

Abstract
An enhanced tonic GABAA inhibition in the thalamus plays a crucial role in experimental absence seizures and has been attributed, on the basis of indirect evidence, to a dysfunction of the astrocytic GABA transporter-1 (GAT-1). Here, the GABA transporter current was directly investigated in thalamic astrocytes from a well-established genetic model of absence seizures, the genetic absence epilepsy rats from Strasbourg (GAERS), and its non-epileptic control (NEC) strain. We also characterized the novel form of GABAergic and glutamatergic astrocyte-to-neuron signalling by recording slow outward currents (SOCs) and slow inward currents (SICs), respectively, in thalamocortical (TC) neurons of both strains. In patch-clamped astrocytes, the GABA transporter current was abolished by combined application of the selective GAT-1 and GAT-3 blocker, NO711 (30 μm) and SNAP5114 (60 μm), respectively, to GAERS and NEC thalamic slices. NO711 alone significantly reduced (41%) the transporter current in NEC, but had no effect in GAERS. SNAP5114 alone reduced by half the GABA transporter current in NEC, whilst it abolished it in GAERS. SIC properties did not differ between GAERS and NEC TC neurons, whilst moderate changes in SOC amplitude and kinetics were observed. These data provide the first direct demonstration of a malfunction of the astrocytic thalamic GAT-1 transporter in absence epilepsy and support an abnormal astrocytic modulation of thalamic ambient GABA levels. Moreover, while the glutamatergic astrocyte–neuron signalling is unaltered in the GAERS thalamus, the changes in some properties of the GABAergic astrocyte–neuron signalling in this epileptic strain may contribute to the generation of absence seizures.
Key points
Enhanced thalamic tonic GABA inhibition plays a role in experimental absence seizures.
In this study we investigated astrocytic GABA transporter function and gliotransmitter release in an absence seizure rat model.
GAT-1 GABA transporter currents in thalamic astrocytes were reduced in an absence seizure rat model.
Spontaneous phasic astrocytic GABA events displayed kinetic differences between absence seizure model rats and non-epileptic controls.
Spontaneous phasic astrocyte glutamate release was not different in absence seizure model rats and non-epileptic controls.
Introduction
Typical absence seizures are a common feature of many idiopathic generalized epilepsies, and consist of sudden and brief periods of lack of consciousness which are invariably accompanied by a stereotypical EEG activity of generalized spike and wave discharges (Crunelli & Leresche, 2002; Blumenfeld, 2005). Although invasive experimental work (Williams, 1953) and more recent non-invasive imaging analysis in humans (Holmes et al. 2004; Hamandi et al. 2006; Bai et al. 2010) has indicated that these seizures are generated by paroxysmal electrical activity of cortical and thalamic networks (Meeren et al. 2002; Manning et al. 2004; Polack et al. 2007), the underlying abnormalities are still ill-defined (Crunelli & Leresche, 2002; Blumenfeld, 2005; Leresche et al. 2012).
Recently, it has been shown that the increased tonic GABAA receptor-mediated inhibition, which is present in thalamocortical (TC) neurons of both genetic and pharmacological models of absence epilepsy, represents both a necessary and sufficient condition for the generation of these non-convulsive seizures (Cope et al. 2009). This finding has provided an important mechanistic insight of why drugs that increase GABAergic function either worsen and/or induce absence seizures both in humans and animals (Hosford & Wang, 1997; Perucca et al. 1998; Ettinger et al. 1999). Moreover, the same work suggested that in genetic mouse and rat models of absence epilepsy a loss-of-function in one of the GABA transporters, i.e. GAT-1, which in the thalamus of both humans and rodents is exclusively located in astrocytes (De Biasi et al. 1998), may be responsible for the enhanced activity of the peri- and/or extra-synaptic GABAA receptors that mediate the tonic GABAA inhibition (Cope et al. 2009). However, this conclusion was based on indirect evidence and no data are available on the function of astrocytic GABA transporters in this type of non-convulsive epilepsy. Indeed, current evidence on transporter function in absence epilepsy is limited to the glutamatergic system, including a decreased expression of glutamate transporters in cortical astrocytes and thalamic neurons in pre- but not post-seizure animals (Dutuit et al. 2002), and a reduced cortical glutamate uptake (Touret et al. 2007).
Although abnormalities of traditional astrocytic functions, i.e. K+ buffering and glutamate homeostasis, are known to contribute to convulsive epileptic discharges (Coulter & Eid, 2012; Steinhauser et al. 2012), it is only comparatively recently that transient astrocytic glutamate release has been implicated in epilepsy. Different groups have reported increased astrocytic calcium activity in some epilepsy models and also an increase in slow inward current (SIC) frequency caused by Ca2+-dependent vesicular release of glutamate from astrocytes acting on neuronal NMDA receptors (Kang et al. 2005; Tian et al. 2005; Fellin et al. 2006; Ding et al. 2007). Early studies suggested that the increased SIC frequency could cause the paroxysmal events and underlie epileptic discharges (Kang et al. 2005; Tian et al. 2005), whilst later studies indicated no essential role for astrocytic glutamate in epilepsy (Fellin et al. 2006) but rather that astrocytic glutamate release acted to potentiate seizure activity (Gomez-Gonzalo et al. 2010) and contribute to epilepsy-associated neurodegeneration (Ding et al. 2007). However, no information is available on any potential alterations in this novel form of astrocyte–neuron signalling or in the astrocytic-derived GABA-mediated slow outward currents (SOCs) (Kozlov et al. 2006; Jimenez-Gonzalez et al. 2011) in absence epilepsy.
