

Abstract
Our current knowledge of the role of astrocytes in health and disease states supports the view that many physiological brain functions and neurological diseases are finely tuned, and in certain cases fully determined, by the continuous cross-talk between astrocytes and neurons. This novel way of interpreting brain activity as a dynamic and reciprocal interplay between astrocytic and neuronal networks has also influenced our understanding of epilepsy, not only forcing a reinterpretation of old findings, but also being a catalyst for novel experimentation. In this review, we summarize some of the recent studies that highlight these novel distinct contributions of astrocytes to the expression of convulsive and non-convulsive epileptiform discharges and seizures. The emerging picture suggests a general framework based on bilateral signalling between astrocytes and neurons for a fuller understanding of epileptogenic and epileptic mechanisms in the brain network. Astrocytes potentially represent targets for the development of those novel chemical entities with improved efficacy for the treatment of convulsive and non-convulsive epilepsy that expert groups have recognized as one of the key priorities for the management of epilepsy.
Introduction
The last 20 years have seen a remarkable transformation in our understanding of the role of astrocytes in brain physiology. The so-called ‘astrocyte revolution’ started with the observation that glutamate application to cell cultures and brain slices was capable of eliciting intracellular Ca2+ transients in astrocytes which could propagate to neighbouring astrocytes as Ca2+ waves. The current view is one that emphasizes a dynamic and bidirectional interplay between astrocytes and neurons that goes well beyond the traditional passive role of astrocytes in neuronal function. The major findings that have led to this scenario can be briefly summarized as follows: (1) astrocytes possess receptors for various neurotransmitters, including glutamate, GABA, ATP, adenosine, noradrenaline, acetylcholine, dopamine and cannabinoids (von Blankenfeld & Ketterman 1991; Porter & McCarthy 1995; Pasti et al. 1997; Khan et al. 2001; Araque et al. 2002; Navarrete & Araque 2008); (2) astrocytes release glutamate (Parpura et al. 1994; Pasti et al. 1997) as well as other gliotransmitters including GABA (Kozlov et al. 2006; Lee et al. 2010), d-serine (Mothet et al. 2005) and ATP (Arcuino et al. 2002; Newman 2003), through mechanisms that rely, at least in part, on intracellular Ca2+ changes and vesicle fusion events (for reviews, see Evanko et al. 2004; Volterra & Meldolesi 2005; Haydon & Carmignoto 2006); (3) astrocytic glutamate and GABA activate neurons with a characteristic electrical signature, the slow inward (Parri et al. 2001; Angulo et al. 2004; Fellin et al. 2004; Lee et al. 2007; Shigetomi et al. 2008) and outward currents (Kozlov et al. 2006; Jimenez-Gonzalez et al. 2011) (SICs and SOCs), respectively; and (4) gliotransmitters exert a fine tuning on synaptic efficacy in both the short- and the long-time domain (Araque et al. 1998; Kang et al. 1998; Brockhaus & Deitmer 2002; Zhang et al. 2003; Bowser & Khakh 2004; Pascual et al. 2005; Panatier et al. 2006, 2011; Serrano et al. 2006; Jourdain et al. 2007; Di Castro et al. 2011; Shigetomi et al. 2011; Torres et al. 2012), affect neuronal excitability (Parpura et al. 1994; Pasti et al. 1997, 2001; Araque et al. 1999; Perea & araque 2007) and enhance neuronal synchronies (Fellin et al. 2004; D’Ascenzo et al. 2007). Besides the above-mentioned neurotransmitters, astrocytes can also release interleukin-1β (IL-1β), BDNF, GDNF, neurosteroids, nitric oxide, TNFα and TGFβ, which can all affect neuronal network activity (for review, see Volterra & Meldolesi 2005). It is not surprising, therefore, that the continuous dynamic cross-talk between single astrocytes and neurons as well as among astrocytic and neuronal networks has been shown to contribute to many physiological functions.
This novel way of interpreting brain activity as a dynamic and reciprocal interplay between astrocytic and neuronal networks has clearly influenced our understanding of neurological diseases, including Alzheimer's disease, epilepsy, inflammation and Parkinson's disease, where ‘traditional’ roles for astrocytes had already been established. Thus, the ‘astrocyte revolution’ has not only led to a reinterpretation of old findings, but also been a catalyst for novel experimentation. This has been particularly true for epilepsy, where in addition to traditional astrocytic dysfunctions related to abnormal extracellular glutamate and potassium homeostasis (Heinemann et al. 2000), the bi-directional astrocyte–neuron signalling may contribute to the generation of a hyper-excitable brain network and ultimately of convulsive seizures (Binder & Steinhauser 2006; Seifert et al. 2010). Similarly in absence seizures, the presence of a localized expression of astrocytic IL-1β in the putative cortical initiation site (Akin et al. 2011), the sensitivity of absence seizures to IL-1β (Kovács et al. 2006; Akin et al. 2011) and the astrocyte-dependent gain-of-function of thalamic GABAA receptors that underlie the tonic GABAA inhibition (Cope et al. 2009; Errington et al. 2011b) have provided new perspectives for the interpretation of these genetically determined, non-convulsive seizures and novel targets for their treatment.
In this review, we summarize recent studies that highlight these and other distinct contributions of astrocytes to the expression of convulsive and non-convulsive epileptiform discharges and seizures (for more comprehensive reviews into astrocytic (dys)functions in epilepsy, see Seifert et al. (2010), Carmignoto & Haydon (2012), Losi et al. (2012) and Steinhäuser et al. (2012)). The emerging picture suggests a general framework based on bilateral signalling between astrocytes and neurons for a fuller understanding of epileptogenic and epileptic mechanisms in the brain networks, and identifies clear astrocytic targets for the development of novel avenues of pharmacological interventions in convulsive and non-convulsive epilepsy.
Astrocytes and absence seizures
Absence seizures consist of sudden and brief periods of lack of consciousness which are invariably accompanied by stereotypical, generalized and synchronous spike and wave discharges (SWDs) in the EEG (Panayiotopoulos 1997; Crunelli & Leresche 2002; Blumenfeld 2005; Avoli 2012). They are present, alone or most commonly in association with other convulsive seizures, in many idiopathic generalized epilepsies and are known to be generated by paroxysmal activity within cortical and thalamic networks with little or no involvement of other brain regions (Williams 1953; Vergnes & Marescaux 1992; Crunelli & Leresche 2002; Bai et al. 2010). However, the abnormalities underlying absence seizures and the pathophysiological mechanisms that lead to the expression of these non-convulsive seizures are still not fully understood (Crunelli & Leresche 2002; Blumenfeld 2005).
