

Abstract
Genetic mutations causing dysfunction of both voltage- and ligand-gated ion channels make a major contribution to the cause of many different types of familial epilepsy. Key mechanisms comprise defective Na+ channels of inhibitory neurons, or GABAA receptors affecting pre- or postsynaptic GABAergic inhibition, or a dysfunction of different types of channels at axon initial segments. Many of these ion channel mutations have been modelled in mice, which has largely contributed to the understanding of where and how the ion channel defects lead to neuronal hyperexcitability. Animal models of febrile seizures or mesial temporal epilepsy have shown that dendritic K+ channels, hyperpolarization-activated cation channels and T-type Ca2+ channels play important roles in the generation of seizures. For the latter, it has been shown that suppression of their function by pharmacological mechanisms or in knock-out mice can antagonize epileptogenesis. Defects of ion channel function are also associated with forms of acquired epilepsy. Autoantibodies directed against ion channels or associated proteins, such as K+ channels, LGI1 or NMDA receptors, have been identified in epileptic disorders that can largely be included under the term limbic encephalitis which includes limbic seizures, status epilepticus and psychiatric symptoms. We conclude that ion channels and associated proteins are important players in different types of genetic and acquired epilepsies. Nevertheless, the molecular bases for most common forms of epilepsy are not yet clear, and evidence to be discussed indicates just how much more we need to understand about the complex mechanisms that underlie epileptogenesis.
 |
Holger Lerche (left) is Clinical Director and Head of the Department of Neurology and Epileptology at the Hertie Institute of Clinical Brain Research at the University of Tübingen, Germany. His main research interest is to unravel the genetics and pathophysiology of inherited epilepsies and related paroxysmal disorders using a combination of genetic and neurophysiological tools. He is also interested in molecular ion channel function, their specific roles in the brain and their pharmacology. After graduating from the University of Munich (LMU), he worked as a postdoc in neurophysiology and as a resident and consultant in neurology and epileptology at the Institute of Applied Physiology and the Department of Neurology of the University of Ulm. He undertook clinical and research fellowships in Bonn/Germany, London/UK and Melbourne/Australia. Mala Shah (right) did her PhD at University College London (UCL, UK) under the supervision of Dr Dennis Haylett. She then obtained a Wellcome Prize Travel Research Fellowship to work in the laboratories of Professors Daniel Johnston at Baylor College of Medicine (Houston, USA) and David Brown at UCL (UK). She subsequently received a lectureship at UCL School of Pharmacy (UK) where she is currently a Reader in Neuroscience. Her research interests include understanding how voltage-gated ion channels activated at sub-threshold membrane potentials affect hippocampal and cortical cell excitability under physiological as well as epileptogenic conditions.
Introduction
The epilepsies are disorders of neuronal network excitability. They can be divided into two major groups. In the first group, which is called ‘symptomatic’, an acquired or inborn structural or metabolic defect of the brain can be identified as the underlying cause of the disease. These forms of epilepsies have a mainly focal origin meaning that the seizures start from a point around the structural lesion. The clinical presentation of the resulting epileptic seizures depends on the respective brain region in which the seizures start and spread, and can vary from light symptoms such as a strange feeling in the stomach or paresthesia in a certain body area, to loss of consciousness and severe convulsions. Typical examples for epileptogenic lesions are tumours, stroke or hippocampal sclerosis, the latter causing mesial temporal lobe epilepsy, one of the most frequent and often pharmacoresistant forms of focal epilepsy. An example of increasing clinical importance is given by epilepsies with antibodies directed against proteins involved in membrane excitability such as ion channels. The second group, termed ‘idiopathic’, is genetically determined and characterized by the lack of structural or other predisposing causes. Both focal and generalized forms of epilepsy can be caused by genetic defects and the resulting epileptic phenotypes can range from mild seizures occurring only in neonates or infants, to severe epileptic encephalopathies with mental retardation, pharmacoresistant epilepsy and other neurological symptoms. The most common disease entity is ‘idiopathic generalized epilepsy’ (IGE) comprising the well-known absence, myoclonic and primary generalized tonic–clonic seizures. The detection of mutations causing idiopathic forms of epilepsy has dramatically advanced our understanding about the pathophysiology in the last 15 years, which is one major topic covered in this review.
There are three main ways in which ion channels are known to be involved in epilepsy. Firstly, there are specific mutations in familial idiopathic epilepsies; secondly, there are specific antibodies in acquired seizure-related disorders; and thirdly, there are changes of ion channel expression and function associated with modification of seizure activity which may contribute to all forms of epilepsy. Here we review these rapidly developing areas using tables to provide more details. For the sake of brevity we will focus on disorders with the main symptom of epilepsy. Recent other developments, for example, increasing research into epileptic seizures in Alzheimer's disease or connections to autism, are not covered here.