In this study, we have directly measured the GABA transporter current in thalamic astrocytes from a well-established genetic model of absence seizures, the genetic absence epilepsy rats from Strasbourg (GAERS) and their non-epileptic control (NEC) strain. Our results conclusively demonstrate that the astrocytic GAT-1 transporter current is absent in thalamic GAERS astrocytes, whereas the properties of SOCs and SICs are slightly changed and unaffected, respectively, compared with NEC. Some of these results were reported in preliminary form (Pirttimaki et al. 2010).
Methods
Ethical approval
All procedures were in accordance with UK Home Office legislation and Animals (Scientific Procedure) Act 1986, and were approved by the Ethics Committees at Aston and Cardiff Universities. Care was taken in planning the experiments to minimize the number of animals.
Slice preparation
Acute brain slices were obtained from 18- to 25-day-old male and female GAERS and NEC rats, which were anaesthetised with isoflurane and killed by cervical dislocation. Horizontal slices (300 μm thick) containing the VB thalamus were cut using an HM650V vibration microtome (Microm International GmbH, Germany), in continuously oxygenated (95% O2–5% CO2) artificial CSF (aCSF) containing (in mm): NaCl 85, NaHCO3 26, MgCl2 2, CaCl2 2, KCl 2.5, NaH2PO4 1.25, glucose 10, sucrose 73.6, indomethacin 0.045 and kynurenic acid 3, as previously described in Cope et al. (2009). Slices were then stored at 33 ± 1°C in an oxygenated incubation chamber containing the above aCSF, before it was gradually replaced with aCSF containing (in mm): NaCl 126, NaHCO3 26, MgCl2 2, CaCl2 2, KCl 2.5, NaH2PO4 1.25 and glucose 10 (Cope et al. 2009).
Neuronal recordings
Slices were viewed with an upright microscope (Olympus, UK) equipped with a water-immersion lens of ×40 magnification, attached to a charge-coupled device (CCD) camera (Olympus U-CMAD-2, Japan) for differential interface contrast (DIC) imaging. Slices were kept in submerged conditions and continuously perfused with recording aCSF of composition (in mm): NaCl 126, NaHCO3 26, MgCl2 1, CaCl2 2, KCl 2.5, NaH2PO4 1.25 and glucose 10, at +33 ± 1°C with a flow rate of 2–3 ml min−1 (as described previously in Cope et al. 2009). In some experiments (see below), Mg2+ was omitted from the aCSF (Pirttimaki et al. 2011). Low-resistance (3–5 MΩ) patch electrodes were prepared from borosilicate GC120F-10 glass micropipettes (1.2 mm OD, 0.69 mm ID, Harvard Apparatus Ltd, UK) using a P-97 Flaming/Brown micropipette puller (Sutter instrument Co., Novato, CA, USA).
Recordings of SICs were made from TC neurons using an internal solution of composition (in mm): KMeSO4 120, Hepes 10, EGTA 0.1, Na2ATP 4, GTP 0.5, pH adjusted to 7.3–7.4 with KOH, osmolarity 285–290 mosmol l−1. SOCs were recorded using a chloride-based solution, comprising (in mm): KCl 125, NaCl 10, MgCl2 2, CaCl2 1, Hepes 10, EGTA 10, Na2ATP 4, GTP 0.3, pH adjusted to 7.3–7.4 with KOH. To block possible contamination by N-methyl-d-aspartate (NMDA) receptor-mediated SICs, MK801 (1 mm) was included in the internal solution when recordings SOCs. All voltage clamp recordings were made at −60 mV and TC neurons with ≥20% change in access resistance were excluded. Currents were sampled at 10 kHz with a Digidata 1440A interface (Axon Instruments, Union City, CA, USA) and filtered at 1 kHz with a computer-controlled MultiClamp 700A (Molecular Devices, Sunnyvale, CA, USA). Data were acquired and viewed online using AxoScope 10.0 (Molecular Devices). All drugs were included in the recording aCSF as indicated in the text. Hypo-osmotic stimulation was induced by omitting 26 mm NaCl from the aCSF.
SICs and SOCs were analysed using the Event Detection protocols in the Clampfit routine of pCLAMP and exported to Sigmaplot (Jandel) for further analysis. Events were accepted as SICs if their amplitude was >20 pA and their time to peak was >20 ms. These criteria were used to retain consistency with our previous studies of SICs in the thalamus (Parri et al. 2010; Pirttimaki et al. 2011; Pirttimaki & Parri, 2012). In the hippocampus, different groups have used different selection criteria with some accepting smaller, more rapid events (Angulo et al. 2004; Perea & Araque, 2005). However applying similar criteria (10 ms time to peak, 10 pA amplitude) to our thalamic recordings only increased the number of SICs by 8%, resulting in a non-significant change in frequency (from 0.11 ± 0.06 SICs min−1 to 0.15 ± 0.07 SICs min−1, n = 4; P > 0.05). Thus, including smaller events may be crucial for hippocampal but not thalamic SICs. Events were accepted as SOCs if their amplitude was >20 pA and their time to peak was >10 ms, based upon experimental data derived in this present study (Fig. 1).
Figure 1. GABA transporter currents.
