

Abstract
Epilepsy is characterised by the propensity of the brain to generate spontaneous recurrent bursts of excessive neuronal activity, seizures. GABA-mediated inhibition is critical for restraining neuronal excitation in the brain, and therefore potentiation of GABAergic neurotransmission is commonly used to prevent seizures. However, data obtained in animal models of epilepsy and from human epileptic tissue suggest that GABA-mediated signalling contributes to interictal and ictal activity. Prolonged activation of GABAA receptors during epileptiform bursts may even initiate a shift in GABAergic neurotransmission from inhibitory to excitatory and so have a proconvulsant action. Direct targeting of the membrane mechanisms that reduce spiking in glutamatergic neurons may better control neuronal excitability in epileptic tissue. Manipulation of brain pH may be a promising approach and recent advances in gene therapy and optogenetics seem likely to provide further routes to effective therapeutic intervention.
 |
Ivan Pavlov works at the UCL Institute of Neurology where he held an Epilepsy Research UK Fellowship to study tonic inhibition in epilepsy. He is currently a Pewterers’ Research Fellow. Kai Kaila is professor at the University of Helsinki. His lab works on neuronal signaling, with a special focus on ionic mechanisms of GABAergic transmission during neuronal development, plasticity and disease. Dimitri Kullmann is professor of neurology at the Department of Clinical and Experimental Epilepsy, UCL Institute of Neurology (London). Richard Miles works in the ICM neuroscience research centre at the Pitié-Salpêtrière hospital in Paris. They share a common interest in GABA-mediated signalling in the brain.
Introduction
Epilepsy is a chronic neurological disorder characterised by the propensity of the brain to generate spontaneous recurrent seizures. Classically this aberrant activity has been attributed to a shift in the balance of excitation and inhibition towards excitation. Early observations showed that antagonists of GABA, the main inhibitory neurotransmitter in the brain, have strong ictogenic effects (Schwartzkroin & Prince, 1977; Gutnick et al. 1982; Connors, 1984). It is also supported by a large (albeit not universal) body of experimental evidence that the number of interneurons is reduced in chronically epileptic hippocampal and neocortical tissue, leading to a reduction in the number of inhibitory synapses in the affected regions (Maglóczky & Freund, 2005). The observation of potentiated GABAergic synapses, sprouting of inhibitory axons, and increased interneuronal excitability in epileptic tissue may reflect compensatory effects (e.g. Zhang et al. 2009). But are such changes anti-epileptic? Boosting GABAergic neurotransmission may seem an effective way to alleviate a predisposition to seizures. However, an altered GABAergic signalling is known to participate in the generation of human epileptiform discharges (Schwartzkroin & Haglund, 1986; Köhling et al. 1998; Cohen et al. 2002; D’Antuono et al. 2004; Avoli et al. 2005), and potentiating GABA-mediated signalling is ineffective in some patients. GABA may also exert paradoxical pro-epileptic effects in neonates (Perucca et al. 1998).
Although impairing inhibition facilitates epileptiform activity, seizures can also be readily induced in control tissue by facilitating neuronal excitability or increasing network activity (Avoli & de Curtis, 2011). Furthermore, a profound loss of functional inhibition in the epileptic network is difficult to reconcile with the episodic nature of the disease. In humans seizures are typically separated by long seizure-free periods, often with intact cognitive and other behaviour. These observations suggest a far more complex contribution of the GABAergic system to the regulation of network dynamics. It is not surprising therefore that the role of GABAergic signalling in the generation of epileptiform activity is still vigorously debated. GABAA-mediated neurotransmission is clearly proconvulsant in one nocturnal epilepsy syndrome (Klaassen et al. 2006), in human cortical dysplasia (D’Antuono et al. 2004) and in human temporal lobe epilepsy with hippocampal sclerosis (Cohen et al. 2002; Huberfeld et al. 2011).
The uncertainties over the pro- or anti-epileptic roles of GABAergic signalling in focal cortical epilepsies suggest that treatments which target mechanisms that control cell firing and so reduce intrinsic neuronal excitability should be examined. Such treatments may beneficially restrain sudden surges in network activity.
Dynamic change in GABAergic signalling during epileptiform activity
Although epileptiform activity is accompanied by recurrent excitatory barrages like those observed in disinhibited tissue, this excitatory drive masks a massive recruitment of inhibitory neurons. In fact, a recent study has suggested that almost all perisomatically targeting interneurons in the hippocampal CA1 area are recruited during network epileptiform discharges (Marchionni & Maccaferri, 2009). Furthermore, the principle that brain areas receiving increased GABAergic drive extend beyond epileptogenic foci (Prince, 1968) has found experimental support in clinical research and animal studies (Goldensohn & Salazar, 1986; Schwartz & Bonhoeffer, 2001). This ‘inhibitory restraint’ around hyperexcitable areas may prevent or retard seizure spread (Trevelyan et al. 2006, 2007). Such an ‘inhibitory veto’ usually suffices to occlude the excitatory drive underlying generation of ictal-like events in cortical pyramidal neurons (Trevelyan et al. 2006).
The role of GABAergic neurotransmission in ictogenesis is ambiguous for several reasons. First, interneurons are highly interconnected by both chemical and electrical synapses, and their divergent outputs to primary neurons could synchronize large cell populations (discussed by Jiruska and co-authors in this issue). Second, depending on the resting membrane potential and trans-membrane gradient of Cl−, GABA can either hyperpolarize or depolarize a postsynaptic neuron (Kaila, 1994; Farrant & Kaila, 2007). Permeability of GABAA receptor channels to HCO3− (Kaila & Voipio, 1987; Kaila et al. 1993) further contributes to GABA-mediated depolarization in particular after excessive GABAergic neurotransmission. In addition, prolonged activity of GABAA receptors enhances extracellular K+ via cotransporter actions, thus initiating a prolonged non-synaptic depolarisation (Kaila et al. 1997; Viitanen et al. 2010).