Genetic defects in voltage-gated or ligand-gated ion channels
Table 1 lists gene mutations found in different epilepsies and Figure 1 shows the localization of some of the relevant channel proteins in different neuronal subtypes and compartments. Classical linkage methodology, on large pedigrees with rare monogenic syndromes, identified mutations in familial idiopathic focal epilepsies: for instance, the genes encoding nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) or potassium channels (KV7) in benign familial neonatal seizures (BFNS). In ‘sporadic’ cases, in which only one member of a family is affected, candidate gene approaches have been successful in identifying de novo pathogenic mutations, the most prominent being SCN1A nonsense mutations in Dravet syndrome A few mutations have also been identified in families with the most common IGEs, childhood and juvenile absence or myoclonic epilepsies, with different GABAA receptor subunit defects being of particular importance (Table 1). However, the substantial complexity of ion channel gene variant profiles among individuals with epilepsy as well as unaffected controls precludes a simple monogenic channelopathy model in the majority of cases, and particularly IGE is considered to be a prototype of a complex genetic disorder in which many – both rare and common – genetic variations play a pathogenic role. Pathophysiological studies demonstrated that two key defects are: (i) a neuronal dysinhibition that can be caused both by loss-of-function defects in different subunits of the postsynaptic GABAA receptor and presynaptic loss-of-function defects of the sodium channel NaV1.1 expressed specifically in inhibitory interneurons, or (ii) dysfunction of axon initial segments, the neuronal structure in which action potentials are generated and in which many of the described channels (for example, NaV1.2 sodium and KV7 potassium channel subunits) are mainly localized. In addition, these clinically originated studies identified novel genes, defined their neuronal functions and sometimes established new physiological principles (such as NaV1.1 as the major sodium channel in GABAergic interneurons). Moreover, KV7 channels have proven to be a novel therapeutic target (reviewed by Weber & Lerche, 2008; Reid et al. 2009).
Table 1.
Genes and proteins mutated in idiopathic epilepsies and epileptic encephalopathies
| Abbreviation | Gene | Protein | References | |
|---|---|---|---|---|
| Idiopathic focal epilepsies | ||||
| Benign familial neonatal seizures | BFNS1/EBN1 | KCNQ2 | KV7.2 (K+ channel) | Biervert et al. 1998; Singh et al. 1998 |
| BFNS2/EBN2 | KCNQ3 | KV7.3 (K+ channel) | Charlier et al. 1998 | |
| Benign familial neonatal–infantile seizures | BFNIS | SCN2A | NaV1.2 (Na+ channel) | Heron et al. 2002; Berkovic et al. 2004 |
| Autosomal dominant nocturnal frontal lobe epilepsy | ADNFLE | CHRNA4 | α4 subunit (nACh) receptor | Steinlein et al. 1995 |
| CHRNB2 | β2 subunit (nACh) receptor | De Fusco et al. 2000 | ||
| CHRNA2 | α2 subunit (nACh) receptor | Aridon et al. 2006 | ||
| Idiopathic generalized epilepsies and associated syndromes | ||||
| Childhood absence epilepsy with febrile seizures | CAE+FS | GABRG2 | γ2 subunit (GABAA receptor) | Wallace et al. 2001 |
| Absence epilepsy and episodic ataxia | CAE+EA2 | CACNA1A | CaV2.1 (Ca2+ channel) | Jouvenceau et al. 2001; Imbrici et al. 2004 |
| Juvenile myoclonic epilepsy | JME | GABRA1 | α1 subunit (GABAA) receptor | Cossette et al. 2002 |
| EFHC1 | EF hand motif protein | Suzuki et al. 2004 | ||
| Genetic (generalized epilepsy with febrile seizures plus (GEFS+) | SCN1A | NaV1.1 (Na+ channel) | Escayg et al. 2000 | |
| GEFS+ | SCN1B | β1 subunit (nACh receptor) | Wallace et al. 1998 | |
| GABRG2 | γ2 subunit (GABAA) receptor | Baulac et al. 2001 | ||
| Generalized epilepsy and paroxysmal dyskinaesia | GEPD | KCNMA1 | KCa1.1 (K+ channel) | Du et al. 2005 |
| Epileptic encephalopathies | ||||
| Dravet syndrome (severe myoclonic epilepsy of infancy) | SMEI | SCN1A | NaV1.1 (Na+ channel) | Claes et al. 2001 |
| Other syndromes | ||||
| Focal epilepsy and episodic ataxia | EA1+FE | KCNA1 | KV1.1 (K+ channel) | Zuberi et al. 1999 |
The table lists part of the affected ion channel genes in humans illustrating how different clinical syndromes arise from inherited disorders of ion channels. Only unequivocally proven genetic defects that have been described more than once in the literature are included. Full references for Table 1 can be found in Supplementary information. The neuronal localization of some of the channel proteins is shown in Figure 1.
Figure 1. Neuronal localization of some relevant voltage- and ligand-gated ion channels.
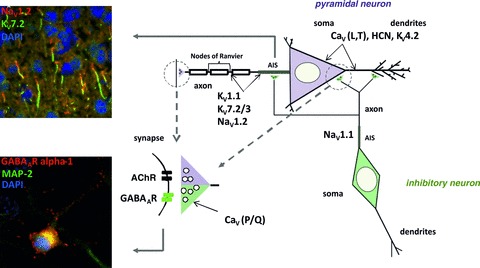
Shown is a schematic view of an excitatory pyramidal (purple) and an inhibitory (green) neuron and their synaptic connections. Distinctive intracellular compartments are targeted by different populations of ion channels, examples of which as mentioned in this review are shown here: in the somatodendritic compartment, CaV (L- and T-type), HCN and some KV channels; at axon initial segments (AIS) and nodes of Ranvier in pyramidal neurons, KV1.1, NaV1.2, KV7.2, KV7.3; at AIS of inhibitory neurons, NaV1.1; in the presynaptic terminals, CaV P/Q type; in the postsynaptic compartment, GABAA and acetylcholine receptors. Upper insert: Colocalization of KV7.2 and NaV1.2 channels at AIS of cortical neurons in an adult mouse brain, as revealed by immunofluorescent staining using an anti-KV7.2 (green) and an anti-NaV1.2 (red) antibody of sections obtained from an unfixed brain; DAPI (blue) was used to mark the nuclei. Lower insert: Distribution of GABAA receptors in a primary cultured hippocampal neuron shown by immunofluorescent staining using an anti-GABAAR alpha-1 subunit antibody (red). An anti-MAP2 antibody (green) was used as a somatodendritic and DAPI (blue) as a nuclear marker (Figure kindly provided by Dr. Snezana Maljevic, modified after Maljevic and Lerche, 2011).