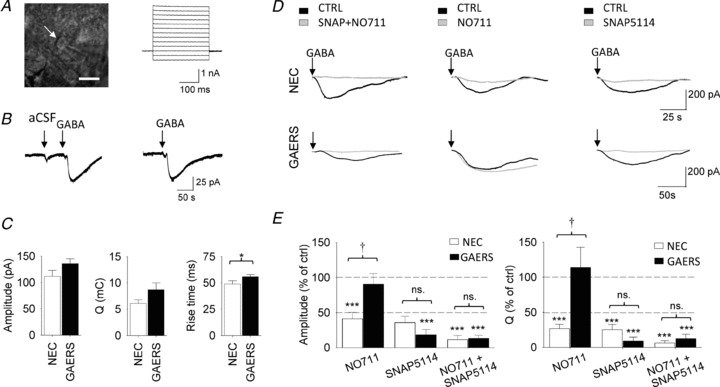
A, left, DIC image of an astrocyte (white arrow) in the VB thalamus with the recording patch electrode visible on the right hand side of the picture (scale bar, 10 μm). Right, current responses to 10 mV voltage steps from the imaged astrocyte. B, left current trace illustrates the typical response of a thalamic astrocytes to focal application of 100 μl aCSF without (aCSF) and with 10 mm GABA (GABA). The current trace to the right illustrates the response of the same astrocyte to a second focal application of GABA a few minutes after the one shown on the left. C, properties of GABA transporter currents in NEC and GAERS. D, GABA transporter currents in control condition (CTRL) and during application of GAT-1 (NO711, 30 μm) and GAT-3 (SNAP5114, 60 μm) blockers. Top and bottom panels show representative currents from NECs and GAERS, respectively. E, amplitude and charge transfer (Q) of the GABA transporter currents in NEC (n = 6) and GAERS (n = 7) expressed as % of those under control conditions. * Indicates significance for each group against control conditions (*P < 0.05, **P < 0.01 or ***P < 0.005); † indicates significant difference (P < 0.05) between NEC and GAERS.
Astrocyte recordings
Recording pipettes (5–8 MΩ) were filled with a solution containing (in mm): caesium methanesulphonate 120, Hepes 10, EGTA 5, Na2ATP 4, KCl 5, GTP 0.5 MgCl2 1 (pH 7.4, osmolarity 280 mosmol l−1). Astrocytes were held at −80 mV and access resistance was monitored throughout experiments. Results from astrocytes where series resistance changed by >20% or was >20 MΩ were discarded. Currents were recorded using an Axopatch 200B preamplifier (Molecular Devices), digitized with a Digidata 1440A, and acquired and analysed using pCLAMP 9.0.
GABA transporter currents were recorded in the presence of TTX (0.5 μm), kynurenic acid (3 mm), TBOA (100 μm), picrotoxin (40 μm), CGP55845 (20 μm) and NPPB (100 μm). GABA transporter currents were evoked by rapid manual focal application of 100 μl of a solution containing 10 mm GABA using a pipette positioned close to the recorded astrocyte. Application of 100 μl of vehicle aCSF without GABA elicited an artifact with time to peak of 5.5 ± 0.7 ms and total charge (Q) of 0.16 ± 0.05 mC (n = 6 astrocytes). These values are orders of magnitude smaller than the time course and Q of the transporter current elicited by 10 mm GABA (see Results), and show that the application time is rapid and that the artifact does not contribute to transporter current measurements. GABA transporter blockers were added to the perfusing solution as required, and were only applied once to a slice. All drug solutions were prepared as previously described (Cope et al. 2009). Charge transfer of GABA transporter current was determined using the Event detection protocols in pCLAMP 9.
Statistics
All quantitative data in the text and figures are presented as mean ± SEM unless otherwise stated. Significance was calculated using unpaired or paired Student's t test as appropriate. A Kolmogorov–Smirnov (K–S) test was used for population distribution comparisons.
Chemicals
Drugs were obtained from the following sources: kynu-renic acid from Sigma–Aldrich (Poole, Dorset, UK); 6-imino-3-(4-methoxyphenyl)-1-(6H)-pyridazinebutanoic acid hydrobromide (gabazine), (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxyl-propyl]-(phenyl-methyl) phosphinic acid (CGP55845), 1[2[tris(methoxy-phenyl)methoxy]-ethyl]-(S)-3-piperidinecarboxylic acid ((S)-SNAP5114), 1-(2-[([diphenylmethylene]imino)-oxy]ethyl)-1,2,5,6-tetrahydro-3-pyri-dinecarboxylic acid hydrochloride (NO711), 5-nitro-2-(3-phenylpropyl-amino)benzoic acid (NPPB) and dl-threo-β-benzyloxy-aspartic acid (dl-TBOA) from Tocris Bioscience (Bristol, UK). d-AP5, picrotoxin and tetrodotoxin (TTX) were obtained from Ascent (Weston-super-Mare, UK).
Results
Astrocytic GABA transporter current
Thalamic astrocytes in the VB were initially identified visually on the basis of their size and morphology (Fig. 1A), followed by electrophysiological characterization which included a linear I–V relationship (Fig. 1A), low input resistance (28.5 ± 0.9 MΩ, n = 87), hyperpolarized resting membrane potential (−75.7 ± 0.6 mV), and lack of action potential currents following depolarizing voltage commands (cf. Parri et al. 2010). To isolate the GABA transporter current, the GABAA and GABAB receptor blockers picrotoxin (40 μm) and CGP55845 (20 μm), respectively, TTX (0.5 μm), the glutamate receptor antagonist kynurenic acid (3 mm), the glutamate transporter blocker TBOA (100 μm) and the anion channel blocker NPPB (100 μm) were included in the perfusate. Under these experimental conditions (hereafter referred to as the control condition), focal application of GABA (100 μl of an aCSF solution containing 10 mm GABA) reliably evoked in the patched astrocytes a substantial inward current (hereafter referred to as the GABA transporter current), which was much larger than that evoked by a similar focal application of 100 μl aCSF (amplitude in NEC: 25 ± 5.6 pA; Q in NEC: 0.16 ± 0.05 mC, n = 6 astrocytes) that did not contain GABA (Fig. 1B) (see Methods). This response was highly reproducible in the same astrocytes (Fig. 1B) as indicated by the similarity in transporter current amplitude, charge transfer (Q) and rise time between the first and second GABA application in slices from both NEC and GAERS (amplitude of 2nd response in NEC: 96 ± 5% of 1st response, n = 7 astrocytes, P = 0.4; in GAERS: 98 ± 9%, n = 8, P = 0.8; Q in NEC: 101 ± 11%, P = 0.9; in GAERS: 118 ± 16%, P = 0.3; rise time in NEC: 102 ± 12%, P = 0.8; in GAERS: 132 ± 18%, P = 0.1).