Extensive genetic analysis of many families affected by idiophatic generalized epilepsies with absence seizures has highlighted a number of mutations in a number of voltage-dependent and transmitter-gated channels, including Ca2+ channels and GABAA receptors (Wallace et al. 2001; Kananura et al. 2002; Maljevic et al. 2006; Macdonald et al. 2010; Lachance-Touchette et al. 2011). Based on the classical view that epilepsy originates from either an enhanced glutamatergic transmission, or a decreased GABAergic transmission, or both, the mutations in GABAA receptor genes had been interpreted as leading to a widespread loss-of-function in GABAA receptor-mediated synaptic transmission in absence seizures. Unfortunately, this expectation turned out not to be completely true, since in transgenic mice carrying one of this human GABAA receptor point mutations (i.e. the R43Q in the γ subunit) (Wallace et al. 2001) abnormalities in GABAergic transmission (i.e. decreased IPSC frequency) were found only in cortical but not in thalamic reticular or thalamocortical neurons (Tan et al. 2007).
This finding is not surprising if one considers that many pieces of independent evidence support the view that in thalamocortical neurons of sensory thalamic nuclei of many absence epilepsy models the GABAergic function is not decreased, but is either increased or unchanged: (1) during spontaneous absence seizures in cats as well as in Genetic Absence Epilepsy Rats from Strasbourg (GAERSs), a well established genetic model of absence seizures (Danober et al. 1998) the majority of thalamocortical neurons shows rhythmic bursts of GABAA IPSPs (Steriade & Contreras 1995; Pinault et al. 1998); (2) the direct intrathalamic injection of either penicillin (Kostopoulos 2000) or bicuculline (Steriade & Contreras 1995) fails to elicit absence seizures; (3) the overwhelming majority of data from in vivo models indicate either no change or an increase in phasic GABAAR-mediated inhibition (i.e. IPSPs or IPSCs) in thalamocortical neurons compared to their respective non-epileptic control strains (Caddick et al. 1999; Bessaih et al. 2006; Tan et al. 2008; Cope et al. 2009); (4) GABAA IPSPs in thalamocortical neurons of GABAAR-β3 subunit KO mice, which have absence seizures, are unchanged compared to wild-type littermates (Huntsman et al. 1999); (5) the ambient GABA level in the thalamus of GAERSs is higher compared to that in the non-epileptic control strain (Richards et al. 1995); and (6) GABAB receptor agonists induce absence seizures in naïve animals and aggravate them in different models of this non-convulsive epilepsy (Snead 1992; Aizawa et al. 1997; Danober et al. 1998). Importantly, a number of studies has reported that drugs that increase GABA levels, i.e. vigabatrin and tiagabine, induce absence seizures in animals and in humans, as well as aggravate them in animal models of, and in patients suffering from, absence epilepsy (Hosford & Wang 1997; Danober et al. 1998; Ettinger et al. 1999; Panayiotopoulos 2001; Perucca et al. 1998).
In line with these findings, it has recently been conclusively demonstrated that an increased tonic GABAA receptor mediated inhibition in thalamocortical neurons is a common feature of mouse and rat genetic and pharmacological models of absence epilepsy, including GAERS, stargazer, lethargic and succinic semialdheide dehydrogenase-KO animals as well as in the γ-hydroxybutyric acid (GHB) and 4,5,6,7-tetrahydroisoxazolo-[5,4-C]pyridine-3-ol (THIP) models (Cope et al. 2009; Errington et al. 2011b). This finding, together with data showing a block of absence seizures in GAERSs following thalamic injection of a GABAA receptor δ subunit-specific antisense oligodeoxynucleotide and the inability to elicit absence seizures in GABAAδ subunit KO mice (Cope et al. 2009), suggests a potential therapeutic role for inverse agonists at peri/extrasynaptic δ subunit-containing GABAA receptors in absence epilepsy (Errington et al. 2011a). Importantly, the enhanced tonic GABAA receptor mediated inhibition that is observed in GAERSs and stargazer mice may be of epileptogenic significance, since it is present before the onset of seizures in these two models (Cope et al. 2009).
The increased thalamic tonic GABAA current that is present in thalamocortical neurons of the genetic mouse and rat models of absence epilepsy does not results from an increased GABA release or from an increased expression in the levels or activity of peri/extrasynaptic δ subunit containing GABAA receptors. Instead, it is due to a loss-of-function of one of the GABA transporters, GAT-1 (Fig. 1; Cope et al. 2009), that in the thalamus of both humans and rodents is exclusively located in astrocytes (Borden 1996; De Biasi et al. 1998; Pow et al. 2005). This conclusion is based both on indirect evidence (i.e. measurements of the tonic GABAA current in GAERSs and stargazer mice) and direct evidence showing that the GABA transporter current measured from patch-clamped astrocytes in GAERS thalamic slices is not affected by NO-711 (a selective blocker of GAT-1) but is abolished by SNAP5114 (a selective blocker of GAT-3) (Pirttimaki et al. 2010, 2012). In contrast, in thalamic astrocytes from non-epileptic control rats, each of NO-711 and SNAP5114 decreases the GABA transporter current by half, and co-application of the two drugs is required for a full block of this current (Pirttimaki et al. 2010, 2012).
Figure 1. Astrocytic molecular players in absence and temporal lobe epilepsy.
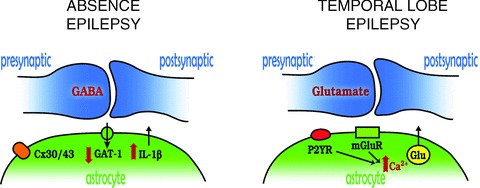
Absence epilepsy: in GABAergic synapses of the sensory thalamic nuclei, the function of the astrocytic GABA transporter GAT-1 is reduced, while in the somatosensory cortex interleukin-1β (IL-1 β) levels are increased. A clear contribution of astrocytic thalamic gap junctions, i.e. connexin (Cx) 30 and 43, to the generation of absence seizures has also been shown. Temporal lobe epilepsy: hyperactive cortical glutamatergic neurons release ATP and glutamate that activate metabotropic purinergic receptors (P2YR) and glutamate receptors (mGluR) in astrocytes. Activation of these receptors is coupled to intracellular [Ca2+] increases, which in turn trigger glutamate release from glial cells thus engaging an excitatory loop with neurons.