Novel genetic technologies allowing sequencing of the whole coding genomic DNA (whole exome) or even the whole genome now allow identification of new mutations and involvement of loci that could not be efficiently explored before by classical methods. As an example for another ion channel alteration in epilepsy, a de novo mutation was recently identified in the sodium channel gene SCN8A in a patient with a severe epileptic encephalopathy. This mutation leads to a dramatic gain-of-function with increased sodium inward current and membrane hyperexcitability (Veeramah et al. 2012).
Ion channel defects modelled in mice
Transgenic and spontaneously mutant mice have proven crucial for identifying novel molecular pathways for epilepsy, validating the causality of candidate genes isolated from human pedigrees, and for unravelling their pathogenic mechanisms. Beginning with the first spontaneous single gene murine model of epilepsy, the mutant mouse tottering, mutations in at least 17 distinct genes have been reported that produce spontaneous electrographic seizures accompanied by behavioural manifestations (Table 2). Along with these spontaneous models, genetic engineering has allowed the creation of an expanding list of targeted null alleles, transgenic overexpression using selected or endogenous promoters, and knock-in of human mutant point mutations.
Table 2.
Genes for voltage-gated ion channel subunits with spontaneous epilepsy phenotypes in mice
| Gene | Current/protein | Seizure type | Mutation Ko, knock-out Ki, knock-in | Reference |
|---|---|---|---|---|
| Sodium | ||||
| Scna1a | Nav1.1 α subunit | convulsive | Ko | Yu et al. 2006; Ogiwara et al. 2007 |
| Scna1a | Targeted R1648H human Ki | Martin et al. 2010 | ||
| Scna1b | Ko | Chen et al. 2004 | ||
| Scn2a | Nav1.2 α subunit | convulsive | transgenic GAL879-881QQQ | Kearney et al. 2001 |
| Calcium | ||||
| Cacna1a | Cav2.1 P/Q-type α subunit | absence | Spontaneous alleles (tottering, leaner, rocker roller, tg4J, tg5J) | Fletcher et al. 1996; Zwingman et al. 2001; Mori et al. 2000; Miki et al. 2008 |
| Cacna1a | Cav2.1 P/Q-type | absence | Cerebellar selective PCP2-Cre promoter Ko | Mark et al. 2011 |
| Cacna1a | Cav2.1 P/Q-type | absence | Ko | Llinas et al. 2007 |
| Cacnb4 | β4 regulatory subunit | absence | Spontaneous (lethargic) | Burgess et al. 1997 |
| Cacna1g | Cav3.1 T-type α subunit | absence | Bac transgene overexpression | Ernst et al. 2009 |
| Cacna2d2 | α2δ regulatory subunit | absence | Spontaneous several alleles | Barclay et al. 200; Brill et al. 2004 |
| Potassium | ||||
| Kcna1 | Kv1.1 | convulsive | Ko | Smart et al. 1998 |
| Kcna1 | Kv1.1 | absence | Overexpression | Sutherland et al. 1999 |
| Kcna2 | Kv1.2 | convulsive | Ko | Brew et al. 2007 |
| Kcnh3 | Kv12.2 | non-convulsive | Ko | Shang et al. 2010 |
| Kcnmb4 | β4 regulatory subunit calcium-activated | non-convulsive | Ko | Brenner et al. 2005 |
| Kcnc2 | Kv3.2 | convulsive | Ko | Lau et al. 2000 |
| KcnJ6 | Girk2 ATP sensitive | convulsive | Ko | Signorini et al. 1997 |
| Kcnq1 | Kv7.1 | convulsive | Human Ki T311I A340E | Goldman et al. 2009 |
| Kcnq2 | Kv7.2 | convulsive | Human Ki A306T | Singh et al. 2008 |
| Kcnq3 | Kv7.3 | convulsive | Human Ki G311V | Singh et al. 2008 |
| Cyclic nucleotide gated | ||||
| Hcn2 | Ih hyperpolarization-activated cyclic nucleotide-gated | absence | Ko spontaneous apathetic | Ludwig et al. 2003; Chung et al. 2009 |
| Chloride | ||||
| Clcn3 | Ichloride | convulsive | Ko | Dickerson et al. 2002 |
Full references can be found in Supplementary information. The neuronal localization of some of the channel proteins is shown in Figure 1.
Unexpected pathogenic mechanisms are emerging from comparative analysis of current defects in excitatory and inhibitory networks. For instance, as already mentioned above, different types of sodium channels expressed in both glutamatergic and GABAergic cell types play unequal roles in excitability, providing an explanation for the network disinhibition arising from Scna1 deletion (Yu et al. 2006; Ogiwara et al. 2007). Another example is how mutations of calcium Cacna1a and Cacnb4 channel subunit genes that decrease P/Q-type currents may lead to the ‘acquired’ downstream enhancement of low-voltage T-type calcium currents sufficient to produce thalamocortical spike-wave epilepsy (Zhang et al. 2002, Ernst et al. 2009).