It should be stressed, however, that GABAA-mediated postsynaptic ‘depolarization’ does not necessarily mean ‘excitation’ as the shunting effects of GABAA receptor activation still tend to oppose firing. In some neuronal types, such as dentate granule cells and layer 5 pyramidal cells, the GABAA reversal potential (EGABA) is positive to the resting membrane potential, even if still negative to firing threshold (Staley & Mody, 1992; Gulledge & Stuart, 2003; Sauer et al. 2012). Activation of GABAA receptors in these neurons may mediate an effective inhibition due to shunting effects. However, in these cells depolarizing IPSPs sum with spatially and temporally separate excitatory inputs (Gulledge & Stuart, 2003; Chiang et al. 2012), so promoting or inhibiting cell firing depending on the timing and cellular localisation of the GABAergic event. Although important for neuronal signal integration properties (Pavlov et al. 2011b), it remains to be determined whether these effects contribute to seizure generation.
The situation may however be very different when interneuronal firing increases during epileptiform activity. Prolonged activity at GABAergic synapses can significantly load Cl− extrusion mechanisms (Payne et al. 2003; Blaesse et al. 2009) in postsynaptic neurons leading to intracellular Cl− accumulation and a consequent depolarizing shift of EGABA. This is aggravated by GABAA-receptor coupled K+ transients. All these changes can readily convert the GABAergic drive from inhibitory to excitatory. Even in control tissue GABA may become excitatory during repeated stimulation (Staley et al. 1995; Kaila et al. 1997). Whether in chronically epileptic tissue such a transition is favoured is not clear. On the one hand, the Cl− extrusion mechanisms are impaired (Rivera et al. 2002; Jin et al. 2005; Pathak et al. 2007). On the other hand, decreased KCC2 expression will tend to decrease excitatory extracellular K+ transients (Viitanen et al. 2010). Possibly fast activity-dependent down-regulation of KCC2 (Rivera et al. 2004; Glykys et al. 2009; Lee et al. 2011; Puskarjov et al. 2012) is a neuroprotective adaptation rather than a maladaptive reactive change.
In addition to fast synaptic neurotransmission high-affinity peri- or extrasynaptic GABAA receptors mediate a slower, ‘tonic’, form of inhibitory signalling (for recent reviews, see Brickley & Mody, 2012; Pavlov & Walker, 2012). These receptors are activated by low ambient concentrations of GABA (Stell & Mody, 2002). Tonic GABAA receptor-mediated conductances are preserved, maybe even increased, in various animal models of epilepsy (Scimemi et al. 2005; Zhang et al. 2007; Zhan & Nadler, 2009; Pavlov et al. 2011a) and are also present in tissue resected from patients with temporal lobe epilepsy (TLE) (Scimemi et al. 2006). Enhanced tonic GABAA conductances would tend to enhance Cl− entry during ictal events, and enhance the load on neuronal Cl− extrusion mechanisms.
Therefore, GABAergic neurotransmission can dynamically change sense during pathological increases in network activity, and the dual nature of GABA-mediated signalling may contribute to ictogenesis (Fig. 1). Clearly measurements of steady-state EGABA levels in quiescent slices are not as informative as data on the capability of a neuron to extrude Cl− under conditions of high Cl− load during the transition to seizure (Khirug et al. 2005; Farrant & Kaila, 2007; Blaesse et al. 2009). Akin to the double faced Janus who presides over beginnings and transitions, GABAergic signalling may switch direction to promote aberrant firing.
Figure 1.
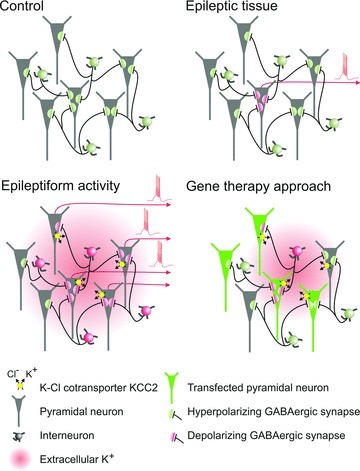
Epilepsy-induced changes in Cl− homeostasis in a subset of neurons may contribute to the spontaneous generation of interictal activity often observed in tissue resected from patients with intractable epilepsy. Massive recruitment and prolonged activation of interneurons during epileptiform activity further increases a load onto neuronal Cl− extrusion mechanism and could shift EGABA to depolarized voltages. This may render GABA excitatory, exacerbating aberrant spiking of glutamatergic cells. In addition, excessive activation of KCC2 results in a transient increase of extracellular K+ so providing additional excitation. Targeting excitability of pyramidal cells using gene transfection techniques may also alleviate undesirable effects of excessive activation of interneurons by reducing their feedback recruitment.
Transition to seizure in temporal lobe epilepsy
Defects in Cl− homeostasis have been linked to epileptiform activity in the adult brain. In temporal lobe tissue obtained from operations on patients with pharmaco-resistant focal epilepsies, the subiculum, downstream from the sclerotic CA1 region, generates a spontaneous interictal-like activity (Cohen et al. 2002). Both glutamatergic and GABAergic transmission are needed for its generation. GABA-mediated synaptic events reverse at depolarized potentials in ∼20% of subicular pyramidal cells. This suggests that an altered Cl− homeostasis in a minority of cells contributes to interictal rhythmogenesis.