The properties of the astrocytic GABA transporter current in control conditions were similar in NEC and GAERS, with only a minor difference in rise time (amplitude in NEC: 112 ± 11 pA, n = 24 astrocytes; in GAERS, 136 ± 9 pA, n = 33, P = 0.08; Q in NEC: 6.1 ± 0.7 mC; in GAERS: 8.7 ± 1.3 mC, P = 0.1; rise time in NEC: 49 ± 3 s; in GAERS: 56 ± 2 s, P = 0.04) (Fig. 1C and D). Changes in the activity of the individual GABA transporters were investigated using the selective GAT-1 and GAT-3 inhibitors, NO711 (30 μm) and SNAP5114 (60 μm), respectively, in both strains. For each patched astrocyte in a slice, after a focal GABA application in control condition, either NO711, SNAP5114 or both were bath applied and focal GABA application repeated. In the presence of NO711, the amplitude of the GABA transporter current in NECs was significantly reduced to 41 ± 9% of control (n = 6 astrocytes, P = 0.002) (Fig. 1D and E). In contrast, NO711 had no effect on the amplitude of the GABA transporter current in GAERS astrocytes (90 ± 1%, n = 7, P = 0.55) (Fig. 1E). Similarly, in the presence of NO711, the charge transfer (Q) of the GABA transporter current was significantly smaller in NECs (27 ± 6% of control, n = 6, P < 0.001), whilst it was unaffected in GAERS (114 ± 27%, n = 7, P = 0.65) (Fig. 1D and E). Application of the GAT-3 blocker SNAP5114 (60 μm) markedly reduced the amplitude and Q of the transporter current in NEC astrocytes (amplitude: 36 ± 9% of control, n = 6, P = 0.001; Q: 25 ± 8%, n = 6, P < 0.001), whereas co-application of NO711 and SNAP5114 nearly abolished it (amplitude: 11 ± 6% of control, n = 6, P < 0.001; Q: 6 ± 3%, n = 6, P < 0.001) (Fig. 1D and E). In contrast, SNAP5114 inhibited the astrocytic GABA transporter current in GAERS to a similar extent (amplitude: 18 ± 6% of control, n = 7, P < 0.001; Q: 8 ± 6%, n = 7, P < 0.001) as the co-application of NO711 and SNAP5114 (amplitude: 13 ± 5% of control, n = 7, P < 0.001; Q: 12 ± 6%, n = 7, P < 0.001) (Fig. 1E). Together, these data clearly demonstrate a dysfunctional GAT-1 activity in GAERS.
Astrocytic-derived GABAergic SOCs
We have previously described the presence and properties of spontaneous astrocytic-derived GABAergic slow outward currents (SOCs) in VB TC neurons of normal Wistar rats (Jimenez-Gonzalez et al. 2011). Because of the experimental conditions used in the present study, GABAergic events were actually recorded, and are displayed as inward currents; however, they will be referred to as slow outward currents for consistency with previous investigations (Kozlov et al. 2006; Jimenez-Gonzalez et al. 2011). We first characterised the rise time for all spontaneous inward currents recorded in TC neurons to investigate differences between SOCs and IPSCs. Currents were recorded in the presence of TTX (1 μm) and kynurenic acid (3 mm) to block SICs and EPSCs. Analysis of the rise time distribution of the inward currents revealed two distinct distributions with rise times <10 ms and >10 ms, respectively (Fig. 2A). Therefore, on a similar basis to previous studies (cf. Kozlov et al. 2006; Jimenez-Gonzalez et al. 2011), events with a rise time >10 ms were classified as SOCs.
Figure 2. SOCs in thalamocortical neurons.
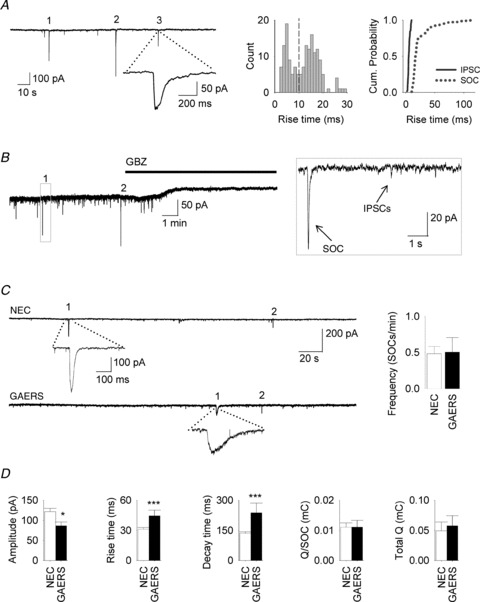
A, left, recording from a NEC TC neuron showing prevalence of outward currents in TTX (1 μm) and kynurenic acid (3 mm) in 1 mm Mg2+ at 33°C. Three SOCs are numbered (1, 2, 3) with one expanded. Middle, distribution of the rise time of all inward currents recorded in the presence of TTX (1 μm) and kynurenic acid (3 mm) (bin size: 1 ms). Note the presence of two populations of events (dotted line at 10 ms), which is also better illustrated by the cumulative probability plot on the right. B, representative trace from a NEC TC neuron (in 1 μm TTX and 3 mm kynurenic acid) showing recorded currents sensitivity to gabazine (GBZ, 25 μm). Boxed area is expanded to the right to show different amplitude and kinetics of SOCs and IPSCs. C, left, representative recording from NEC (top) and GAERS (below) showing SOC frequency. One numbered SOC is expanded below each trace. Right, bar graph showing SOC frequency (NEC, n = 28 neurons; GAERS, n = 21). D, bar graphs of other SOC parameters (Q: average charge transfer of SOCs; Total Q: sum of SOC charge transfer in 10 min).
