

Abstract
The chloride channel CLC-3 is expressed in the brain on synaptic vesicles and postsynaptic membranes. Although CLC-3 is broadly expressed throughout the brain, the CLC-3 knockout mouse shows complete, selective postnatal neurodegeneration of the hippocampus, suggesting a crucial role for the channel in maintaining normal brain function. CLC-3 channels are functionally linked to NMDA receptors in the hippocampus; NMDA receptor-dependent Ca2+ entry, activation of Ca2+/calmodulin kinase II and subsequent gating of CLC-3 link the channels via a Ca2+-mediated feedback loop. We demonstrate that loss of CLC-3 at mature synapses increases long-term potentiation from 135 ± 4% in the wild-type slice preparation to 154 ± 7% above baseline (P < 0.001) in the knockout; therefore, the contribution of CLC-3 is to reduce synaptic potentiation by ∼40%. Using a decoy peptide representing the Ca2+/calmodulin kinase II phosphorylation site on CLC-3, we show that phosphorylation of CLC-3 is required for its regulatory function in long-term potentiation. CLC-3 is also expressed on synaptic vesicles; however, our data suggest functionally separable pre- and postsynaptic roles. Thus, CLC-3 confers Cl− sensitivity to excitatory synapses, controls the magnitude of long-term potentiation and may provide a protective limit on Ca2+ influx.
Key points
The CLC-3 chloride channel is expressed on postsynaptic membranes, where it is spatially and functionally linked to the NMDA receptor.
CLC-3 is phosphorylated by Ca2+/calmodulin kinase II.
Loss of CLC-3 expression or prevention of CLC-3 phosphorylation/gating results in excessive induction of long-term potentiation.
Given that knockout of CLC-3 results in hippocampal neurodegeneration, our results suggest that CLC-3 gating may provide a protective limit on plasticity and Ca2+ influx.
Introduction
The chloride channel CLC-3 is expressed in the brain on synaptic vesicles (Stobrawa et al. 2001) and postsynaptic plasma membranes (Wang et al. 2006). Although CLC-3 is broadly expressed throughout the brain (Kawasaki et al. 1994), the CLC-3 knockout mouse shows complete, selective postnatal neurodegeneration of the hippocampus (and photoreceptors). Similar neurodegeneration has been documented in the three Clcn3−/− mice that have been made and can be seen over a time period of 7 months in the hippocampus of the Clcn3−/− mice in this study. The apparent hippocampal sensitivity to CLC-3 loss, in combination with the synaptic localization and previously demonstrated role in modulating synaptic events, motivated our investigation into the function of CLC-3 in controlling plasticity at the CA3–CA1 synapse (Stobrawa et al. 2001; Dickerson et al. 2002; Yoshikawa et al. 2002).
The Cl− gradient, often through the employment of CLC channels and transporters, is a key determinant of cell excitability over short time scales, such as during bursts of inhibitory network activity, and over extended time scales throughout development (Ben-Ari et al. 1989, 1997; Ben-Ari, 2002). Underscoring the importance of the Cl− ion in cell function, five of the nine CLC Cl− channel family members have so far been linked to human diseases, such as myotonia (CLC-1), Bartter syndrome (CLC-Kb) and Dent's disease (CLC-5). Additionally, decreased expression of K+–Cl− cotransporter KCC2, the primary extruder of Cl− in mature neurons, is associated with ischaemic brain injury and seizures (Jin et al. 2005; Pathak et al. 2007; Papp et al. 2008). Changes in KCC2 expression can be induced by NMDA receptor (NMDAR) activity, resulting in depolarizing GABAA receptor currents (Lee et al. 2011). Although it is apparent that excitatory synaptic activity can indirectly affect inhibition via [Cl−]i, a role for Cl− in directly altering excitatory synaptic responses is relatively unexplored.