Such a depolarized Cl− reversal potential occurs in reactive responses to ischaemia (Pond et al. 2006; Papp et al. 2008), axotomy (Nabekura et al. 2002) and nerve section that induces chronic pain (Coull et al. 2003; Lu et al. 2008). In human epileptic tissue, expression of two K+–Cl− cotransporter molecules, NKCC1 and KCC2, may be altered. Expression of the Na+–K+–2Cl− cotransporter, NKCC1, which usually imports Cl−, seems to be increased, while the Cl−-extruding K+–Cl− cotransporter, KCC2 (Payne et al. 2003; Blaesse et al. 2009), seems to be reduced (Muñoz et al. 2007; Huberfeld et al. 2007; Shimizu-Okabe et al. 2011). It is worth noting here that NKCC1 is also expressed in glia, while KCC2 is neuron specific in brain tissue (Blaesse et al. 2009).
Recent work on juvenile rats has shown that epileptiform activity down-regulates KCC2 both in vivo and in vitro through enhanced activation of the Ca2+-dependent protease calpain (Puskarjov et al. 2012) and, interestingly, calpain expression is increased in pharmaco-resistant human cortical TLE tissue (Feng et al. 2011). Since a wide spectrum of molecules involved in GABAergic signalling are calpain substrates (for references, see Puskarjov et al. 2012), it is possible that erosion of inhibition in epileptic tissue (Cohen et al. 2002; Huberfeld et al. 2007) is at least partly mediated by enhanced constitutive calpain activity.
In patients or animal models of focal epilepsies, interictal activity is interrupted occasionally by a seizure. Most of our understanding of this transition has been obtained from exposure to convulsants in vitro. Seizure-like events in vitro, in intracranial records from patients (Huberfeld et al. 2011) and in EEG records from animals (Bragin et al. 2009) are often preceded by distinct population events. In situ, these events seem to be limited to focal sites of seizure initiation. In vitro, they generate larger fields than interictal events that spread faster and further. They are generated in the subiculum and depend on glutamatergic synapses between subicular pyramidal cells. However while these pre-ictal population events, may be generated purely by recurrent excitation, they induce a strong interneuronal firing.
Mechanisms underlying the transition from pre-ictal discharges to seizure are not well understood. Possibly the pre-ictal bursts that precede the transition may inform on the mechanisms involved or even trigger the seizure. However while pre-ictal events correspond to a glutamate-mediated synchrony, both GABAergic and glutamatergic signalling are active and necessary for ictal-like events (Huberfeld et al. 2011). Recent work has emphasized a glial contribution to focal seizures, and glial control of external levels of both potassium and glutamate is compromised in an epileptic brain (Coulter & Eid, 2012; Steinhäuser et al. 2012). Clearly though interneurons are strongly excited by convulsants and discharges at high frequency during pre-ictal events.
Two consequences of a strong activation of postsynaptic GABAA receptors due to repetitive interneuron firing may help initiate and prolong seizures. First, Cl− extrusion mechanisms may not suffice to maintain homeostasis. The resulting Cl− loading will induce a dynamic shift in the Cl− reversal potential and so in the polarity of inhibitory events in some pyramidal cells. Secondly, even if overwhelmed, the cotransporter KCC2, continues to export not only Cl− but also K+ ions (Viitanen et al. 2010). The increase in external K+ from this route adds to that due to strong neuronal firing, increasing neuronal excitability even further and so prolonging an ictal event. It will also induce water movement into neurons (Lux et al. 1986), so reducing extracellular volume with further pro-ictal effects including enhanced ephaptic interactions (Jefferys, 1995) and increased external glutamate and K+ concentrations (Traynelis & Dingledine, 1989).
Role of brain pH in the generation and treatment of seizures
Evidence dating back several decades has shown that the excitability of neuronal circuits is strongly modulated by changes in pH (for references, see Tolner et al. 2011). In general, an elevation of pH leads to enhanced excitability while an acidosis has the opposite effect (Chesler & Kaila, 1992), and it is interesting to note that there is much evidence pointing to a key role for activity-dependent acidosis as an intrinsic mechanism for self-termination of seizures (de Curtis et al. 1998).
The relevance of pH in controlling network excitability and seizure generation seems to be particularly high in the immature brain. Indeed, seizures (often caused by local trauma, haemorrhages, or birth asphyxia) occur more frequently during the neonatal period than at any other age (Hauser et al. 1993), and spontaneous network events in neonatal hippocampal slices (the so-called giant depolarizing potentials, GDPs) are extremely sensitive to changes in intracellular pH (pHi) (Ruusuvuori et al. 2010). A decrease of pHi by 0.05 units in CA3 pyramidal neurons, induced by application of weak membrane-permeant acids such as l- and d-lactate, propionate or hypercarbia (from 5% to 8% CO2 at constant extracellular pH) led to a transient block of the GDPs. Furthermore, the recovery of GDPs closely paralleled pHi recovery in CA3 pyramidal cells (Ruusuvuori et al. 2010) which act as conditional pacemakers providing the major excitatory drive for GDPs (Sipiläet al. 2005). It has been proposed that the suppressing effect of weak acids on GDPs is due to altered mitochondrial energy metabolism in neonatal slices (Holmgren et al. 2010; Bregestovski & Bernard, 2012). However, a quantitatively similar pHi-dependent suppression of GDP generation occurs in standard physiological solution with 10 mm glucose, whether the weak acid applied is (l-lactate) or is not (d-lactate, propionate) an effective substrate of mitochondrial ATP production, or an end product, such as CO2. Moreover, neuronal mitochondrial membrane potential is unaffected by weak acids, but depends critically on glucose availability (Ruusuvuori et al. 2010).
