, Orsenigo E, Monti L, Falqui L, Uccella S, La Rosa S, Usellini L, Properzi G, Di Carlo V, Del Maschio A, Capella C, Secchi A. 2003. Long-term beneficial effect of islet transplantation on diabetic macro-/microangiopathy in type 1 diabetic kidney-transplanted patients. Diabetes Care 26:1129–1136 [DOI] [PubMed] [Google Scholar]
- 253. Chan KM, Raikwar SP, Zavazava N. 2007. Strategies for differentiating embryonic stem cells (ESC) into insulin-producing cells and development of non-invasive imaging techniques using bioluminescence. Immunol Res 39:261–270 [DOI] [PubMed] [Google Scholar]
- 254. Okita K, Hong H, Takahashi K, Yamanaka S. 2010. Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat Protoc 5:418–428 [DOI] [PubMed] [Google Scholar]
- 255. Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, Hess C, Zweigerdt R, Gruh I, Meyer J, Wagner S, Maier LS, Han DW, Glage S, Miller K, Fischer P, Schöler HR, Martin U. 2009. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 5:434–441 [DOI] [PubMed] [Google Scholar]
- 256. Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. 2009. How I treat ADA deficiency. Blood 114:3524–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Couri CE, Voltarelli JC. 2011. Stem cell-based therapies and immunomodulatory approaches in newly diagnosed type 1 diabetes. Curr Stem Cell Res Ther 6:10–15 [DOI] [PubMed] [Google Scholar]
- 258. Wu XH, Liu CP, Xu KF, Mao XD, Zhu J, Jiang JJ, Cui D, Zhang M, Xu Y, Liu C. 2007. Reversal of hyperglycemia in diabetic rats by portal vein transplantation of islet-like cells generated from bone marrow mesenchymal stem cells. World J Gastroenterol 13:3342–3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Oh SH, Muzzonigro TM, Bae SH, LaPlante JM, Hatch HM, Petersen BE. 2004. Adult bone marrow-derived cells trans-differentiating into insulin-producing cells for the treatment of type I diabetes. Lab Invest 84:607–617 [DOI] [PubMed] [Google Scholar]
- 260. Moriscot C, de Fraipont F, Richard MJ, Marchand M, Savatier P, Bosco D, Favrot M, Benhamou PY. 2005. Human bone marrow mesenchymal stem cells can express insulin and key transcription factors of the endocrine pancreas developmental pathway upon genetic and/or microenvironmental manipulation in vitro. Stem Cells 23:594–603 [DOI] [PubMed] [Google Scholar]
- 261. Li Y, Zhang R, Qiao H, Zhang H, Wang Y, Yuan H, Liu Q, Liu D, Chen L, Pei X. 2007. Generation of insulin-producing cells from PDX-1 gene-modified human mesenchymal stem cells. J Cell Physiol 211:36–44 [DOI] [PubMed] [Google Scholar]
- 262. Sun Y, Chen L, Hou XG, Hou WK, Dong JJ, Sun J, Tang KX, Wang B, Song J, Li H, Wang KX. 2007. Differentiation of bone marrow-derived mesenchymal stem cells from diabetic patients into insulin-producing cells in vitro. Chin Med J (Engl) 120:771–776 [PubMed] [Google Scholar]
- 263. Karnieli O, Izhar-Prato Y, Bulvik S, Efrat S. 2007. Generation of insulin-producing cells from human bone marrow mesenchymal stem cells by genetic manipulation. Stem Cells 25:2837–2844 [DOI] [PubMed] [Google Scholar]
- 264. Xie QP, Huang H, Xu B, Dong X, Gao SL, Zhang B, Wu YL. 2009. Human bone marrow mesenchymal stem cells differentiate into insulin-producing cells upon microenvironmental manipulation in vitro. Differentiation 77:483–491 [DOI] [PubMed] [Google Scholar]