The Cl− ion is also critically involved in volume regulation and is the primary charge shunt conductance utilized in intracellular organelles. In the brain, presynaptic CLC-3 was shown to aid synaptic vesicle acidification (Stobrawa et al. 2001; Riazanski et al. 2011); however, the necessity of CLC-3 as the glutamatergic vesicular shunt pathway is debated owing to an apparent Cl− flux through the glutamate transporter VGLUT1 (Schenck et al. 2009). Conversely, at inhibitory synapses CLC-3 serves as the primary charge shunt pathway to facilitate neurotransmitter loading (Riazanski et al. 2011). Vesicular CLC-3 activation is likely to be gated by changes in intravesicular pH, driven by the vesicular ATPase (Matsuda et al. 2008b). Plasma membrane CLC-3 is unique among its family members in that it is gated by Ca2+-dependent phosphorylation (Huang et al. 2001; Robinson et al. 2004; Wang et al. 2006). NMDAR-dependent Ca2+ entry, activation of Ca2+/calmodulin kinase II (CaMKII) and subsequent phosphorylation/gating of CLC-3 by CaMKII link the two channels via a Ca2+-mediated feedback loop. Two major splice variants of CLC-3, CLC-3A and CLC-3B, have differential expression profiles with regard to both tissue type and subcellular localization to organelles or the plasma membrane (Gentzsch et al. 2002; Ogura et al. 2002; Zhao et al. 2007); the reliance on pH versus CaMKII phosphorylation for gating is potentially explained by inherent differences in isoform structure or divergent trafficking. As a result of the shift in the Cl− gradient during development (Ben-Ari et al. 1989, 1997; Ben-Ari, 2002; Jentsch et al. 2005), plasma membrane CLC-3 promotes depolarization in immature neurons and suppresses excitability in mature neurons when Cl− flux is inhibitory (Wang et al. 2006). Thus, in the mature brain, CLC-3 channels serve as a charge shunt pathway, giving rise to membrane hyperpolarization, reduction in excitatory current amplitude and promotion of the block of NMDARs by Mg2+.
Long-term potentiation (LTP) at the Schaffer collateral–CA1 synapse in the hippocampus is dependent on Ca2+ entry through NMDARs (Lynch et al. 1983; Kauer et al. 1988; Malenka et al. 1988; Malenka & Nicoll, 1993). Influx of Ca2+ activates protein kinases, such as CaMKII, that act to increase the sensitivity of the postsynaptic cell to glutamate release via AMPA receptor insertion (Shi et al. 1999; Bredt & Nicoll, 2003; Collingridge et al. 2004), phosphorylation (Benke et al. 1998; Derkach et al. 1999; Lee et al. 2003) or immobilization in the postsynaptic density (PSD; Bats et al. 2007; Opazo et al. 2010). The tight functional and spatial coupling of CLC-3 and synaptic NMDARs demonstrated by Wang et al. (2006) suggests that CLC-3 acts essentially as a biophysical property of the NMDAR; the receptor is functionally sensitive to the Cl− gradient, without a literal Cl− conductance of its own. Neuronal Cl− homeostasis is a fundamental determinant of excitability in the brain; thus, we investigated the ability of CLC-3 to regulate synaptic plasticity in the hippocampus. Given that there is no specific antagonist of CLC-3, we used a Clcn3−/− mouse model for our studies (Dickerson et al. 2002).
Methods
Ethical approval
Studies were performed according to the principles set forth by the Animal Welfare Act and Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996) and were approved by the University of Chicago Institutional Animal Care and Use Committee.
Slice preparation
We used Clcn3−/− (KO) mice (Dickerson et al. 2002) and littermate wild-type (WT) mice as control animals. Genotyping was performed as previously described (Dickerson et al. 2002; Wang et al. 2006) by Transnetyx (Cordova, TN, USA). Mice were anaesthetized using isofluorane and rapidly decapitated using a guillotine. Brains were removed and horizontal hippocampal slices (350 μm) prepared from juvenile mice (postnatal day 19–30) in ice-cold dissection solution. Slices were incubated at room temperature for at least 2 h in oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid containing (mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2 and 10 glucose. In experiments with Tat-peptides, 5 or 10 μm Tat-peptide was added to the incubation chamber. Tat-CLC-3107−116 (YGRKKRRQRRR-INSKKKESAW) and Tat-CLC-3scr (YGRKKRRQRRR-KSIESANKWK) were obtained from GenScript (Piscataway, NJ, USA).
Electrophysiology
Extracellular field recordings were performed in an interface chamber at 32°C using an EPC9 patch-clamp amplifier (HEKA, Bellmore, New York, USA). Extracellular recording pipette (4–6 MΩ) solution contained artificial cerebrospinal fluid as above. Recordings were low-pass filtered at 2.8 kHz with an eight-pole Bessel filter and digitized at 10 kHz. A theta burst stimulation protocol was used to induce LTP (Albensi et al. 2007), which was maintained for at least 50 min after induction.