Genotype–phenotype correlations of inherited ion channel disorders are far from being well understood. For example, gain- and loss-of-function mutations in the same gene, or in related members of the same gene family, can give rise to alternative seizure phenotypes. In addition, even when the electrographic seizures themselves appear similar, epilepsies arising from different ion channel genes may be accompanied by remarkably different behaviours and co-morbidities, such as the absence of hippocampal remodelling (Singh et al. 2008), or sudden unexpected death (Goldman et al. 2009; Glasscock et al. 2010) in the K+ channel family.
Various specific digenic interactions have been explored in mouse models, illustrating the prominent additive (Kearney et al. 2006; Hawkins et al. 2011), and suppressant (Glasscock et al. 2007), effects of ion channel mutations on epilepsy phenotypes. These pairwise combinations are highly informative, but still too simple to explain the complex inheritance of human epilepsy.
Recent analysis of ion channel mutation profiles in idiopathic epilepsy reveals a marked complexity, with no two individuals showing sharing gene variant pattern, but similar channel variant pattern diversity, including ‘deleterious’ variants in known ‘monogenic’ epilepsy genes, in unaffected individuals (Klassen et al. 2011). In mouse models, it is well known, but often not emphasized, that epilepsy phenotypes, as well as induced seizure thresholds (Frankel et al. 2001), are strongly dependent on the genetic background of inbred mouse strains. Thus, the contribution of specific ion channel defects to brain excitability disorders is complex even when they are associated with a clear mendelian disease phenotype in a particular mouse strain or human pedigree. This complexity poses a major challenge to the clinical genetic assessment of the individual's epilepsy risk.
Ion channel plasticity in acquired epilepsy
Temporal lobe epilepsy (TLE), which is the most common form of acquired epilepsy, can be initiated by an insult such as traumatic head injury, febrile seizures or status epilepticus (Engel, 1996), but inflammatory or autoimmune diseases may also be a cause (Bien et al. 2007). Following these insults, there is often a ‘latent period’ before the onset of chronic TLE. The insult preceding TLE can be mimicked in animals by the administration of chemoconvulsants such as kainic acid or pilocarpine to induce status epilepticus (SE), or kindling in which focal seizures are repeatedly induced using electrical stimulation and febrile seizures. Studies both on the mouse tissue and human tissue from TLE patients have provided valuable information on some of the potential mechanisms underlying TLE. In excitatory glutamatergic neurons, the cell surface expression and biophysical properties of numerous voltage-gated ion channels are altered (Table 3), leading to fundamentally altered integrative properties of these neurons.
Table 3.
Voltage-gated ion channels that undergo plasticity during acquired epilepsies
| Channel/current | Form of epilepsy | Nature of change | Impact on cell excitability | Reference |
|---|---|---|---|---|
| HCN channel/ HCN current Ih | Temporal lobe epilepsy (TLE) | Sustained reduction in current density following status epilepticus (SE) induction | Enhanced pyramidal and interneuron excitability | Dugladze et al. 2007; Jung et al. 2007; Marcelin et al. 2009; Shah et al. 2004; Shah et al. 2012; Shin et al. 2008; Wierschke et al. 2010 |
| HCN channels/ HCN current Ih | Febrile seizure-induced epilepsy | Enhanced HCN channel expression and current | Enhanced rebound activity following inhibitory post-synaptic potentials (IPSPs) | Bender et al. 2003; Chen et al. 2001; Dyhrfjeld-Johnsen et al. 2008 |
| HCN current Ih | Fragile X syndrome | Enhanced HCN channel current | Impaired long-term potentiation (LTP in pyramidal neurons) | Brager et al. 2012 |
| CaV3.2 channels/ T-type Ca2+ current | TLE | Transient elevation of expression and current from SE to chronic epilepsy | Enhanced pyramidal cell bursting | Becker et al. 2008; Su et al. 2002 |
| KV4.2 channels/ A-type K+ current | TLE | Reduction 1 week after SE and persisting during chronic TLE | Enhanced pyramidal cell dendritic excitability | Bernard et al. 2004; Monaghan et al. 2008 |
| BK channels | TLE | Reduction in expression during chronic TLE | ?? | Pacheco Otalora et al. 2008 |
| Kir2 channels/ inward rectifier current | Chronic TLE | Enhanced expression | Reduced dentate gyrus granule cell excitability | Young et al. 2009 |
| KCNN1 (SK1) KCNN2 (SK2) and KCNN3 (SK3) channels | TLE | Transient reduction during chronic TLE | Increased number of hippocampal population spikes | Oliveira et al. 2010 |
| Persistent sodium current | TLE | Sustained increase following SE | Enhanced neuronal excitability | Agrawal et al. 2003; Chen et al. 2011; Epsztein et al. 2010; Hargus et al. 2011; Vreugdenhil et al. 2004 |
Full references can be found in Supplementary information. The neuronal localization of some of the channel proteins is shown in Figure 1.
The expression and function of one particular ion channel, the hyperpolarization-activated cyclic nucleotide gated (HCN) channel, is implicated in these changes. HCN channel activity is reduced in the cortex and hippocampus within a few hours of SE induction in animal models (Shah et al. 2004; Jung et al. 2011), and the reduction persists for weeks in the models (Shah et al. 2012). Furthermore, a decrease in HCN mRNA and current function has also been found in cortical and hippocampal tissue obtained from TLE patients (Bender et al. 2003; Wierschke et al. 2010), suggesting that decreased HCN channel current is playing a role in epileptogenesis.