In the presence of TTX (1 μm) and kynurenic acid (3 mm), application of the GABAA antagonist gabazine (25 μm) abolished (0.9 ± 0.9% compared with before gabazine application, n = 8 neurons, P < 0.001) all recorded inward currents events (i.e. IPSCs and SOCs), confirming that they were mediated by GABAA receptors (Fig. 2B). No difference was observed in the frequency of SOCs between NEC and GAERS TC neurons (NEC: 0.48 ± 0.09 SOCs min−1, n = 28 neurons; GAERS: 0.51 ± 0.20 SOCs min−1, n = 21, P = 0.9) (Fig. 2C). The average Q of single SOCs and the total Q (summated SOC Q values recorded in 10 min) were also similar between NEC and GAERS (Q for NEC: 0.011 ± 0.001 mC, n = 28 neurons; for GAERS: 0.011 ± 0.002 mC, n = 21, P = 0.7; total Q for NEC: 0.05 ± 0.01 mC; for GAERS: 0.06 ± 0.02 mC, P = 0.99) (Fig. 2D). However, the SOC amplitude was significantly smaller in GAERS (86 ± 9 pA, n = 161 SOCs in 21 neurons) than in NEC (121 ± 8 pA, n = 402 SOCs in 28 neurons; P = 0.02), whilst the rise time and decay time were increased (rise time in NEC: 31 ± 2 ms; in GAERS: 44 ± 6 ms, P = 0.004; decay time in NEC: 135 ± 7 ms; in GAERS: 237 ± 48 ms, P = 0.002) (Fig. 2D).
Astrocytic-derived glutamatergic SICs are similar in control and epileptic rats
Spontaneous SICs recorded in a nominal 0 mm Mg2+ aCSF at 33 ± 1°C from GAERS TC neurons had similar characteristics (frequency: 0.15 ± 0.03 SICs min−1, n = 25 neurons; amplitude: 80.5 ± 6.9 pA, n = 94 SICs; rise time: 88.8 ± 23.9 ms) to those previously described in VB TC neurons of normal Wistar rats (Parri et al. 2001). They were insensitive to TTX (1 μm) (Fig. 3A) (frequency: 148.8 ± 31.2% of control, n = 10 neurons, P = 0.15; rise time in control: 159 ± 26 ms, n = 60 SICs; in TTX: 120 ± 19 ms, n = 70, P = 0.2) and were abolished by d-AP5 (50 μm) (0% of control, n = 5 neurons, P < 0.001) (Fig. 3B).
Figure 3. SICs in thalamocortical neurons.
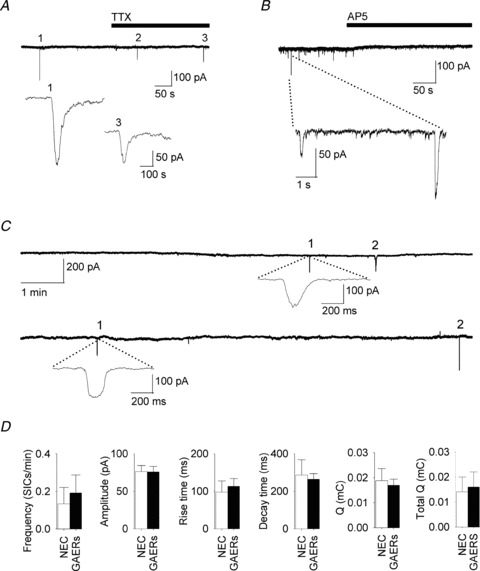
A, current recording from a GAERS TC neuron showing no change in SIC frequency before and during TTX (1 μm) in a Mg2+-free aCSF at 33 ± 1°C. Two SICs (numbered 1 and 3) are expanded below. B, recording from a GAERS TC neuron illustrate the block of SICs by d-AP5 (50 μm) in Mg2+-free aCSF in the presence of TTX (1 μm). C, representative recordings in Mg2+ (1 mm) from NEC (top) and GAERS (below) showing two (numbered) SICs in each trace. One SIC is expanded below each trace. D, bar graph showing different parameters for SICs recorded in NEC (n = 42 SICs in 28 neurons) and GAERS (n = 71/26).