Immunohistochemistry
Brain tissues from two postnatal day 42 WT and Clcn3−/− mice were transcardially perfused and fixed with 4% paraformaldehyde (PFA). Brain tissues were cryoprotected in 30% sucrose solution overnight and further postfixed overnight in 4% PFA at 4°C. The brain was embedded and frozen in cryo-optimal temperature cutting compound (Electron Microscopy Sciences, Hatfield, PA, USA) and cut into serial cryosections (50 μm). For labelling of synaptic proteins, slices were rinsed with PBS and permeabilized with TritonX-100 (0.25%) for 5 min at room temperature. Non-specific binding sites were blocked in PBS blocking solution containing normal goat serum (5%) and bovine serum albumin (1%) for 30 min. Slices were incubated at room temperature for 1 h with the following primary antibodies diluted in blocking solution: rabbit α-CLC-3 (1:100 dilution; Alomone, Jerusalem, Israel) plus mouse monoclonal α-NR1 (1:100 dilution; Abcam, Cambridge, MA, USA) or goat polyclonal α-PSD-95 (1:100 dilution; Abcam). Primary antibody binding was amplified and visualized with Dylight 488-conjugated goat α-rabbit antibody (1:250 dilution; Abcam) for α-CLC3 and Alexa 633-conjugated goat α-mouse or donkey α-goat antibody (1:250 dilution; Abcam) for α-NMDA receptor subunit 1 (α-NR1) and PSD-95, respectively, for 30 min at room temperature. Coverslips were mounted and examined with a Leica SP5 confocal microscope at ×63 magnification with ×4 zoom.
For staining of CLC-3-expressing tsA cells (human embryonic kidney cells transformed with the SV40 Large T-antigen), the stably transfected or mock-transfected cells were washed three times with ice-cold PBS and fixed in 4% paraformaldehyde for 10 min at room temperature. Cells were then permeabilized with 0.2% Tween-20 in PBS for 30 min at room temperature. After 1 h in blocking buffer, cells were incubated with a rabbit α-CLC-3 antibody (Alomone) at 1:100 dilution for 2 h at room temperature. Cells were then washed with PBS and incubated with AlexaFluor 488-conjugated goat α-rabbit IgG (Invitrogen, Grand Island, NY, USA) for 1 h. Mock-transfected cells were used as controls. The coverslips were mounted on a slide and observed using confocal microscopy (Olympus Fluoview).
Cell culture/protein expression
CLC-3 expressing tsA cells and mock-transfected tsA cells were used according to Huang et al. (2001). Stably transfected clonal cell lines were maintained using zeocin (Invitrogen) at 200 μg ml−1.
Preparation of autonomous CaMKII
Ten microlitres of CaMKII (1 mg ml−1; purified from rodent brain; gift from Dr Andrew Hudman at the University of Indiana) were incubated with 2 μl calmodulin (1 mg ml−1) and 8 μl autophosphorylation cocktail (2 mm CaCl2, 25 mm MgCl2, 2 mm ATPγS and 125 mm Tris–HCl, pH 7.4) at room temperature for 10 min. For peptide phosphorylation studies, the mixture was dialysed with 1× CaMKII reaction buffer using 30,000 molecular weight cut-off centrifugal filter apparatus. Buffer exchange was repeated three times to eliminate autophosphorylation cocktail and calmodulin.
Phosphorylation of synthetic peptides by CaMKII
Reaction mixtures were prepared in triplicate and consisted of 1× reaction buffer plus 50 μm peptide, 200 μm Mg-ATP, 125 μCi ml−1[γ-32P]ATP (PerkinElmer, Waltham, Massachusetts, USA) and 2 μl diluted kinase (added last). The total reaction volume was 30 μl. Upon addition of kinase, tubes were incubated for 30 min at 30°C. After incubation, reaction mixtures were spotted onto labelled P81 phosphocellulose squares (Millipore, Billerica, MA, USA) and dropped into ice-cold 75 mm phosphoric acid wash buffer to terminate the reaction and wash away unbound ATP. The P81 squares were washed with wash buffer, dipped briefly in acetone, dried, and then placed in individual scintillation vials. Radioactivity was measured in a scintillation counter (Cherenkov counting).
Immunoprecipitation and Western blot
tsA cells stably transfected with CLC-3 were solubilized in modified RIPA buffer (150 mm NaCl, 10 mm NaPO4, pH 7.4, 1% Triton X-100, 0.1% SDS and 2 mm EDTA) with protease inhibitors (1:200 dilution; Sigma-Aldrich, St. Louis, MO, USA). Fifty microlitres of protein A-conjugated magnetic Dynabeads (Invitrogen) was incubated with 5 μl α-CLC-3 (Alomone) for 10 min at room temperature with rotation and used to immunoprecipitate CLC-3 from the cell lysate at room temperature for 1 h. Following three washes with PBS, CLC-3 was eluted with Western blot sample buffer (200 mm Tris-HCl, pH 6.8, 8% SDS, 40% glycerol, 50 mm EDTA and 100 mm DTT). For phosphorylation experiments, the precipitated CLC-3 was incubated with 50 μl of CaMKII reaction mixture (2 μg CaMKII, 5 mm CaCl2, 3 μg calmodulin and 5 mm Mg-ATP) at room temperature for 30 min before elution. The phosphorylation reaction was terminated, protein eluted, and resolved by 4–20% SDS-PAGE (Bio-Rad, Hercules, CA, USA). The resulting bands on the gel were detected using HRP-conjugated secondary antibody with enhanced chemiluminescence (Thermo Scientific, Waltham, MA, USA).