A major clinical problem is that neonatal seizures are frequently unresponsive to anti-epileptic drugs such as phenobarbital and benzodiazepines (Rennie & Boylan, 2007; Bonifacio et al. 2011) which enhance the inhibitory actions mediated by GABAA receptors in adults (Rogawski & Löscher, 2004). In fact, such pro-GABA drugs can even potentiate neonatal seizures even in full term babies because the neuronal damage associated with epileptiform activity can induce a relatively fast positive shift in EGABA (see e.g. Nardou et al. 2011). Hence, the search for novel antiepileptic drugs and other therapeutic strategies is particularly important for neonatal seizures. Manipulating neuronal pHi might be a successful way to control epileptiform activity in neonates and infants (Schuchmann et al. 2006; Helmy et al. 2011).
In a recent study, based on a novel model of birth asphyxia, seizures were found to be triggered by a brain alkalosis (Helmy et al. 2011). These data suggest that in human post-asphyxia neonates, the standard practice of fast restoration of normocapnia leads to a pathophysiological alkaline overshoot of brain pH which will promote seizures. Consequently, a novel resuscitation approach, ‘graded restoration of normocapnia’, was put forward. This technique abolished the post-asphyxic alkaline ‘overshoot’ of brain pH and, consequently, seizure induction was strongly suppressed (Helmy et al. 2011, 2012).
Febrile seizures (FSs) are the most common type of epileptiform events in humans and the majority of FSs take place between 6 months and 5 years of age, peaking at 16–18 months (Shinnar & Glauser, 2002). Experiments based on direct measurements of cortical pH in a rat pup model of FS showed that seizures are triggered by hyperventilation and the consequent respiratory alkalosis (Schuchmann et al. 2006). Notably, exposure of the rat pups to 5% ambient CO2 blocked FSs in the rat pups within 20 s. A possible role for respiratory alkalosis in FS generation in children was examined in a large, retrospective study on age-, fever- and sex-matched children with respiratory tract infections or gastroenteritis (Schuchmann et al. 2011). Blood acid–base data from children hospitalised for FSs, showed a respiratory alkalosis; and that the low systemic pH caused by gastroenteritis seems to prevent FSs. Moreover, a subset of data showed that FSs did not occur in FS-susceptible individuals with fever caused by gastroenteritis. Thus, our study (Schuchmann et al. 2011) indicated that a respiratory alkalosis is involved in triggering FSs in children. This raises the intriguing possibility that the standard therapeutic effect of benzodiazepines on FSs (McIntyre et al. 2005) is, at least in part, caused by suppression of breathing by these drugs.
Breathing 5% CO2 inhibits seizures in the adult rat, macaque and human brain (Tolner et al. 2011). In this study, the human epilepsy patients were under presurgical monitoring, and the effect of 5% CO2 could be examined only after seizure generalization (needed for localizing the ictogenic area). However, a clear anticonvulsant effect was observed. Obviously, an earlier time point of CO2 application would have been even more effective. In addition to acute seizure suppression, the action of CO2 on brain pH is so fast that it might be used in anticipation of a seizure episode by patients with chronic epilepsy.
Targeting intrinsic properties of excitatory neurons using gene therapy to prevent seizure generation
Epilepsy-induced changes that facilitate generation of epileptiform activity include alterations in active membrane conductances and have been implicated, for example, in the conversion of regular spiking pyramidal cells into burst spiking neurons (Beck & Yaari, 2008). Such a bursting phenotype of glutamatergic neurons may then initiate synchronous network behaviour (Tryba et al. 2011). The glutamate-mediated pre-ictal discharges detected before initiation of an ictal event may also be promoted by an increased neuronal excitability. Changes in several ion channels have been reported in experimental epilepsy models. These include channels underlying the hyperpolarization-activated conductance (Shah et al. 2004), A-type K+ channels (Castro et al. 2001; Bernard et al. 2004), and T-type Ca2+ channels (Su et al. 2002). There is also evidence for an enhancement in persistent Na+ currents (Chen et al. 2011), which may result from a change in accessory subunits (Aman et al. 2009) or splicing (Fletcher et al. 2011). Most of these studies have concentrated on principal cells, but some evidence exists that a pathway upstream of Kv1.1 in fast-spiking interneurons is altered in experimental epilepsy (Li et al. 2012).
Several clinically useful anti-epileptic drugs are thought to suppress neuronal excitability via membrane ion channels. However, the scope for discovery of novel small molecule anticonvulsants may be limited. Different cell populations, in many brain structures, often express similar channels, so even drugs with perfect molecular specificity may have side effects. Pharmacotherapy may also be limited if up-regulation of drug transporters prevents anti-epileptic drugs from reaching their targets (Schmidt & Löscher, 2005). An alternative approach is to use gene therapy, which has already had some success in treating retinal degeneration and inherited immune deficiency disorders (e.g. Gaspar et al. 2011; Jacobson et al. 2012). This can in principle be targeted to the epileptogenic zone in focal epilepsy.