Two key questions arise from these studies. Firstly, what is the nature of the GAT-1 abnormality? GAT-1 expression levels in GAERSs and stargazer mice are similar to those of their respective non-epileptic control strains, and no mutation is present in the GAT-1 gene of these two genetic models (Cope et al. 2009). Thus, it may be that GAT-1 remains as an immature intracellular protein or that the phosphorilation of this transporter is compromised. Secondly, why is astrocytic GAT-3, the only other GABA transporter that is present in the thalamus (Borden 1996; De Biasi et al. 1998; Pow et al. 2005), unable to compensate for the malfunctioning GAT-1? One possibility might be that GAT-3 is located close to the synaptic sites and thus away from the peri/extrasynaptic GABAA receptors that elicit the tonic GABAA current. However, a recent investigation of GABAB receptor IPSCs in thalamocortical neurons concluded that GAT-1 is ‘primarily localized near GABAergic synapses whereas GAT-3 is localized both near and far away from synapses’ (Beenhakker & Huguenard 2010). A detailed electron microscopy study of the relative position of these transporters with respect to synaptic and to δ-containing peri/extrasynaptic GABAA receptors may provide some conclusive results on this issue.
Possibly as a consequence of the drastically reduced activity of GAT-1 in the thalamus, some properties of the slow inward currents (SOCs), the characteristic signature of the GABAergic astrocyte-to-neuron signalling (Kozlov et al. 2006), are altered in GAERS thalamocortical neurons, i.e. small changes in amplitude, rise time and decay time (Pirttimaki et al. 2010, 2012). Although it is difficult at present to ascribe a precise mechanistic significance for these changes in thalamic SOCs to the pathophysiological processes occurring during absence seizures, it is interesting that vigabatrin, which elicits and/or exacerbates absence seizures in animals and humans (see above), has been shown to increase the frequency of SOCs in thalamic neurons (Jimenez-Gonzalez et al. 2011). It remains to be seen whether similar changes in the activity of GABA transporters and SOCs are present in cortical territories, in particular in the putative ‘cortical initiation site’ of typical absence seizures that has been identified in rat genetic models (Meeren et al. 2002; Polack et al. 2007), and in humans suffering from this form of epilepsy (Holmes et al. 2004; Westmijse et al. 2009; Bai et al. 2010; Moeller et al. 2010). As far as the slow inward currents (SICs), the characteristic signature of the glutamatergic astrocyte-to-neuron signalling (Angulo et al. 2004; Fellin et al. 2004), are concerned, no differences are observed in the properties of thalamic SICs between GAERSs prior to seizure onset and age-matched non-epileptic rats (Pirttimaki et al. 2010, 2012) and no change in astrocytic glutamate transporters has been reported at this age in the thalamus of this genetic model (Dutuit et al. 2002). In cortical neurons, on the other hand, one might expect to see alterations in SICs, since glutamate uptake is reduced in this brain region of pre-seizure GAERSs (Touret et al. 2007) as a result of a decreased expression of the astrocytic glutamate transporters GLT-1 and GLAST (Dutuit et al. 2002).
Another key finding that provides a potential epileptogenic role of astrocytes in the expression of absence seizures stems from the original observation of Dutuit et al. (2000), who described increased expression of glial fibrillary protein levels in cortical and thalamic astrocytes of GAERS, both prior to the expression of the first seizure and in adulthood. A recent investigation has now shown the selective induction of IL-1β in activated astrocytes within the putative ‘cortical initiation site’ of absence seizures (i.e. the peri-oral region of the somatosensory cortex), but not in other cortical regions or in the thalamus of GAERSs prior to seizure onset. In adulthood, IL-1β is detected in the entire somatosensory cortex, but again not in the remaining cortex or thalamus of this absence model (Fig. 1) (Akin et al. 2011). The increased cortical expression of IL-1β is not simply an epiphenomenon, since systemic injection of a specific blocker of IL-1β synthesis markedly reduces absence seizures in this model (Akin et al. 2011), and injection of lipopolysaccharide, an inducer of IL-1β, increases the number of absence seizures in WAG/Rij rats (Kovács et al. 2006), another well-established model of absence epilepsy (Coenen & Van Luijtelaar 2003).
It is important to comment on the ability of the gap-juction blocker carbenoxolone to drastically reduce absence seizures in both rat (WAG/Rij) and mouse (lethargic) models when it is injected intrathalamically or systemically, respectively (Gareri et al. 2005). This anti-absence effect of carbenoxolone has been explained to result from a block of neuronal gap-juctions: indeed, (1) in the reticular thalalmic nucleus there is a strong immunosignal for the exclusively neuronal connexin 36 (Nagy & Rash 2000) and the presence of electrical coupling among neurons of this thalamic nucleus has been directly demonstrated (Landisman et al. 2002); and (2) a weak expression of the neuronal (and astrocytic) connexin 45 (Nagy & Rash 2000; Söhl et al. 2005) as well as electrophysiological and freeze-fracture evidence of gap junctions among neurons of the sensory thalamic nuclei have been reported (Hughes et al. 2004, 2011). However, both the reticular thalamic nucleus and the sensory thalamic nuclei also exhibit strong immunoreactivity for the astroglia-specific connexin 30 and connexin 43 (Fig. 1) (Nagy & Rash 2000). Thus, astrocytic gap junctions are as likely to be involved as neuronal gap juctions in the anti-absence effect of carbenoxolone and in the mechanisms underlying the expression of absence seizures.
In summary, the work described above has highlighted a number of novel astrocytic targets that may lead to potential therapeutic avenues (Fig. 1). In particular, novel substances that can either decrease gap-junction communications (among astrocytes as well as neurons), negatively interfere with the IL-1β pathway, increase GAT-1 function or reduce the activity of extrasynaptic GABAA receptors may prove useful in the pharmacological rescue of clinical epileptic phenotypes of absence seizures. A potential mechanistic explanation of how the loss of function of astrocytic GAT-1 and the resulting increase in tonic GABAA inhibition lead to the expression of absence seizures is depicted in Fig. 2.
Figure 2. Astrocytic contribution to the mechanisms underlying temporal lobe and absence epilepsy.