HCN channels are cation channels that are activated at potentials more hyperpolarized to −40 mV (Biel et al. 2009). They are predominantly expressed in cortical and hippocampal pyramidal cell dendrites where, intriguingly, they reduce pyramidal cell excitability by restricting synaptic integration and excitability (Shah et al. 2010). HCN channels, however, are also expressed in certain interneurons (Aponte et al. 2006; Dugladze et al. 2007; Matt et al. 2011) as well as pre-synaptically (Bender et al. 2003; Aponte et al. 2006; Huang et al. 2011).
Thus, does a reduction in HCN channel function per se facilitate the onset of chronic TLE? Four HCN subunits (HCN1–4) have been cloned to date. HCN1 subunits are predominantly expressed in the cortex and hippocampus (Biel et al. 2009) and HCN1 null mice are more susceptible to seizures induced by chemoconvulsants or kindling, suggesting that a loss of HCN channel function is likely to enhance epileptogenesis (Huang et al. 2009; Santoro et al. 2010). Further, transiently restoring HCN channel expression by disrupting the interaction between the neuron-restrictive silence factor (NRSF) and HCN1, delays the onset of spontaneous seizure activity (McClelland et al. 2011), and several anti-convulsants used for TLE, such as lamotrigine, augment HCN channel function (Poolos et al. 2002).
By contrast, in some forms of acquired epilepsy such as that following febrile seizures, HCN channel expression and function is increased in hippocampal pyramidal neurons (Chen et al. 2001; Brewster et al. 2002; Dyhrfjeld-Johnsen et al. 2008). Moreover, dendritic HCN current, Ih, is enhanced in hippocampal pyramidal neurons in mice with targeted deletions in the fragile X FMR1 gene (Brager et al. 2012), although FMR1 knock-out mice do not exhibit spontaneous seizures but are more susceptible to audiogenic seizures (Musumeci et al. 2000). Similarly, only about a third of the rodents subjected to febrile seizures develop chronic epilepsy (Walker & Kullmann, 1999; Dube et al. 2009). Hence, whether Ih upregulation under these conditions is a homeostatic change or epileptogenic remains to be further investigated.
In addition to HCN channels, the mRNA levels and activity of the low-threshold T-type Ca2+, CaV3.2, channel are transiently elevated in the hippocampus following SE induction (Su et al. 2002; Becker et al. 2008). Inhibition of these channels is also likely to benefit TLE treatment as seizure frequency and incidence is significantly lowered in CaV3.2 null mice (Becker et al. 2008). Intriguingly, hippocampal mossy fibre sprouting, a hallmark of TLE, is absent following SE induction in CaV3.2 null mice (Becker et al. 2008), suggesting that Ca2+ entry through CaV3.2 channels may have effects additional to alterations in neuronal excitability.
Although there is considerable evidence that HCN and CaV3.2 channels undergo plasticity very early on in the epileptic process, other channels are likely to be involved in seizure induction. Certainly during chronic TLE, the expression and biophysical properties of a substantial number of K+ and Na+ channels are altered, examples of which are shown in Table 3. A better understanding of how ion channel expression and function contributes to epileptogenesis, is important and may lead to more targeted treatments for TLE and other epilepsies.
Ion channel plasticity contributing to epileptic phenotypes may also occur following inherited lesions in non-ion channel gene mutations. A striking example is the aberrant excitability and electrographic seizures identified in models of amyloid precursor protein beta (Abeta) overexpression (Palop et al. 2007). The seizure activity emanates from the hippocampal and neocortical circuitry and has been described in a wide variety of human and mouse models of Alzheimer's disease (Noebels, 2011). Recent analysis of tissue from Alzheimer's disease patients and a mouse model identified decreased levels of sodium NaV1.1 currents and SCN1A subunit expression in parvalbumin-containing interneurons (Verret et al. 2012). When these currents were restored in the mouse model by transgenic expression, inhibitory synaptic activity and gamma oscillations were restored, hypersynchrony was reduced and memory deficits were ameliorated, indicating that ion channel-mediated disinhibition makes a strong contribution to epileptogenesis in this model.
Antibodies to voltage-gated or ligand-gated ion channels
Antibodies to AMPAR3 were reported in a few patients with Rasmussen's encephalitis, a devastating unihemispheric condition in childhood (Rogers et al. 1994), but they are not found frequently among cases of this rare epilepsy syndrome (Watson et al. 2004). More recently, antibodies to other neuronal surface proteins have been found in patients with seizures presenting as part of a more widespread inflammatory brain disorder (see Vincent et al. 2011a). The ion channel and related proteins that are the targets for these specific antibodies are listed in Table 4.
Table 4.