Thus far, experimental anti-epileptic gene therapy strategies have mainly targeted neurotransmitters and their receptors. Thus, overexpression of galanin, NPY and Y2 receptors have all been shown to attenuate seizures (Haberman et al. 2003; Richichi et al. 2004; Noèet al. 2008; Woldbye et al. 2010). However, these have mainly been studied in rodent models where the viral vector has been delivered prior to a chemoconvulsant stimulus. Hitherto, only one study has shown that gene therapy delivered after the establishment of an epileptic focus can attenuate seizures (Noèet al. 2008). The refinement of adeno-associated virus and lentivirus vectors raises the prospect for stable long-term overexpression of exogenous genes, with minimal neuronal toxicity, and the efficiency of the Camk2a promoter implies that it should be possible to selectively reduce intrinsic excitability in principal cells as a therapeutic strategy.
Of the many genes involved in regulating neuronal excitability, Kcna1, which encodes Kv1.1, is especially interesting, because overexpression in hippocampal cultures both raises the threshold for eliciting action potentials and reduces neurotransmitter release (Heeroma et al. 2009). This strategy has the potential advantage that, even if the synaptic excitation of transduced neurons decreased through a homeostatic ‘synaptic scaling’ mechanism (Turrigiano, 2008), neurotransmitter release from their terminals would still be attenuated.
An alternative approach to reducing neuronal excitability constitutively is to provide the means to suppress neuronal firing ‘on demand’, when a seizure is detected. The light-sensitive prokaryotic Cl− pump halorhodopsin (NpHR) has been used successfully to suppress burst firing in organotypic cultures (Tønnesen et al. 2009), and this strategy could be used in vivo, although the challenges to detect the seizure onset and deliver light of the appropriate wavelength and intensity to the transduced neurons are substantial. A potential disadvantage of NpHR is that it alters the Cl− reversal potential, thereby making GABAA receptors depolarizing. Thus, although the acute effect of photoactivation is to hyperpolarize neurons, fast GABAA receptor-mediated inhibition may be compromised (Raimondo et al. 2012). The proton pump Arch, in principle, avoids this disadvantage because pH shifts are rapidly buffered (Chow et al. 2010).
Conclusions and further directions
There is a strong need for new drug targets in resistant TLEs. The lack of mechanistic data on the exact events that initiate a seizure remains a major obstacle to the design of efficient antiepileptic therapies. Might pathways controlling Cl− homeostasis be a useful target? There has been interest in the diuretic molecule bumetanide which can block the Cl− importing cotransporter, NKCC1, without affecting the exporting transporter, KCC2. The possible antiepileptic actions of bumetanide may also include an increase in the extracellular volume fraction and a consequent decrease in ephaptic synchronization of neuronal spiking (Hochman, 2012). Notably, however, preclinical trials have not provided conclusive evidence for antiepileptic actions of bumetanide, in the absence of drugs which potentiate GABAergic transmission (for review, see Löscher et al. 2012).
In the neonatal period, when excitability of the brain is particularly sensitive to changes in pH, strategies that reduce alkaline shifts may be more efficient than those that potentiate GABAergic neurotransmission. Another promising approach that can help to reduce the propensity of the brain to generate seizures is emerging from developments in gene therapy. In this case the excitability of neurons is manipulated by transfecting them with certain genes using virus injections (Fig. 1). Will this approach become a routine in treatment of human epilepsy? Many gene therapy-based clinical trials are currently underway, and while the answer to this question is yet uncertain, improvements in safe gene delivery to target cells give rise for a cautious optimism for the future. Devising more effective treatment strategies, however, will still depend on the individual circumstances and a better understanding of the mechanisms underlying aberrant neuronal activity.
Acknowledgments
The authors’ research is supported by: Epilepsy Research UK (Fellowship A0832 to I.P.), Letten Foundation, the Academy of Finland, the Sigrid Juselius Foundation, the Jane and Aatos Erkko Foundation (K.K.), the European Research Council, the Wellcome Trust and the MRC (D.M.K.), the ANR and INSERM (R.M.).
References
- Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci. 2009;29:2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, de Curtis M. GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol. 2011;95:104–132. doi: 10.1016/j.pneurobio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Pumain R, Köhling R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog Neurobiol. 2005;77:166–200. doi: 10.1016/j.pneurobio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Bonifacio SL, Glass HC, Peloquin S, Ferriero DM. A new neurological focus in neonatal intensive care. Nat Rev Neurol. 2011;7:485–494. doi: 10.1038/nrneurol.2011.119. [DOI] [PubMed] [Google Scholar]