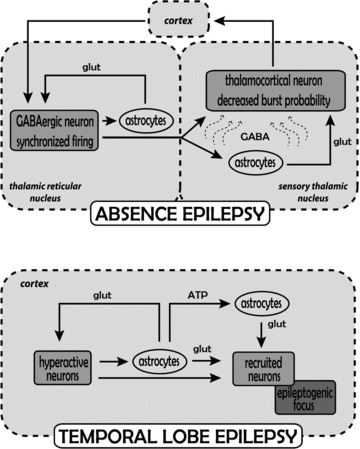
Absence epilepsy: the generation of absence seizures involves abnormal firing activity among cortical cells and neurons in the thalamic reticular and sensory thalamic nuclei. During absence seizures increased firing in GABAergic neurons of the thalamic reticular nucleus (which is driven by the synchronized cortical input) leads to enhanced GABA levels (curved arrows) in sensory thalamic nuclei and the resulting increase in tonic GABAA current in thalamocortical neurons since GAT-1 transporter function is diminished (see Fig. 1). The increased tonic GABAA current reduces, but does not abolish, firing in the thalamocortical neurons, while at the same time blocks faithful transmission of sensory inputs and reduces the effect of any potential increase in astrocytically released glutamate on these neurons. On the other hand, the putative increase in astrocytically released glutamate re-enforces synchronized firing in reticular thalamic neurons. It is unlikely that any increased astrocytically released GABA affects reticular thalamic neurons since they show no tonic GABAA current. It remains to be determined whether similar alterations occur in the cortical territory. Temporal lobe epilepsy: the generation of temporal lobe seizures involves abnormal firing activity initially restricted to a group of cortical neurons. Increased glutamate release activates local astrocytes, which by releasing glutamate signal back to neurons, thus enhancing their hyperactivity, and also signal to other astrocytes and neurons in nearby regions increasing the probability of neuronal recruitment into the epileptogenic focus.
Astrocytes and temporal lobe epilepsy
Among the most common and severe forms of convulsive epilepsies is the temporal lobe epilepsy (TLE). The clinical manifestation of this disorder is recurrent, unprovoked seizures that arise as an intense, synchronous discharge in a relatively high number of neurons from a restricted region of the medial or lateral temporal lobe, i.e. the epileptogenic focus, and eventually generalize to both temporal lobes and extratemporal structures through a progressive recruitment of other neuronal populations (Traub & Wong 1982; Jefferys 1990; Avoli et al. 2002; Pinto et al. 2005; Trevelyan et al. 2006).
Several different factors that are directly or indirectly linked to neurons, such as ion channel mutations, defects in cortical development and brain injuries, are known to result in the excessive excitability of neurons that characterizes a brain network prone to seizures. Non-synaptic factors can also contribute. A role for the non-neuronal cell astrocyte as a modulator of epiletogenesis was proposed over 20 years ago and for a long time this role was linked to the ability of astrocytes to buffer both extracellular K+ and glutamate that are released in excess during epileptic discharges (Heinemann et al. 1977; Heinemann 1986; Demarque et al. 2004; Xu et al. 2009). Several studies performed in both animal models and human epilepsy demonstrated, indeed, that a defective K+ buffering by astrocytes generates a hyperexcitable neuronal network that can enhance seizure generation (Wallraff et al. 2006; Djukic et al. 2007). The demonstration that astrocytes can respond to neuronal signals with Ca2+ elevations and signal back to neurons by releasing different gliotransmitters including glutamate, d-serine, GABA and ATP hinted at a more intriguing hypothesis of a direct role of this form of astrocyte-to-neuron communication in the generation of epileptiform activities. Among gliotransmitters, glutamate has been revealed to have the potential to be significantly involved in this action. The first, indirect clues for such a role came from a number of different studies performed in brain slice preparations. These studies revealed that Ca2+-dependent glutamate release from astrocytes modifies the probability of neurotransmitter release (thus increasing the frequency of spontaneous events and potentiating the evoked excitatory synaptic response) by acting on presynaptic type I metabotropic receptors (mGluRs) (Fiacco & McCarthy 2004; Perea & Araque 2007) or N-methyl-d-aspartate receptors (NMDARs) (Jourdain et al. 2007). Glutamate released from activated astrocytes also potentiates or depresses inhibitory transmission (Liu et al. 2004) via activation of presynaptic kainate or mGlu type II and III receptor, respectively. An additional target of astrocytic glutamate is the postsynaptic membrane. Studies in cell cultures (Araque et al. 1998) and brain slice preparations (Angulo et al. 2004; Fellin et al. 2004) revealed that upon stimuli that trigger Ca2+ elevations in astrocytes very slow inward currents (SICs) could be recorded in pyramidal neurons. The SICs that resulted were typically insensitive to tetrodotoxin (TTX) and mediated by extrasynaptically located N-methyl-d-aspartate glutamate receptors (NMDARs) (Angulo et al. 2004; Fellin et al. 2004). The astrocytic origin of SICs was demonstrated by a series of experiments which included photolysis of a Ca2+-caged compound in single astrocytes (Fellin et al. 2004). Beside in the hippocampus, SICs have been described in other brain regions (Fellin 2009), including thalamus (Parri et al. 2001), cortex (Ding et al. 2007), nucleus accumbens (D’Ascenzo et al. 2007; Fellin et al. 2007), olfactory bulb (Kozlov et al. 2006) and brainstem (Reyes-Haro et al. 2010). In the context of a possible involvement of this astrocyte-to-neuron signal in the generation of epileptiform activities, noteworthy is that: (1) SICs can depolarize the neuronal membrane sufficiently to elicit bursts of action potentials (Fellin et al. 2006); (2) SICs can occur synchronously in two neurons when their cell bodies are within about 100 μm of one another (Fellin et al. 2004); (3) the ability of astrocytes to release d-serine, which is probably the endogenous ligand on the so-called ‘glycine site’ of the NMDAR (Schell et al. 1995; Mothet et al. 2000), can be also be important in SIC generation; (4) the Ca2+ elevation that accompanies SICs occurs simultaneously in small groups of adjacent pyramidal neurons suggesting that astrocytic glutamate can favour synchronous neuronal discharges (Fellin et al. 2004). Therefore, the release of glutamate from activated astrocytes represents a non-neuronal source of excitation in the neuronal network and a non-synaptic mechanism of neuronal synchrony (Carmignoto & Zonta 2008) that have the potential to enhance the generation epileptiform activities.