Comparison of SIC properties recorded in NEC and GAERS in 1 mm Mg2+ at 33°C revealed no significant differences, indicating that the glutamatergic astrocyte-to-neuron signalling is not altered in this genetic model of absence seizures (Fig. 3C and D). Thus, the SIC frequency was 0.13 ± 0.09 SICs min−1 (n = 28 neurons) in NEC and 0.19 ± 0.09 SICs min−1 in GAERS (n = 26, P = 0.66). Moreover, the other SIC properties were similar between the two strains (amplitude in NEC: 76.3 ± 7.9 pA, n = 42 SICs/28 neurons; in GAERS: 75.8 ± 7.3 ms, n = 71/26, P = 0.98; rise time in NEC: 97.6 ± 30.1 ms; in GAERS, 113.1 ± 20.6 ms, P = 0.66; decay time in NEC: 286.2 ± 81.7 ms; in GAERS, 264.3 ± 29.3 ms, P = 0.76; Q in NEC: 0.019.6 ± 0.005 mC; in GAERS: 0.017 ± 0.003 mC, P = 0.71; total Q in NEC: 0.014 ± 0.006 mC; in GAERS: 0.016 ± 0.06 mC, P = 0.3) (Fig. 3C and D).
Discussion
The main findings of this study are that in the thalamus of GAERS, a well-established genetic model of absence seizures, (1) GAT-1 is functionally deficient in VB astrocytes, (2) the properties of SOCs, i.e. the hallmark of the inhibitory astrocyte-to-neuron signalling, are slightly affected, and (3) the excitatory astrocyte-to-neuron signalling, i.e. SICs, is similar to that of non-epileptic rats.
GAT-1 malfunction in thalamic astrocytes
Our previous work (Cope et al. 2009), where the neuronal tonic GABAA current was used as an indirect read-out of the activity of the two main thalamic GABA transporters, GAT-1 and GAT-3, indicated a loss-of-function of GAT-1 in GAERS and other genetic models of absence seizures. Here, by recording the GABA transporter current from thalamic astrocytes, we show that in GAERS this current is unaffected by the block of GAT-1 (with NO711) and abolished by blocking GAT-3 (with SNAP5114), providing direct and conclusive evidence of an abnormal GAT-1 function in experimental absence epilepsy. It is unlikely that our results are compromised either by (i) the presence of an additional neuronal GABA uptake system, since in contrast to other brain regions, GAT-1 and GAT-3 are the main GABA transporters in the thalamus and are largely, if not exclusively, located in astrocytes in the rat and human thalamus (De Biasi et al. 1998); (Vitellaro-Zuccarello et al. 2003), and/or (ii) by the pharmacological profile of the transporter blockers since their selectivity for each GABA transporter is well established at the concentrations used in this study (Keros & Hablitz, 2005).
In the investigation of the tonic GABAA current in TC neurons, GAT-3 was unable to compensate for the malfunctioning GAT-1, i.e. the tonic GABAA current of these neurons was larger in GAERS, and other genetic absence models, than in their respective control animals (Cope et al. 2009). In contrast, the GABA transporter current of GAERS thalamic astrocytes in control condition was shown here to be similar to that in NEC, indicating an increased functional expression of GAT-3 in GAERS. This apparent discrepancy is not surprising if one considers the different experimental conditions used in these two studies. Thus, the former investigation measured the effect of the physiological ambient GABA as set by the balance between its neuronal release and astrocytic uptake, while in the present study we were challenging the transporter function in response to a focal application of GABA, which most probably affected the entire astrocytic surface. Indeed, the inability of GAT-3 to compensate for GAT-1 in the former but not the latter condition suggests that GAT-1 and GAT-3 have different locations, with GAT-1 probably positioned relatively closer than GAT-3 to the extrasynaptic δ-containing GABAA receptors that mediate the tonic current, or possibly closer to the source of GABA release mediating the tonic current. Unfortunately, we were unable to reliably record physiological (i.e. synaptically evoked) GABA transporter currents by electrically stimulating the thalamic reticular nucleus afferents, the only GABAergic input to TC neurons in the rat VB, and thus to test directly whether under these conditions GAT-3 would have been unable to compensate for GAT-1 in GAERS. Importantly, a recent confocal microscopy analysis of the relative position of GAT-1 and GAT-3 with respect to GABA synaptic proteins in the thalamus has concluded that GAT-1 is ‘primarily localized near GABAergic synapses whereas GAT-3 is localized both near and far away from synapses’ (Beenhakker & Huguenard, 2010). In the absence of any investigation at the ultrastructural level on the relative location of these transporters with respect to postsynaptic δ- and non-δ-containing GABAA receptors and to postsynaptic GABAB receptors in the thalamus, or any other brain regions, it is difficult at present to explain these potentially contradictory results.
The inability of NO711 to affect the transporter current evoked by focal GABA application in GAERS thalamic astrocytes strongly indicate that the enhanced tonic GABAA current in TC neurons of this absence model is not due to a simple misplacement of GAT-1 along the astrocytic plasma membrane. Thus, since both GAT-1 mRNA and protein levels are similar in GAERS and NEC thalamus and only a silent mutation is present in the GAT-1 gene of GAERS and stargazer mouse (another model of absence epilepsy), it is likely that the GAT-1 malfunction in these models of non-convulsive epilepsy could be due to either its impaired translocation to the astrocytic plasma membrane (i.e. increased internalization) and/or to an abnormal phosphorylation. Interestingly, no GAT-1 abnormality is present in the dentate gyrus granule cells of GAERS, which do not participate in the expression of typical absence seizures (Cope et al. 2009). This indicates regionally specific transporter deficiencies. It is therefore possible that GAT-1 (or even GAT-3) astrocytic deficiencies compounding the absence phenotype may be present in other regions of the thalamocortical loop.