- Bragin A, Azizyan A, Almajano J, Engel J. The cause of the imbalance in the neuronal network leading to seizure activity can be predicted by the electrographic pattern of the seizure onset. J Neurosci. 2009;29:3660–3671. doi: 10.1523/JNEUROSCI.5309-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregestovski P, Bernard C. Excitatory GABA: how a correct observation may turn out to be an experimental artifact. Front Pharmacol. 2012;3:65. doi: 10.3389/fphar.2012.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Mody I. Extrasynaptic GABAA receptors: Their function in the CNS and implications for disease. Neuron. 2012;73:23–34. doi: 10.1016/j.neuron.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro PA, Cooper EC, Lowenstein DH, Baraban SC. Hippocampal heterotopia lack functional Kv4.2 potassium channels in the methylazoxymethanol model of cortical malformations and epilepsy. J Neurosci. 2001;21:6626–6634. doi: 10.1523/JNEUROSCI.21-17-06626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Su H, Yue C, Remy S, Royeck M, Sochivko D, Opitz T, Beck H, Yaari Y. An increase in persistent sodium current contributes to intrinsic neuronal bursting after status epilepticus. J Neurophysiol. 2011;105:117–129. doi: 10.1152/jn.00184.2010. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- Chiang P-H, Wu P-Y, Kuo T-W, Liu Y-C, Chan C-F, Chien T-C, Cheng J-K, Huang Y-Y, Chiu C-D, Lien C-C. GABA is depolarizing in hippocampal dentate granule cells of the adolescent and adult rats. J Neurosci. 2012;32:62–67. doi: 10.1523/JNEUROSCI.3393-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, Boyden ES. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Connors BW. Initiation of synchronized neuronal bursting in neocortex. Nature. 1984;310:685–687. doi: 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- Coull JAM, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis M, Manfridi A, Biella G. Activity-dependent pH shifts and periodic recurrence of spontaneous interictal spikes in a model of focal epileptogenesis. J Neurosci. 1998;18:7543–7551. doi: 10.1523/JNEUROSCI.18-18-07543.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antuono M, Louvel J, Köhling R, Mattia D, Bernasconi A, Olivier A, Turak B, Devaux A, Pumain R, Avoli M. GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex. Brain. 2004;127:1626–1640. doi: 10.1093/brain/awh181. [DOI] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABAA receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hao J, Ye L, Dayao C, Yan N, Yan Y, Chu L, Shi F. Overexpression of μ-calpain in the anterior temporal neocortex of patients with intractable epilepsy correlates with clinicopathological characteristics. Seizure. 2011;20:395–401. doi: 10.1016/j.seizure.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EV, Kullmann DM, Schorge S. Alternative splicing modulates inactivation of type 1 voltage-gated sodium channels by toggling an amino acid in the first S3-S4 linker. J Biol Chem. 2011;286:36700–36708. doi: 10.1074/jbc.M111.250225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, Bjorkegren E, Bayford J, Brown L, Davies EG, Veys P, Fairbanks L, Bordon V, Petropolou T, Kinnon C, Thrasher AJ. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011;3:97ra80. doi: 10.1126/scitranslmed.3002716. [DOI] [PubMed] [Google Scholar]
- Glykys J, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ. Differences in cortical versus subcortical GABAergic signalling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–672. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldensohn ES, Salazar AM. Temporal and spatial distribution of intracellular potentials during generation and spread of epileptogenic discharges. Adv Neurol. 1986;44:559–582. [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Excitatory actions of GABA in the cortex. Neuron. 2003;37:299–309. doi: 10.1016/s0896-6273(02)01146-7. [DOI] [PubMed] [Google Scholar]
- Gutnick MJ, Connors BW, Prince DA. Mechanisms of neocortical epileptogenesis in vitro. J Neurophysiol. 1982;48:1321–1335. doi: 10.1152/jn.1982.48.6.1321. [DOI] [PubMed] [Google Scholar]
- Haberman RP, Samulski RJ, McCown TJ. Attenuation of seizures and neuronal death by adeno-associated virus vector galanin expression and secretion. Nat Med. 2003;9:1076–1080. doi: 10.1038/nm901. [DOI] [PubMed] [Google Scholar]
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia. 1993;34:453–468. doi: 10.1111/j.1528-1157.1993.tb02586.x. [DOI] [PubMed] [Google Scholar]
- Heeroma JH, Henneberger C, Rajakulendran S, Hanna MG, Schorge S, Kullmann DM. Episodic ataxia type 1 mutations differentially affect neuronal excitability and transmitter release. Dis Model Mech. 2009;2:612–619. doi: 10.1242/dmm.003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmy MM, Tolner EA, Vanhatalo S, Voipio J, Kaila K. Brain alkalosis causes birth asphyxia seizures, suggesting therapeutic strategy. Ann Neurol. 2011;69:493–500. doi: 10.1002/ana.22223. [DOI] [PubMed] [Google Scholar]
- Helmy MM, Ruusuvuori E, Watkins PV, Voipio J, Kanold PO, Kaila K. 2012. Acid extrusion via blood-brain barrier causes brain alkalosis and seizures after neonatal asphyxia. Brain (in press) [DOI] [PMC free article] [PubMed]
- Hochman DW. The extracellular space and epileptic activity in the adult brain: explaining the antiepileptic effects of furosemide and bumetanide. Epilepsia. 2012;53(Suppl 1):18–25. doi: 10.1111/j.1528-1167.2012.03471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren CD, Mukhtarov M, Malkov AE, Popova IY, Bregestovski P, Zilberter Y. Energy substrate availability as a determinant of neuronal resting potential, GABA signalling and spontaneous network activity in the neonatal cortex in vitro. J Neurochem. 2010;112:900–912. doi: 10.1111/j.1471-4159.2009.06506.x. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Menendez de la Prida L, Pallud J, Cohen I, Le Van Quyen M, Adam C, Clemenceau S, Baulac M, Miles R. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat Neurosci. 2011;14:627–634. doi: 10.1038/nn.2790. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signalling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ, Calcedo R, Pang JJ, Erger KE, Olivares MB, Mullins CL, Swider M, Kaushal S, Feuer WJ, Iannaccone A, Fishman GA, Stone EM, Byrne BJ, Hauswirth WW. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthal. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferys JG. Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions. Physiol Rev. 1995;75:689–723. doi: 10.1152/physrev.1995.75.4.689. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J Physiol. 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–165. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- Khirug S, Huttu K, Ludwig A, Smirnov S, Voipio J, Rivera C, Kaila K, Khiroug L. Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur J Neurosci. 2005;21:899–904. doi: 10.1111/j.1460-9568.2005.03886.x. [DOI] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–19457. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling R, Lücke A, Straub H, Speckmann EJ, Tuxhorn I, Wolf P, Pannek H, Oppel F. Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain. 1998;121:1073–1087. doi: 10.1093/brain/121.6.1073. [DOI] [PubMed] [Google Scholar]