This hypothesis was specifically addressed in a number of subsequent studies but the results obtained were controversial and fuelled an intense debate on the role of astrocytes in epileptogenesis (D’Ambrosio 2006; Seifert et al. 2006; Wetherington et al. 2008). In the first of these studies, Tian et al. (2005) used different in vitro models of epilepsy to show that the paroxysmal depolarising shifts (PDSs), i.e. the cellular correlate of interictal events recorded between seizures, are resistant to both TTX and different Ca2+ channel blockers that inhibit neuronal action potential firing and synaptic release, respectively, but are sensitive to antagonists of the glutamate receptors AMPA and NMDA. These results suggested that PDSs could be generated by a non-neuronal source of glutamate, such as the astrocyte, and this possibility was confirmed by the finding that Ca2+ elevations triggered by photolysis of Ca2+-caged compounds in individual astrocytes were associated with events reminiscent of the PSDs recorded in the field potentials and could reflect, or be caused by, NMDAR-mediated SICs (Tian et al. 2005). In contrast, results obtained by Fellin et al. (2006) in the low Mg2+/picrotoxin model showed that SICs evoked by astrocytic glutamate are not required for epileptic discharges because both the interictal, i.e. the PDSs, and the ictal events were detected, although at a reduced frequency, in the presence of the NMDAR antagonist d-AP5. In the same model, after astrocytes’ Ca2+ elevations were drastically reduced by slice perfusion with the mGluR antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP), the frequency of interictal events was observed to increase with respect to controls while the frequency and duration of ictal events were reduced (Fig. 1) (Gomez-Gonzalo et al. 2010). These results suggest that astrocytic glutamate per se does not mediate interictal events and rather that it may have a modulatory role in the generation of ictal discharges (Fig. 2) (Fellin & Haydon 2005; Fellin et al. 2006). Our recent work in an entorhinal cortex (EC) slice model of focal epilepsy provides support for such a role (Gomez-Gonzalo et al. 2010). In this model a propagating, focal seizure-like, ictal discharge was observed to develop in the presence of the proconvulsant 4-amino pyridine and low Mg2+ after a local NMDA stimulation of a small group of layer V–VI neurons (Losi et al. 2010). We first found that a double, but not a single, episode of NMDA stimulation regularly evoked an ictal discharge and that the ictal discharge was preceded by a massive Ca2+ elevations in astrocytes. When these early Ca2+ elevations in astrocytes were inhibited by BAPTA (introduced in the astrocyte syncytium by patching an individual astrocyte with a BAPTA-containing pipette), the episode of neuronal hyperactivity induced by double NMDA stimulations failed to generate an ictal discharge. Thus, the early activation of the astrocytes was not a mere consequence of the increased neuronal activity and it rather had a causative role in the generation of focal ictal discharges. It appears that when astrocytes are consistently engaged by an episode of hyperactivity in a group of neurons, they generate a feed-back signal, i.e. Ca2+-dependent release of glutamate and/or d-serine, that causes a larger population of neurons to be recruited into a coherent synchronous activity. If this feedback signal operates on a brain network prone to seizures, it contributes to drive neurons towards the ictal discharge threshold (Gomez-Gonzalo et al. 2010; Losi et al. 2010). The initiation of an ictal discharge at the epileptogenic focus may be thus represented also by neurons that are secondarily activated in a recruitment process that involves astrocytes. In support of this view, when the neuronal network in the EC is rendered hyperexcitable by slice perfusion with low Mg2+/picrotoxin, a selective stimulation of astrocytes by the peptide Thr-Phe-Leu-Leu-Arg-NH2 (TFLLR) was sufficient to initiate a propagating ictal discharge (Gomez-Gonzalo et al. 2010). TFLLR is known to evoke glutamate release and neuronal SICs by activating the thrombin protease activated receptor-1 (PAR-1) (Lee et al. 2007; Shigetomi et al. 2008; Gomez-Gonzalo et al. 2010). All in all, this study essentially shows that: (1) neuronal activity is critical for the generation of a neuronal network prone to seizures and that (2) astrocytes established with neurons a recurrent excitatory loop that lowers the threshold for the focal generation of seizure-like ictal discharges (Fig. 2).
Conclusions
The findings reviewed here highlight novel key elements of the astrocytic involvement in absence epilepsy, including GAT-1, connexin 30- and 43-based gap-junctions and the IL-1β pathway, and in temporal lobe epilepsy, including mGlu and the P2Y receptor-mediated pathways. Shifting the focus from neurons to astrocyte–neuron interactions (Steinhäuser et al. 2012) has allowed us to ‘further understand the neurobiology the epileptogenic brain and the mechanisms underlying the emergence of seizures’ (Baulac & Pitkanen 2009), and thus to discover novel, astrocyte-based, targets for the potential development of those (fourth generation) antiepileptic drugs with increased efficacy that expert groups have identified as one of the key priorities for an improved clinical management of convulsive and non-convulsive epilepsy (Baulac & Pitkanen 2009; Löscher & Schmidt 2011; Galanopoulou et al. 2012; Simonato et al. 2012).
Acknowledgments
We would like to thank Dr Micaela Zonta for her critical comments on the manuscript and invaluable help with the figures. The original work described in this review was supported by European Union HEALTH-F2–2007-202167 to V.C. and G.C., by Telethon Italy (GGP10138B; GGP07274) and Cariparo Foundation to G.C., and by Wellcome Trust (78403) to V.C. The authors have no conflict of interest to declare.