Seizure-related syndromes associated with antibodies to ion channels or receptors
| Target channel | Antibodies to: | Clinical syndrome | Clinical features | Associated features | Key references |
|---|---|---|---|---|---|
| Kv1 complex includes LGI1, CASPR2 | LGI1, CASPR2 | Limbic encephalitis | Amnesia, change in personality or psychosis, temporal lobe and other seizure types, mainly partial complex seizures | Serum hyponatraemia | Vincent et al. 2004; Irani et al. 2010 |
| Kv1 | LGI1 | Faciobrachial dystonic seizures (FBDS) | Brief frequent dystonic seizures usually unilateral often involving the arm and ipsilateral face | Can precede limbic encephalitis Often resistant to AEDs Immunotherapy-responsive | Irani et al. 2011 |
| AMPAR1/2 | AMPAR1/AMPAR2 mainly | Limbic encephalitis | As above but with more evident psychosis | Lai et al. 2009 | |
| AMPAR3 | AMPAR3 in a few reports, otherwise none defined | Rasmussen's encephalitis | Intractable unilateral seizures with hemiplegia | Patients develop epilepsia partialis continua (EPC) | Bien et al. 2004 |
| GABAbR | GABAbR | Limbic encephalitis | As above but often dominated by TLE | Lancaster et al. 2010 | |
| NMDAR | NR1 | Psychiatric features and seizures | Seizures are part of the presentation but are not well defined. | Most patients progress over days or weeks to a complex encephalopathy | Dalmau et al. 2011; Niehusmann et al. 2009 |
Full references can be found in Supplementary information. The neuronal localization of some of the channel proteins is shown in Figure 1.
Most of the patients have a form of limbic encephalitis – a syndrome that includes amnesia, seizures and psychiatric or behavioural changes, and MRI or cerebrospinal fluid (CSF) evidence of inflammation (Vincent et al. 2004). Antibodies that immunoprecipitate dendrotoxin-labelled KV1 channels extracted from rodent brain tissue, and bind to LGI1, a secreted protein tightly associated with these channels in situ (see also Table 1), are also found in a recently described seizure type termed faciobrachial dystonic seizures. These involve brief (a few seconds), very frequent (up to 200 per day), usually unilateral dystonic epileptic events. These seizures may precede the onset of limbic encephalitis, and are frequently anti-epileptic drug (AED) resistant, but seizure frequency is markedly reduced following immunotherapies (Irani et al. 2011). Antibodies that immunoprecipitate the KV1 complexes are also found in a proportion (around 10–15%) of patients with idiopathic forms of epilepsy (McKnight et al. 2005; Vincent et al. 2011b; Quek et al. 2012).
Most antibodies reduce the surface expression of their target by binding divalently to adjacent cell-surface proteins and causing their internalisation – a mechanism that has been shown to apply to NMDAR antibodies binding to hippocampal neurons in culture (Hughes et al. 2010). Direct functional effects on ion channel function have not yet been demonstrated, but purified IgG from one patient with KV1/LGI1 antibodies increased the release probability at the mossy fibre–CA3 synapse, similar to that of dendrotoxin, and suggesting that the antibodies modify KV1 channel function (Lalic et al. 2011). It is still early days and these disorders present many challenges including how and where the antibodies access the brain parenchyma, whether they induce permanent damage with possible compensatory changes, or changes that are fully reversible, and the relative roles of serum and CSF antibodies.
Challenges and questions for the future
Although a large amount of the data on ion channel defects shed light on the aetiology of seizures in both genetic and acquired epilepsies, there is still a lack of understanding of the precise mechanisms underlying epileptogenesis leading to a chronically epileptic brain. In addition to adding to the cellular and molecular changes that occur in epilepsy, it will be necessary to ask new qualitatively different questions.
How complex are the genetic epilepsies and what is the role of genetic background in defining the disease phenotype?
Is it justified to call the familial forms of epilepsy with known gene mutations monogenic? There is no doubt that many single genes are responsible for the respective epilepsy syndromes, as summarised in Table 1, but all these syndromes also show considerable phenotypic variability. For example, mutations with complete loss of function of SCN1A are found more frequently in Dravet syndrome than in generalized epilepsy with febrile seizures plus (GEFS+), which is more often caused by missense mutations; however, the opposite relationship has also been described (http://www.molgen.vib-ua.be/SCN1AMutations/). The inconsistant relationship between the phenotype and a single monogenic mutation may be explained in part by other co-expressed ion channel variants in the genomic background, as seen in mouse models. Large sequencing efforts in groups with different phenotypes might be able to detect such variability in humans, but proof-of-concept studies are so far lacking.
Despite the existence of monogenic or major gene effects in some patients (Table 1), in the majority such defects have not been found (Heinzen et al. 2012). Search for individual patterns of variability among ion channels and related proteins has not yet provided clear differences between patients and controls (Klassen et al. 2011). However, more detailed bioinformatic analyses may demonstrate pathways that are affected more commonly in cases than in controls. In addition, the genetic variation in ion channel genes is only one piece of the puzzle. Variations in copy numbers of several chromosomal regions are significantly more frequent in IGE patients than in controls (Sisodiya & Mefford, 2011), suggesting non-ion channel genetic influences. Time will tell whether whole exome or genome sequencing efforts will be able to clarify a significant part of the genetic origin of complex genetic epilepsies.
Are there neuronal compartments that have been insufficiently examined with respect to epilepsy-related changes?
While a large number of beautiful studies have addressed changed properties of somatodendritic compartments, we currently know very little about the local integrative properties of small-calibre dendrites, even though these processes receive the majority of excitatory inputs in most excitatory neurons. It will be necessary to apply techniques suitable for the analysis of small-calibre dendrites such as multiphoton glutamate uncaging and imaging, along with novel techniques for obtaining patch-clamp recording from these processes, to study local integration at these sites. Similar approaches will be required to interrogate other critical excitability compartments, including the axon initial segment and presynaptic terminals. The properties of aberrantly sprouted axons and their collateral terminals involved in seizure-induced neosynaptogenesis also require exploration.