- Lee HHC, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K-X, Lu Y-M, Xu Z-H, Zhang J, Zhu J-M, Zhang J-M, Cao S-X, Chen X-J, Chen Z, Luo J-H, Duan S, Li X-M. Neuregulin 1 regulates excitability of fast-spiking neurons through Kv1.1 and acts in epilepsy. Nat Neurosci. 2012;15:267–273. doi: 10.1038/nn.3006. [DOI] [PubMed] [Google Scholar]
- Lu Y, Zheng J, Xiong L, Zimmermann M, Yang J. Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with down-regulation of the chloride transporter KCC2 in rat. J Physiol. 2008;586:5701–5715. doi: 10.1113/jphysiol.2008.152348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux HD, Heinemann U, Dietzel I. Ionic changes and alterations in the size of the extracellular space during epileptic activity. Adv Neurol. 1986;44:619–639. [PubMed] [Google Scholar]
- Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2012 doi: 10.1016/j.neuropharm.2012.05.045. (in press) [DOI] [PubMed] [Google Scholar]
- Maglóczky Z, Freund TF. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005;28:334–340. doi: 10.1016/j.tins.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Marchionni I, Maccaferri G. Quantitative dynamics and spatial profile of perisomatic GABAergic input during epileptiform synchronization in the CA1 hippocampus. J Physiol. 2009;587:5691–5708. doi: 10.1113/jphysiol.2009.179945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre J, Robertson S, Norris E, Appleton R, Whitehouse WP, Phillips B, Martland T, Berry K, Collier J, Smith S, Choonara I. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet. 2005;366:205–210. doi: 10.1016/S0140-6736(05)66909-7. [DOI] [PubMed] [Google Scholar]
- Muñoz A, Méndez P, DeFelipe J, Alvarez-Leefmans FJ. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. 2007;48:663–673. doi: 10.1111/j.1528-1167.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardou R, Yamamoto S, Chazal G, Bhar A, Ferrand N, Dulac O, Ben-Ari Y, Khalilov I. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain. 2011;134:987–1002. doi: 10.1093/brain/awr041. [DOI] [PubMed] [Google Scholar]
- Noè F, Pool A-H, Nissinen J, Gobbi M, Bland R, Rizzi M, Balducci C, Ferraguti F, Sperk G, During MJ, Pitkänen A, Vezzani A. Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain. 2008;131:1506–1515. doi: 10.1093/brain/awn079. [DOI] [PubMed] [Google Scholar]
- Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–689. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu F-C, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I, Huusko N, Drexel M, Kirchmair E, Sperk G, Pitkänen A, Walker MC. Progressive loss of phasic, but not tonic, GABAA receptor-mediated inhibition in dentate granule cells in a model of post-traumatic epilepsy in rats. Neuroscience. 2011a;194:208–219. doi: 10.1016/j.neuroscience.2011.07.074. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Scimemi A, Savtchenko L, Kullmann DM, Walker MC. Ih-mediated depolarization enhances the temporal precision of neuronal integration. Nat Commun. 2011b;2:199. doi: 10.1038/ncomms1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I, Walker MC. Tonic GABAA receptor-mediated signalling in temporal lobe epilepsy. Neuropharmacology. 2012 doi: 10.1016/j.neuropharm.2012.04.003. (in press; doi: 10.1016/j.neuropharm.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation–chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5–17. doi: 10.1111/j.1528-1157.1998.tb01268.x. [DOI] [PubMed] [Google Scholar]
- Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD. The chloride transporter Na+-K+-Cl− cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci. 2006;26:1396–1406. doi: 10.1523/JNEUROSCI.1421-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA. Inhibition in ‘epileptic’ neurons. Exp Neurol. 1968;21:307–321. doi: 10.1016/0014-4886(68)90043-5. [DOI] [PubMed] [Google Scholar]
- Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo JV, Kay L, Ellender TJ, Akerman CJ. Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat Neurosci. 2012;15:1102–1104. doi: 10.1038/nn.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie J, Boylan G. Treatment of neonatal seizures. Arch Dis Child Fetal Neonatal Ed. 2007;92:F148–150. doi: 10.1136/adc.2004.068551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richichi C, Lin E-JD, Stefanin D, Colella D, Ravizza T, Grignaschi G, Veglianese P, Sperk G, During MJ, Vezzani A. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. 2004;24:3051–3059. doi: 10.1523/JNEUROSCI.4056-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- Ruusuvuori E, Kirilkin I, Pandya N, Kaila K. Spontaneous network events driven by depolarizing GABA action in neonatal hippocampal slices are not attributable to deficient mitochondrial energy metabolism. J Neurosci. 2010;30:15638–15642. doi: 10.1523/JNEUROSCI.3355-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J-F, Struber M, Bartos M. Interneurons provide circuit-specific depolarization and hyperpolarization. J Neurosci. 2012;32:4224–4229. doi: 10.1523/JNEUROSCI.5702-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Löscher W. Drug resistance in epilepsy: putative neurobiologic and clinical mechanisms. Epilepsia. 