References
- Aizawa M, Ito Y, Fukuda H. Pharmacological profiles of generalized absence seizures in lethargic, stargazer and gamma-hydroxybutyrate-treated model mice. Neurosci Res. 1997;29:17–25. doi: 10.1016/s0168-0102(97)00066-7. [DOI] [PubMed] [Google Scholar]
- Akin D, Ravizza T, Maroso M, Carcak N, Eryigit T, Vanzulli I, Aker RG, Vezzani A, Onat FY. IL-1b is induced in reactive astrocytes in the somatosensory cortex of rats with genetic absence epilepsy at the onset of spike-and-wave discharges, and contributes to their occurrence. Neurobiol Dis. 2011;44:259–269. doi: 10.1016/j.nbd.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Martín ED, Perea G, Arellano JI, Buño W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Arcuino G, Lin JH-C, Takano T, Liu C, Jiang L, Gao Q, Kang J, Nedergaard M. Intercellular calcium signaling mediated by point-source burst release of ATP. Proc Natl Acad Sci U S A. 2002;99:9840–9845. doi: 10.1073/pnas.152588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, D’Antuono M, Louvel J, Kohling R, Biagini G, Pumain R, D’Arcangelo G, Tancredi V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68:167–207. doi: 10.1016/s0301-0082(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Avoli M. A brief history on the oscillating roles of thalamus and cortex in absence seizures. Epilepsia. 2012;53:779–789. doi: 10.1111/j.1528-1167.2012.03421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Vestal M, Berman R, Negishi M, Spann M, Vega C, Desalvo M, Novotny EJ, Constable RT, Blumenfeld H. Dynamic time course of typical childhood absence seizures: EEG, behavior, and functional magnetic resonance imaging. J Neurosci. 2010;30:5884–5893. doi: 10.1523/JNEUROSCI.5101-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac M, Pitkanen A. Research priorities in epilepsy for th next decade. Epilepsia. 2009;50:571–583. [Google Scholar]
- Beenhakker MP, Huguenard JR. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci. 2010;30:15262–15276. doi: 10.1523/JNEUROSCI.3243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessaih T, Bourgeais L, Badiu CI, Carter DA, Toth TI, Ruano D, Lambolez B, Crunelli V, Leresche N. Nucleus-specific abnormalities of GABAergic synaptic transmission in a genetic model of absence seizures. J Neurophysiol. 2006;96:3074–3081. doi: 10.1152/jn.00682.2006. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54:358–368. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H. Cellular and network mechanisms of spike-wave seizures. Epilepsia. 2005;46(Suppl 9):21–33. doi: 10.1111/j.1528-1167.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- Borden LA. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem Int. 1996;29:335–356. doi: 10.1016/0197-0186(95)00158-1. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci. 2004;24:8606–8620. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhaus J, Deitmer JW. Long-lasting modulation of synaptic input to Purkinje neurons by Bergmann glia stimulation in rat brain slices. J Physiol. 2002;545:581–593. doi: 10.1113/jphysiol.2002.028423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caddick SJ, Wang C, Fletcher CF, Jenkins NA, Copeland NG, Hosford DA. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4lh) and tottering (Cacna1atg) mouse thalami. J Neurophysiol. 1999;81:2066–2074. doi: 10.1152/jn.1999.81.5.2066. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Zonta M. Physiological and pathological roles of astrocyte-mediated neuronal synchrony. In: Parpura V, Haydon PG, editors. Astrocytes in (Patho)Physiology of the Nervous System. USA: Springer; 2008. pp. 552–553. [Google Scholar]
- Carmignoto G, Haydon P. Astrocyte calcium signaling and epilepsy. Glia. 2012;60:1227–1230. doi: 10.1002/glia.22318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen AM, Van Luijtelaar EL. Genetic animal models for absence epilepsy: a review of the WAG/Rij strain of rats. Behav Genet. 2003;33:635–655. doi: 10.1023/a:1026179013847. [DOI] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R. Does glutamate released by astrocytes cause focal epilepsy. Epilepsy Curr. 2006;6:173–176. doi: 10.1111/j.1535-7511.2006.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, Moss SJ, Haydon PG. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci U S A. 2007;104:1995–2000. doi: 10.1073/pnas.0609408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol. 1998;55:27–57. doi: 10.1016/s0301-0082(97)00091-9. [DOI] [PubMed] [Google Scholar]
- De Biasi S, Vitellaro-Zuccarello L, Brecha NC. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization. Neuroscience. 1998;83:815–828. doi: 10.1016/s0306-4522(97)00414-4. [DOI] [PubMed] [Google Scholar]
- Demarque M, Villeneuve N, Manent JB, Becq H, Represa A, Ben-Ari Y, Aniksztejn L. Glutamate transporters prevent the generation of seizures in the developing rat neocortex. J Neurosci. 2004;24:3289–3294. doi: 10.1523/JNEUROSCI.5338-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, Tiret P, Volterra A. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci. 2011;14:1276–1284. doi: 10.1038/nn.2929. [DOI] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutuit M, Didier-Bazès M, Vergnes M, Mutin M, Conjard A, Akaoka H, Belin MF, Touret M. Specific alteration in the expression of glial fibrillary acidic protein, glutamate dehydrogenase, and glutamine synthetase in rats with genetic absence epilepsy. Glia. 2000;32:15–24. doi: 10.1002/1098-1136(200010)32:1<15::aid-glia20>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Dutuit M, Touret M, Szymocha R, Nehlig A, Belin M-F, Didier-Blazès M. Decreased expression of glutamate transporters in genetic absence epilepsy rats before seizure occurrence. J Neurochem. 2002;80:1029–1038. doi: 10.1046/j.0022-3042.2002.00768.x. [DOI] [PubMed] [Google Scholar]
- Errington AC, Cope DW, Crunelli V. Augmentation of tonic GABAA inhibition in absence epilepsy: therapeutic value of inverse agonists at extrasynaptic GABAA receptors. Adv Pharmacol Sci. 2011a;2011:790590. doi: 10.1155/2011/790590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington AC, Gibson KM, Crunelli V, Cope DW. Aberrant GABAA receptor-mediated inhibition in cortico-thalamic networks of succinic semialdehyde dehydrogenase deficient mice. PLoS One. 2011b;6:e19021. doi: 10.1371/journal.pone.0019021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AB, Bernal OG, Andriola MR, Bagchi S, Flores P, Just C, Pitocco C, Rooney T, Tuominen J, Devinsky O. Two cases of nonconvulsive status epilepticus in association with tiagabine therapy. Epilepsia. 1999;40:1159–1162. doi: 10.1111/j.1528-1157.1999.tb00835.x. [DOI] [PubMed] [Google Scholar]
- Evanko DS, Zhang Q, Zorec R, Haydon PG. Defining pathways of loss and secretion of chemical messengers from astrocytes. Glia. 2004;47:233–240. doi: 10.1002/glia.20050. [DOI] [PubMed] [Google Scholar]