How are changes integrated at the mesoscale (i.e. the level of micronetworks and assemblies of these hundreds of neurons)?
The manifestations of all neurological disorders, such as seizures in the case of epilepsy, rely on a disturbed interplay of different neuron types in the CNS. However, in both genetic and acquired epilepsies, we know surprisingly little about the precise changes in excitability and synaptic integration in different types of principal neurons and interneurons. In the case of some genetic epilepsy models, changes in interneurons appear to be crucial in mediating hyperexcitability, but these studies have so far not dissected the role of specific interneuron types (Yu et al. 2006; Ogiwara et al. 2007). It will undoubtedly be important to understand disease-related modifications in different, well-defined cell types in the CNS. This applies to genetic epilepsy models, the effects of putative disease-related autoantibodies, as well as to acquired epilepsies. Furthermore, it will be necessary to apply and further develop the techniques to record and manipulate the activity of large-scale networks of principal cells and interneurons in normal and diseased brain in order to understand how the different neuron types interact in the epileptic brain to cause aberrant synchronization (Feldt et al. 2011).
What is the in vivo relevance for changes in ion channels observed in vitro?
The manifold changes in ion channels, described in part above, predict powerful changes of synaptic integration and neuronal input–output behaviour. However, it would be highly desirable to identify how neuronal firing behaviour in vivo changes in chronic epilepsy. Studies using juxtacellular multielectrode arrays, single-unit, or direct intracellular and patch-clamp recordings as well as in vivo imaging techniques will be required to probe the in vivo relevance of in vitro membrane excitability findings.
What is the role of homeostasis in defining the disease phenotype?
Neurons, and most probably all neural cells, have an immense inherent capability for homeostasis, manifested as the ability to conserve functional properties in the face of continuous environmental perturbation and turnover of their proteolipid components. The arsenal of neuronal homeostatic mechanisms includes regulation of both synaptic and intrinsic properties to maintain neuronal functions. In view of this fact, it is perhaps surprising that in epilepsy the underlying ion channel defects are not homeostatically compensated. It will be important to understand fully which homeostatic mechanisms are active under normal excitability conditions in order to identify the conditions under which they fail.
Glossary
- IGE
idiopathic generalized epilepsy
- SE
status epilepticus
- TLE
temporal lobe epilepsy
Supplementary material
Supplementary information
References
- Aponte Y, Lien CC, Reisinger E, Jonas P. Hyperpolarization-activated cation channels in fast-spiking interneurons of rat hippocampus. J Physiol. 2006;574:229–243. doi: 10.1113/jphysiol.2005.104042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AJ, Pitsch J, Sochivko D, Opitz T, Staniek M, Chen CC, et al. Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J Neurosci. 2008;28:13341–13353. doi: 10.1523/JNEUROSCI.1421-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Soleymani SV, Brewster AL, Nguyen ST, Beck H, Mathern GW, Baram TZ. Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci. 2003;23:6826–6836. doi: 10.1523/JNEUROSCI.23-17-06826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Bien CG, Urbach H, Schramm J, Soeder BM, Becker AJ, Voltz R, et al. Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology. 2007;69:1236–1244. doi: 10.1212/01.wnl.0000276946.08412.ef. [DOI] [PubMed] [Google Scholar]
- Brager DH, Akhavan AR, Johnston D. Impaired dendritic expression and plasticity of h-channels in the fmr1−/y mouse model of fragile X syndrome. Cell Rep. 2012;1:225–233. doi: 10.1016/j.celrep.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, Baram TZ. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev. 2009;31:366–371. doi: 10.1016/j.braindev.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugladze T, Vida I, Tort AB, Gross A, Otahal J, Heinemann U, et al. Impaired hippocampal rhythmogenesis in a mouse model of mesial temporal lobe epilepsy. Proc Natl Acad Sci U S A. 2007;104:17530–17535. doi: 10.1073/pnas.0708301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Morgan RJ, Foldy C, Soltesz I. Upregulated H-current in hyperexcitable CA1 dendrites after febrile seizures. Front Cell Neurosci. 2008;2:2. doi: 10.3389/neuro.03.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JJ. Introduction to temporal lobe epilepsy. Epilepsy Res. 1996;26:141–150. doi: 10.1016/s0920-1211(96)00043-5. [DOI] [PubMed] [Google Scholar]
- Ernst WL, Zhang Y, Yoo JW, Ernst SJ, Noebels JL. Genetic enhancement of thalamocortical network activity by elevating α1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J Neurosci. 2009;29:1615–1625. doi: 10.1523/JNEUROSCI.2081-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldt S, Bonifazi P, Cossart R. Dissecting functional connectivity of neuronal microcircuits: experimental and theoretical insights. Trends Neurosci. 2011;34:225–236. doi: 10.1016/j.tins.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Frankel WN, Taylor L, Beyer B, Tempel BL, White HS. Electroconvulsive thresholds of inbred mouse strains. Genomics. 2001;74:306–312. doi: 10.1006/geno.2001.6564. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis. 2011;41:655–660. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen EL, Depondt C, Cavalleri GL, Ruzzo EK, Walley NM, Need AC, et al. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am J Hum Genet. 2012;91:293–302. doi: 10.1016/j.ajhg.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC, Shah MM. Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci. 2011;14:478–486. doi: 10.1038/nn.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Walker MC, Shah MM. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009;29:10979–10988. doi: 10.1523/JNEUROSCI.1531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30:5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69:892–900. doi: 10.1002/ana.22307. [DOI] [PubMed] [Google Scholar]