2005;46:858–877. doi: 10.1111/j.1528-1167.2005.54904.x. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Hauck S, Henning S, Grüters-Kieslich A, Vanhatalo S, Schmitz D, Kaila K. Respiratory alkalosis in children with febrile seizures. Epilepsia. 2011;52:1949–1955. doi: 10.1111/j.1528-1167.2011.03259.x. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Schmitz D, Rivera C, Vanhatalo S, Salmen B, Mackie K, Sipilä ST, Voipio J, Kaila K. Experimental febrile seizures are precipitated by a hyperthermia-induced respiratory alkalosis. Nat Med. 2006;12:817–823. doi: 10.1038/nm1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TH, Bonhoeffer T. In vivo optical mapping of epileptic foci and surround inhibition in ferret cerebral cortex. Nat Med. 2001;7:1063–1067. doi: 10.1038/nm0901-1063. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Haglund MM. Spontaneous rhythmic synchronous activity in epileptic human and normal monkey temporal lobe. Epilepsia. 1986;27:523–533. doi: 10.1111/j.1528-1157.1986.tb03578.x. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Prince DA. Penicillin-induced epileptilorm activity in the hippocampal in vitro preparation. Ann Neurol. 1977;1:463–469. doi: 10.1002/ana.410010510. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Andersson A, Heeroma JH, Strandberg J, Rydenhag B, McEvoy AW, Thom M, Asztely F, Walker MC. Tonic GABAA receptor-mediated currents in human brain. Eur J Neurosci. 2006;24:1157–1160. doi: 10.1111/j.1460-9568.2006.04989.x. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Semyanov A, Sperk G, Kullmann DM, Walker MC. Multiple and plastic receptors mediate tonic GABAA receptor currents in the hippocampus. J Neurosci. 2005;25:10016–10024. doi: 10.1523/JNEUROSCI.2520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Okabe C, Tanaka M, Matsuda K, Mihara T, Okabe A, Sato K, Inoue Y, Fujiwara T, Yagi K, Fukuda A. KCC2 was downregulated in small neurons localized in epileptogenic human focal cortical dysplasia. Epilepsy Res. 2011;93:177–184. doi: 10.1016/j.eplepsyres.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Shinnar S, Glauser TA. Febrile seizures. J Child Neurol. 2002;17(Suppl 1):S44–52. doi: 10.1177/08830738020170010601. [DOI] [PubMed] [Google Scholar]
- Sipilä ST, Huttu K, Soltesz I, Voipio J, Kaila K. Depolarizing GABA acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J Neurosci. 2005;25:5280–5289. doi: 10.1523/JNEUROSCI.0378-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J, Mody I. Shunting of excitatory input to dentate granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J Neurophysiol. 1992;68:197–212. doi: 10.1152/jn.1992.68.1.197. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. 2012;60:1192–1202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- Stell BM, Mody I. Receptors with different affinities mediate phasic and tonic GABAA conductances in hippocampal neurons. J Neurosci. 2002;22:RC223. doi: 10.1523/JNEUROSCI.22-10-j0003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, Beck H. Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci. 2002;22:3645–3655. doi: 10.1523/JNEUROSCI.22-09-03645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolner EA, Hochman DW, Hassinen P, Otáhal J, Gaily E, Haglund MM, Kubová H, Schuchmann S, Vanhatalo S, Kaila K. Five percent CO2 is a potent, fast-acting inhalation anticonvulsant. Epilepsia. 2011;52:104–114. doi: 10.1111/j.1528-1167.2010.02731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol. 1989;61:927–938. doi: 10.1152/jn.1989.61.5.927. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–12455. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Yuste R. Feedforward inhibition contributes to the control of epileptiform propagation speed. J Neurosci. 2007;27:3383–3387. doi: 10.1523/JNEUROSCI.0145-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryba AK, Kaczorowski CC, Ben-Mabrouk F, Elsen FP, Lew SM, Marcuccilli CJ. Rhythmic intrinsic bursting neurons in human neocortex obtained from pediatric patients with epilepsy. Eur J Neurosci. 2011;34:31–44. doi: 10.1111/j.1460-9568.2011.07746.x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tønnesen J, Sørensen AT, Deisseroth K, Lundberg C, Kokaia M. Optogenetic control of epileptiform activity. Proc Natl Acad Sci U S A. 2009;106:12162–12167. doi: 10.1073/pnas.0901915106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitanen T, Ruusuvuori E, Kaila K, Voipio J. The K+-Cl− cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol. 2010;588:1527–1540. doi: 10.1113/jphysiol.2009.181826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldbye DPD, Angehagen M, Gøtzsche CR, Elbrønd-Bek H, Sørensen AT, Christiansen SH, Olesen MV, Nikitidou L, Hansen TVO, Kanter-Schlifke I, Kokaia M. Adeno-associated viral vector-induced overexpression of neuropeptide Y Y2 receptors in the hippocampus suppresses seizures. Brain. 2010;133:2778–2788. doi: 10.1093/brain/awq219. [DOI] [PubMed] [Google Scholar]
- Zhan R-Z, Nadler JV. Enhanced tonic GABA current in normotopic and hilar ectopic dentate granule cells after pilocarpine-induced status epilepticus. J Neurophysiol. 2009;102:670–681. doi: 10.1152/jn.00147.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei W, Mody I, Houser CR. Altered localization of GABAA receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J Neurosci. 2007;27:7520–7531. doi: 10.1523/JNEUROSCI.1555-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yamawaki R, Wen X, Uhl J, Diaz J, Prince DA, Buckmaster PS. Surviving hilar somatostatin interneurons enlarge, sprout axons, and form new synapses with granule cells in a mouse model of temporal lobe epilepsy. J Neurosci. 2009;29:14247–14256. doi: 10.1523/JNEUROSCI.3842-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]