- Fellin T. Communication between neurons and astrocytes: relevance to the modulation of synaptic and network activity. J Neurochem. 2009;108:533–544. doi: 10.1111/j.1471-4159.2008.05830.x. [DOI] [PubMed] [Google Scholar]
- Fellin T, D’Ascenzo M, Haydon PG. Astrocytes control neuronal excitability in the nucleus accumbens. ScientificWorldJournal. 2007;7:89–97. doi: 10.1100/tsw.2007.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci. 2006;26:9312–9322. doi: 10.1523/JNEUROSCI.2836-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Haydon PG. Do astrocytes contribute to excitation underlying seizures. Trends Mol Med. 2005;11:530–533. doi: 10.1016/j.molmed.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanopoulou AS, Buckmaster PS, Staley KJ, Moshé SL, Perucca E, Engel J, Jr, Löscher W, Noebels JL, Pitkänen A, Stables J, White HS, O’Brien TJ, Simonato M. Identification of new epilepsy treatments: Issues in preclinical methodology. Epilepsia. 2012;53:571–582. doi: 10.1111/j.1528-1167.2011.03391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareri P, Condorelli D, Belluardo N, Citraro R, Barresi V, Trovato-Salinaro A, Mudo G, Ibbadu GF, Russo E, De Sarro G. Antiabsence effects of carbenoxolone in two genetic animal models of absence epilepsy (WAG/Rij rats and lh/lh mice) Neuropharmacology. 2005;49:551–563. doi: 10.1016/j.neuropharm.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, de Curtis M, Ratto GM, Carmignoto G. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. 2010;8:e1000352. doi: 10.1371/journal.pbio.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Heinemann U. Excitatory amino acids and epilepsy-induced changes in extracellular space size. Adv Exp Med Biol. 1986;203:449–460. doi: 10.1007/978-1-4684-7971-3_34. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Gabriel S, Jauch R, Schulze K, Kivi A, Eilers A, Kovacs R, Lehmann TN. Alterations of glial cell function in temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S185–189. doi: 10.1111/j.1528-1157.2000.tb01579.x. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD, Gutnick MJ. Extracellular free calcium and potassium during paroxsmal activity in the cerebral cortex of the cat. Exp Brain Res. 1977;27:237–243. doi: 10.1007/BF00235500. [DOI] [PubMed] [Google Scholar]
- Holmes MD, Brown M, Tucker DM. Are “generalized” seizures truly generalized? Evidence of localized mesial frontal and frontopolar discharges in absence. Epilepsia. 2004;45:1568–1579. doi: 10.1111/j.0013-9580.2004.23204.x. [DOI] [PubMed] [Google Scholar]
- Hosford DA, Wang Y. Utility of the lethargic (lh/lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures. Epilepsia. 1997;38:408–414. doi: 10.1111/j.1528-1157.1997.tb01729.x. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Lorincz M, Cope DW, Blethyn KL, Kekesi KA, Parri HR, Juhasz G, Crunelli V. Synchronized oscillations at alpha and theta frequencies in the lateral geniculate nucleus. Neuron. 2004;42:253–268. doi: 10.1016/s0896-6273(04)00191-6. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Lorincz ML, Blethyn K, Kekesi KA, Juhasz G, Turmaine M, Parnavelas JG, Crunelli V. Thalamic gap junctions control local neuronal synchrony and influence macroscopic oscillation amplitude during EEG alpha rhythms. Front Psychol. 2011;2:193. doi: 10.3389/fpsyg.2011.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Jefferys JG. Basic mechanisms of focal epilepsies. Exp Physiol. 1990;75:127–162. doi: 10.1113/expphysiol.1990.sp003390. [DOI] [PubMed] [Google Scholar]
- Jimenez-Gonzalez C, Pirttimaki T, Cope DW, Parri HR. Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets delta-subunit-containing GABAA receptors. Eur J Neurosci. 2011;33:1471–1482. doi: 10.1111/j.1460-9568.2011.07645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, Rebstock J, Heils A, Steinlein OK. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Khan ZU, Koulen P, Rubinstein M, Grandy DK, Goldman-Rakic PS. An astroglia-linked dopamine D2-receptor action in prefrontal cortex. Proc Natl Acad Sci U S A. 2001;98:1964–1969. doi: 10.1073/pnas.98.4.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostopoulos GK. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: the continuing development of a hypothesis. Clin Neurophysiol. 2000;111(Suppl 2):S27–38. doi: 10.1016/s1388-2457(00)00399-0. [DOI] [PubMed] [Google Scholar]
- Kovács Z, Kekesi KA, Szilagyi N, Abraham I, Szekacs D, Kiraly N, Papp E, Csaszar I, Szego E, Barabas K, Peterfy H, Erdei A, Bartfai T, Juhasz G. Facilitation of spike-wave discharge activity by lipopolysaccharides in Wistar Albino Glaxo/Rijswijk rats. Neuroscience. 2006;140:731–742. doi: 10.1016/j.neuroscience.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci U S A. 2006;103:10058–10063. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H, Lortie A, Carmant L, Bedford F, Bowie D, Cossette P. Novel α1 and γ2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. 2011;34:237–249. doi: 10.1111/j.1460-9568.2011.07767.x. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL, Connors BW. Electrical synapses in the thalamic reticular nucleus. J Neurosci. 2002;22:1002–1009. doi: 10.1523/JNEUROSCI.22-03-01002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, Traynelis SF. Astrocytic control of synaptic NMDA receptors. J Physiol. 2007;581:1057–1081. doi: 10.1113/jphysiol.2007.130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ. Channel-mediated tonic GABA release from glia. Science. 2010;330:790–796. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- Liu Q-S, Xu Q, Arcuino G, Kang J, Nedergaard M. Astrocyte-mediated activation of neuronal kainate receptors. Proc Natl Acad Sci U S A. 2004;101:3172–3177. doi: 10.1073/pnas.0306731101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: Ways out of the current dilema. Epilepsia. 2011;52:657–678. doi: 10.1111/j.1528-1167.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- Losi G, Cammarota M, Chiavegato A, Gomez-Gonzalo M, Carmignoto G. A new experimental model of focal seizures in the entorhinal cortex. Epilepsia. 2010;51:1493–1502. doi: 10.1111/j.1528-1167.2009.02472.x. [DOI] [PubMed] [Google Scholar]
- Losi G, Cammarota M, Carmignoto G. The role of astroglia in the epileptic brain. Front Pharmacol. 2012;3:132. doi: 10.3389/fphar.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010;588:1861–1869. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, Schlesinger F, Ursu D, Melzer W, Cossette P, Bufler J, Lerche H, Heils A. A mutation in the GABAA receptor α1-subunit is associated with absence epilepsy. Ann Neurol. 2006;59:983–987. doi: 10.1002/ana.20874. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller F, LeVan P, Muhle H, Stephani U, Dubeau F, Siniatchkin M, Gotman J. Absence seizures: individual patterns revealed by EEG-fMRI. Epilepsia. 2010;51:2000–2010. doi: 10.1111/j.1528-1167.2010.02698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci U S A. 2005;102:5606–5611. doi: 10.1073/pnas.0408483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Rash JE. Connexins and gap junctios of astrocytes and oligodendrocytes in the CNS. Brain Res Revs. 2000;32:29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- Navarrete M, Araque A. Endocannabinoids mediate neuron-astrocyte communication. Neuron. 2008;57:883–893. doi: 10.1016/j.neuron.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. 2003;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet J-P, Toquet B, Pollegioni L, Poulain DA, Oliet SHR. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Panatier A, Vallee J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–798. doi: 10.1016/j.cell.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Panayiotopoulos CP. Absences epilepsies. In: Engel JJ, Pedley TA, editors. Epilepy: A Comprehensive Textbook. Philadelphia: Lippicott-Raven; 1997. pp. 2327–2346. [Google Scholar]