- Jung S, Warner LN, Pitsch J, Becker AJ, Poolos NP. Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci. 2011;31:14291–14295. doi: 10.1523/JNEUROSCI.1148-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JA, Yang Y, Beyer B, Bergren SK, Claes L, Dejonghe P, Frankel WN. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum Mol Genet. 2006;15:1043–1048. doi: 10.1093/hmg/ddl019. [DOI] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalic T, Pettingill P, Vincent A, Capogna M. Human limbic encephalitis serum enhances hippocampal mossy fiber-CA3 pyramidal cell synaptic transmission. Epilepsia. 2011;52:121–131. doi: 10.1111/j.1528-1167.2010.02756.x. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Lerche H. Epileptogenesis in idiopathic epilepsy. In: Shorvon S, Andermann F, Guerrini R, editors. Cambridge University Press; 2011. The Causes of Epilepsy, Chapter 2. [Google Scholar]
- McClelland S, Flynn C, Dube C, Richichi C, Zha Q, Ghestem A, et al. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol. 2011;70:454–464. doi: 10.1002/ana.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight K, Jiang Y, Hart Y, Cavey A, Wroe S, Blank M, et al. Serum antibodies in epilepsy and seizure-associated disorders. Neurology. 2005;65:1730–1736. doi: 10.1212/01.wnl.0000187129.66353.13. [DOI] [PubMed] [Google Scholar]
- Matt L, Michalakis S, Hofmann F, Hammelmann V, Ludwig A, Biel M, Kleppisch T. HCN2 channels in local inhibitory interneurons constrain LTP in the hippocampal direct perforant path. Cell Mol Life Sci. 2011;68:125–137. doi: 10.1007/s00018-010-0446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, Oostra BA. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 2000;41:19–23. doi: 10.1111/j.1528-1157.2000.tb01499.x. [DOI] [PubMed] [Google Scholar]
- Noebels J. A perfect storm: Converging paths of epilepsy and Alzheimer's dementia intersect in the hippocampal formation. Epilepsia. 2011;52:39–46. doi: 10.1111/j.1528-1167.2010.02909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Ly NB, Yoo JW, Ho1 KO, Yu1 GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012 doi: 10.1001/archneurol.2011.2985. (Epublish ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87:41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science. 1994;265:648–651. doi: 10.1126/science.8036512. [DOI] [PubMed] [Google Scholar]
- Santoro B, Lee JY, Englot DJ, Gildersleeve S, Piskorowski RA, Siegelbaum SA, et al. Increased seizure severity and seizure-related death in mice lacking HCN1 channels. Epilepsia. 2010;51:1624–1627. doi: 10.1111/j.1528-1167.2010.02554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Hammond RS, Hoffman DA. Dendritic ion channel trafficking and plasticity. Trends Neurosci. 2010;33:307–316. doi: 10.1016/j.tins.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Huang Z, Martinello K. HCN and KV7 (M-) channels as targets for epilepsy treatment. Neuropharmacology. 2012 doi: 10.1016/j.neuropharm.2012.03.005. (Epublish ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NA, Otto JF, Dahle EJ, Pappas C, Leslie JD, Vilaythong A, et al. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J Physiol. 2008;586:3405–3423. doi: 10.1113/jphysiol.2008.154971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodiya SM, Mefford HC. Genetic contribution to common epilepsies. Curr Opin Neurol. 2011;24:140–145. doi: 10.1097/WCO.0b013e328344062f. [DOI] [PubMed] [Google Scholar]
- Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, Beck H. Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci. 2002;22:3645–3655. doi: 10.1523/JNEUROSCI.22-09-03645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011a;10:759–772. doi: 10.1016/S1474-4422(11)70096-5. [DOI] [PubMed] [Google Scholar]
- Vincent A, Buckley C, Schott JM, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701–712. doi: 10.1093/brain/awh077. [DOI] [PubMed] [Google Scholar]
- Vincent A, Irani SR, Lang B. Potentially pathogenic autoantibodies associated with epilepsy and encephalitis in children and adults. Epilepsia. 2011b;52:8–11. doi: 10.1111/j.1528-1167.2011.03224.x. [DOI] [PubMed] [Google Scholar]
- Walker MC, Kullmann DM. Febrile convulsions: a ‘benign’ condition. Nat Med. 1999;5:871–872. doi: 10.1038/11308. [DOI] [PubMed] [Google Scholar]
- Watson R, Jiang Y, Bermudez I, Houlihan L, Clover L, McKnight K, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology. 2004;63:43–50. doi: 10.1212/01.wnl.0000132651.66689.0f. [DOI] [PubMed] [Google Scholar]
- Weber YG, Lerche H. Genetic mechanisms in idiopathic epilepsies. Dev Med Child Neurol. 2008;50:648–654. doi: 10.1111/j.1469-8749.2008.03058.x. [DOI] [PubMed] [Google Scholar]
- Wierschke S, Lehmann TN, Dehnicke C, Horn P, Nitsch R, Deisz RA. Hyperpolarization-activated cation currents in human epileptogenic neocortex. Epilepsia. 2010;51:404–414. doi: 10.1111/j.1528-1167.2009.02275.x. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mori M, Burgess DL, Noebels JL. Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J Neurosci. 2002;22:6362–6371. doi: 10.1523/JNEUROSCI.22-15-06362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.