Astrocyte–neuron signalling in absence epilepsy
The dysfunction in GAT-1 might indicate a general astrocytic deficiency, which would affect other astrocytic mechanisms such as gliotransmitter release and the consequent astrocyte–neuron signalling cascade. This was in part the case since the GABAergic SOCs had slower kinetics and a smaller amplitude in GAERS than in NEC. Whether the changes in kinetics, and in particular the slower decay time of SOCs in GAERS, are a direct consequence of the GAT-1 malfunction remains to be determined. However, these differences in kinetics and amplitude compensate each other in such a way that the Q of the SOCs, i.e. the amount of astrocytic GABA-mediated drive sensed by a TC neuron, was similar in the two strains.
Although SOCs were shown to be able to synchronously inhibit neighbouring neurons in the olfactory bulb (Kozlov et al. 2006) and to induce hyperpolarisations of VB TC neurons (Jimenez-Gonzalez et al. 2011), their physiological role is still unclear. This makes it difficult to ascribe a precise pathological significance to the slower rise and decay times and the smaller amplitude of SOCs in GAERS. In the VB thalamus, however, SOCs were shown to target the δ-subunit-containing GABAA receptors that underlie the tonic GABAA current (Jimenez-Gonzalez et al. 2011). Therefore, whilst we do not yet know whether SOCs may be involved in synchronizing TC neurons, their function in the thalamus may more likely be that of a slowly acting, continuous contribution to thalamic tonic GABAA-mediated inhibition. Interestingly, the GABA transaminase inhibitor vigabatrin, which elicits and/or exacerbates absence seizures in animals and humans (Lortie et al. 1993), has been shown to increase in a time-dependent manner the frequency of SOCs in VB TC neurons (Jimenez-Gonzalez et al. 2011). This may indirectly suggest a potential astrocytic GABA release mechanism that contributes to thalamic tonic GABAA inhibition and ultimately to absence seizure generation.
In contrast to studies in models of convulsive epilepsy that have reported differences in SICs and calcium-dependent astrocytic glutamate release in hippocampal and cortical astrocytes (Kang et al. 2005; Tian et al. 2005; Fellin et al. 2006; Ding et al. 2007), we found no differences in the amplitude, frequency or kinetics of SICs recorded from GAERS and NEC TC neurons. Since astrocytic GABA is metabolized via the TCA cycle to glutamine, which in turn is essential for sustaining glutamatergic neurotransmission, these data demonstrate that despite a potential reduction in glutamine resulting from the deficient GABA uptake in GAERS astrocytes due to the malfunctioning GAT-1, the releasable glutamate pool in the astrocytes is not affected. Compared with convulsive epileptic discharges, the lack of differences in SIC properties in GAERS is not surprising since, while during the paroxysmal discharges of convulsive epilepsy hippocampal and cortical neurons show strong action potential discharges, inhibition is the hallmark of neuronal activity in TC neurons during non-convulsive absence seizures (Pinault et al. 1998; Cope et al. 2005). In this respect, it is more likely that changes in SIC properties may occur in other brain regions, i.e. cortex, that shows increased excitation during, and are pivotal for the initiation of, absence seizures in GAERS and other genetic models of this type of non-convulsive epilepsy.
The frequency of recorded SICs in the VB thalamus was similar to previous studies (Parri et al. 2010; Pirttimaki et al. 2011; Pirttimaki & Parri, 2012), but lower than some studies in hippocampal CA1 neurons. Modifying our analysis criteria did not drastically change the apparent SIC frequency (see Methods): therefore, our selection criteria do not account for the apparent low thalamic incidence of these events. Two reasons may explain this result: inherently different spontaneous SIC frequency in different brain areas (this may indicate different functional astrocytic contributions), or that different local experimental conditions affect SIC detection. The former is supported by data showing different frequencies between different cellular areas by the same group (Le Meur et al. 2012), while the latter is suggested by different frequencies within the same brain area as reported by different groups. Because of the continuing debate on the physiological roles of glial–neuron signalling, with some groups reporting a lack of gliotransmitter effect (Agulhon et al. 2010), it will be important to further investigate these issues and perhaps standardise experimental and selection criteria for gliotransmitter events.
In summary, our results directly demonstrate a malfunction of the GABA transporter GAT-1 in thalamic astrocytes of a well-established genetic model of absence epilepsy, and indicate that this abnormality is not a reflection of a generalised pathology affecting all astrocytic functions. These data have strong potential applications for the development of novel therapeutic avenues for human absence seizures.
Acknowledgments
This work was supported by the European Union (grant HEALTH F2-2007-202167).
Glossary
- GAT
GABA transporter
- GAERS
genetic absence epilepsy rats from Strasbourg
- NEC
non-epileptic controls
- SIC
slow inward current
- SOC
slow outward current
- TC
thalamocortical
- VB
ventrobasal
Author contributions
Experiments were performed at Cardiff University (V.C. laboratory) and Aston University (H.R.P. laboratory). Conception and design of the experiments by T.P., H.R.P. and V.C. Experiments conducted by T.P. and H.R.P. Analysis and interpretation of data by T.P., H.R.P. and V.C. Drafting of article T.P., H.R.P and V.C. All authors approved the final version of the manuscript.
Author's present address
T. Pirttimaki: Department of Neurobiology, A.I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, 70211 Kuopio, Finland.