- Panayiotopoulos CP. Treatment of typical absence seizures and related epileptic syndromes. Paediatr Drugs. 2001;3:379–403. doi: 10.2165/00128072-200103050-00006. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5–17. doi: 10.1111/j.1528-1157.1998.tb01268.x. [DOI] [PubMed] [Google Scholar]
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol. 1998;509:449–456. doi: 10.1111/j.1469-7793.1998.449bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto DJ, Patrick SL, Huang WC, Connors BW. Initiation, propagation, and termination of epileptiform activity in rodent neocortex in vitro involve distinct mechanisms. J Neurosci. 2005;25:8131–8140. doi: 10.1523/JNEUROSCI.2278-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Cope DW, Parri HR, Crunelli V. Glial signalling in the ventrobasal thalamus of rats with absence seizures. Soc Neurosci Abstr. 2010 255.3. [Google Scholar]
- Pirttimaki TM, Parri HR, Crunelli V. Astrocytic GAt-1 dysfunction in experimental absence seizures. J Physiol. 2013;591:823–833. doi: 10.1113/jphysiol.2012.242016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. GFAP-positive hippocampal astrocytes in situ respond to glutamatergic neuroligands with increases in [Ca2+]i. Glia. 1995;13:101–112. doi: 10.1002/glia.440130204. [DOI] [PubMed] [Google Scholar]
- Pow DV, Sullivan RK, Williams SM, Scott HL, Dodd PR, Finkelstein D. Differential expression of the GABA transporters GAT-1 and GAT-3 in brains of rats, cats, monkeys and humans. Cell Tissue Res. 2005;320:379–392. doi: 10.1007/s00441-004-0928-0. [DOI] [PubMed] [Google Scholar]
- Reyes-Haro D, Muller J, Boresch M, Pivneva T, Benedetti B, Scheller A, Nolte C, Kettenmann H. Neuron-astrocyte interactions in the medial nucleus of the trapezoid body. J Gen Physiol. 2010;135:583–594. doi: 10.1085/jgp.200910354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Lemos T, Whitton PS, Bowery NG. Extracellular GABA in the ventrolateral thalamus of rats exhibiting spontaneous absence epilepsy: a microdialysis study. J Neurochem. 1995;65:1674–1680. doi: 10.1046/j.1471-4159.1995.65041674.x. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Molliver ME, Snyder SH. D-Serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci U S A. 1995;92:3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, Carmignoto G, Steinhauser C. Astrocyte dysfunction in epilepsy. Brain Res Rev. 2010;63:212–221. doi: 10.1016/j.brainresrev.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitabilty have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Tong X, Kwan KY, Corey DP, Khakh BS. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci. 2011;15:70–80. doi: 10.1038/nn.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonato M, Löscher W, Cole AJ, Dudek E, Engel J, Jr, Kaminski RM, Loeb JA, Scharfman H, Staley KJ, Velíšek L, Klitgaard H. Finding a better drug for epilepsy: Preclinical screening strategies and experimental trial design. Epilepsia. 2012;53:1860–1867. doi: 10.1111/j.1528-1167.2012.03541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead OC., 3rd Evidence for GABAB-mediated mechanisms in experimental generalized absence seizures. Eur J Pharmacol. 1992;213:343–349. doi: 10.1016/0014-2999(92)90623-c. [DOI] [PubMed] [Google Scholar]
- Söhl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci. 2005;6:191–200. doi: 10.1038/nrn1627. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Seifert G, Bednes P. Astrocyte dysfunction in temporal lope epilepsy: K+ channels and gap junction coupling. Glia. 2012;60:1192–1202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci. 1995;15:623–642. doi: 10.1523/JNEUROSCI.15-01-00623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Chiu C, Jones MV, Petrou S. Increased thalamic inhibition in the absence seizure prone DBA/2J mouse. Epilepsia. 2008;49:921–925. doi: 10.1111/j.1528-1167.2008.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, Jones MV, Seeburg PH, Sakmann B, Berkovic SF, Sprengel R, Petrou S. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A. 2007;104:17536–17541. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G-F, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres A, Wang F, Xu Q, Fujita T, Dobrowolski R, Willecke K, Takano T, Nedergaard M. Extracellular Ca2+ acts as a mediator of communication from neurons to glia. Sci Signal. 2012;5:ra8. doi: 10.1126/scisignal.2002160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touret M, Parrot S, Denoroy L, Belin M-F, Didier-Blazès M. Glutamatergic alterations in the cortex of genetic absence epilepsy rats. BCM Neurosci. 2007;8:69. doi: 10.1186/1471-2202-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Wong RK. Cellular mechanism of neuronal synchronization in epilepsy. Science. 1982;216:745–747. doi: 10.1126/science.7079735. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–12455. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C. Cortical and thalamic lesions in rats with genetic absence epilepsy. J Neural Transm Suppl. 1992;35:71–83. doi: 10.1007/978-3-7091-9206-1_5. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- von Blankenfeld G, Kettenmann H. Glutamate and GABA receptors in vertebrate glial cells. Mol Neurobiol. 1991;5:31–43. doi: 10.1007/BF02935611. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABAA receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmijse I, Ossenblok P, Gunning B, van Luijtelaar G. Onset and propagation of spike and slow wave discharges in human absence epilepsy: A MEG study. Epilepsia. 2009;50:2538–2548. doi: 10.1111/j.1528-1167.2009.02162.x. [DOI] [PubMed] [Google Scholar]
- Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58:168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. A study of thalamic and cortical rhythms in petit mal. Brain. 1953;76:50–69. doi: 10.1093/brain/76.1.50. [DOI] [PubMed] [Google Scholar]
- Xu L, Zeng LH, Wong M. Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2009;34:291–299. doi: 10.1016/j.nbd.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J-m, Wang H-k, Ye C-q, Ge W, Chen Y, Jiang Z-l, Wu C-p, Poo M-m, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]