References
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Vestal M, Berman R, Negishi M, Spann M, Vega C, Desalvo M, Novotny EJ, Constable RT, Blumenfeld H. Dynamic time course of typical childhood absence seizures: EEG, behavior, and functional magnetic resonance imaging. J Neurosci. 2010;30:5884–5893. doi: 10.1523/JNEUROSCI.5101-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci. 2010;30:15262–15276. doi: 10.1523/JNEUROSCI.3243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H. Consciousness and epilepsy: why are patients with absence seizures absent. Prog Brain Res. 2005;150:271–286. doi: 10.1016/S0079-6123(05)50020-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- De Biasi S, Vitellaro-Zuccarello L, Brecha NC. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization. Neuroscience. 1998;83:815–828. doi: 10.1016/s0306-4522(97)00414-4. [DOI] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutuit M, Touret M, Szymocha R, Nehlig A, Belin MF, Didier-Bazes M. Decreased expression of glutamate transporters in genetic absence epilepsy rats before seizure occurrence. J Neurochem. 2002;80:1029–1038. doi: 10.1046/j.0022-3042.2002.00768.x. [DOI] [PubMed] [Google Scholar]
- Ettinger AB, Bernal OG, Andriola MR, Bagchi S, Flores P, Just C, Pitocco C, Rooney T, Tuominen J, Devinsky O. Two cases of nonconvulsive status epilepticus in association with tiagabine therapy. Epilepsia. 1999;40:1159–1162. doi: 10.1111/j.1528-1157.1999.tb00835.x. [DOI] [PubMed] [Google Scholar]
- Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci. 2006;26:9312–9322. doi: 10.1523/JNEUROSCI.2836-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, de Curtis M, Ratto GM, Carmignoto G. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. 2010;8:e1000352. doi: 10.1371/journal.pbio.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamandi K, Salek-Haddadi A, Laufs H, Liston A, Friston K, Fish DR, Duncan JS, Lemieux L. EEG-fMRI of idiopathic and secondarily generalized epilepsies. NeuroImage. 2006;31:1700–1710. doi: 10.1016/j.neuroimage.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Holmes MD, Brown M, Tucker DM. Are “generalized” seizures truly generalized? Evidence of localized mesial frontal and frontopolar discharges in absence. Epilepsia. 2004;45:1568–1579. doi: 10.1111/j.0013-9580.2004.23204.x. [DOI] [PubMed] [Google Scholar]
- Hosford DA, Wang Y. Utility of the lethargic (lh/lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures. Epilepsia. 1997;38:408–414. doi: 10.1111/j.1528-1157.1997.tb01729.x. [DOI] [PubMed] [Google Scholar]
- Jimenez-Gonzalez C, Pirttimaki T, Cope DW, Parri HR. Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets delta-subunit-containing GABAA receptors. Eur J Neurosci. 2011;33:1471–1482. doi: 10.1111/j.1460-9568.2011.07645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang N, Xu J, Xu Q, Nedergaard M, Kang J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;94:4121–4130. doi: 10.1152/jn.00448.2005. [DOI] [PubMed] [Google Scholar]
- Keros S, Hablitz JJ. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol. 2005;94:2073–2085. doi: 10.1152/jn.00520.2005. [DOI] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci U S A. 2006;103:10058–10063. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Meur K, Mendizabal-Zubiaga J, Grandes P, Audinat E. GABA release by hippocampal astrocytes. Front Comput Neurosci. 2012;6:59. doi: 10.3389/fncom.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leresche N, Lambert RC, Errington AC, Crunelli V. From sleep spindles of natural sleep to spike and wave discharges of typical absence seizures: is the hypothesis still valid. Pflugers Arch. 2012;463:201–212. doi: 10.1007/s00424-011-1009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortie A, Chiron C, Mumford J, Dulac O. The potential for increasing seizure frequency, relapse and appearance of new seizure types with vigabatrin. Neurology. 1993;43:S24–27. [PubMed] [Google Scholar]
- Manning JP, Richards DA, Leresche N, Crunelli V, Bowery NG. Cortical-area specific block of genetically determined absence seizures by ethosuximide. Neuroscience. 2004;123:5–9. doi: 10.1016/j.neuroscience.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Sensory and cortical activation of distinct glial cell subtypes in the somatosensory thalamus of young rats. Eur J Neurosci. 2010;32:29–40. doi: 10.1111/j.1460-9568.2010.07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005;25:2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5–17. doi: 10.1111/j.1528-1157.1998.tb01268.x. [DOI] [PubMed] [Google Scholar]
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol. 1998;509:449–456. doi: 10.1111/j.1469-7793.1998.449bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Cope DW, Parri HR, Crunelli V. Program No. 255.3. 2010 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience, 2010. Online. 2010. Glial signalling in the ventrobasal thalamus of rats with absence seizures. [Google Scholar]
- Pirttimaki TM, Hall SD, Parri HR. Sustained neuronal activity generated by glial plasticity. J Neurosci. 2011;31:7637–7647. doi: 10.1523/JNEUROSCI.5783-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Parri HR. Glutamatergic input-output properties of thalamic astrocytes. Neuroscience. 2012;205:18–28. doi: 10.1016/j.neuroscience.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. 2012;60:1192–1202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touret M, Parrot S, Denoroy L, Belin MF, Didier-Bazes M. Glutamatergic alterations in the cortex of genetic absence epilepsy rats. BMC Neurosci. 2007;8:69. doi: 10.1186/1471-2202-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitellaro-Zuccarello L, Calvaresi N, De Biasi S. Expression of GABA transporters, GAT-1 and GAT-3, in the cerebral cortex and thalamus of the rat during postnatal development. Cell Tissue Res. 2003;313:245–257. doi: 10.1007/s00441-003-0746-9. [DOI] [PubMed] [Google Scholar]
- Williams D. A study of thalamic and cortical rhythms in petit mal. Brain. 1953;76:50–69. doi: 10.1093/brain/76.1.50. [DOI] [PubMed] [Google Scholar]