Neuron cultures
Hippocampal neurons for testing Tat-peptide uptake, a kind gift from Dr Jeremy Marks at the University of Chicago, were prepared from mice on postnatal day 5. Neurons were sparsely plated (1 × 104 neurons per dish) on no. 1.5 thickness coverglass to allow clear visualization of individual processes. Neurons were maintained in Neurobasal-A Medium supplemented by 2% B-27 Serum-Free Supplement (50×) and 0.5 mm GlutaMAX. Cultures were maintained for 1–2 weeks in culture before experiments were performed. All supplies were from Invitrogen.
Data analysis
Unless otherwise indicated, all data in this study are reported as mean values ± SEM with the number of slices in parentheses. Confidence was assessed by Student's paired or unpaired t test between data sets using OriginPro 8.2 (Northampton, MA, USA) as indicated in the figure legends.
Results
CLC-3 and NMDARs colocalize in the PSD of CA1 neurons
Previous studies have demonstrated that CLC-3 localizes to the postsynaptic plasma membrane of excitatory synapses in cultured hippocampal neurons (Wang et al. 2006). Using confocal microscopy on fixed hippocampal slices, we labelled WT and Clcn3−/− slices with an antibody to CLC-3 (Fig. 1A). CLC-3 was expressed in WT slices throughout the CA1 region of the hippocampus in both the cell body layer and the dendrites, while Clcn3−/− sections showed no specific labelling. We next co-labelled the sections for CLC-3 and either the NR1 subunit of the NMDAR or the postsynaptic density protein PSD-95 (Fig. 1B). We found a high degree of overlap of CLC-3 with NR1 and PSD-95 in the stratum radiatum of CA1 (Fig. 1B). Quantitative analysis of the fluorescence signals revealed correlation coefficients of 0.80 and 0.84 for CLC-3 with PSD-95 and NR1, respectively, reflecting the precise spatial correlation of the proteins.
Figure 1. CLC-3 colocalizes with postsynaptic density 95 (PSD-95) and NMDA receptors (NMDARs) at postsynaptic sites.
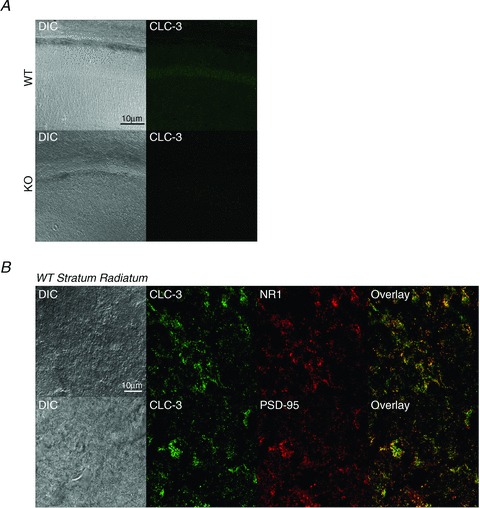
Cryosections were made from the hippocampus of postnatal day 42 wild-type (WT) and Clcn3−/− (KO) mice, stained for desired proteins, and imaged at ×63 magnification with ×4 zoom on a confocal microscope. A, CLC-3 is expressed throughout the CA1 region of the hippocampus in WT but not Clcn3−/− sections. B, CLC-3 immunostaining overlaps extensively with staining for the postsynaptic proteins NR1 and PSD-95 in the WT stratum radiatum. Manders’ coefficients quantifying the degree of overlap are 0.82 for the fraction of NR1 overlapping CLC-3 and 0.70 for CLC-3 overlapping NR1, with a correlation coefficient of 0.84. The fraction of PSD-95 overlapping CLC-3 is 0.67 and CLC-3 overlapping PSD-95 is 0.80, with a correlation coefficient of 0.81. Qualitative analysis of colocalization was done using ImageJ plug-in JACoP (Fabrice et al. 2006).
CLC-3 regulates the magnitude of LTP at Schaffer collateral–CA1 synapses
Long-term potentiation at the Schaffer collateral–CA1 synapse in the hippocampus is dependent on Ca2+ entry through NMDARs (Lynch et al. 1983; Kauer et al. 1988; Malenka et al. 1988; Malenka & Nicoll, 1993). Based on the functional and spatial coupling of CLC-3 with NMDARs, we investigated a role for CLC-3 in modulating LTP in the hippocampus.
Extracellular recording was used to preserve the endogenous Cl− gradient, crucial for interpreting the role of ECl in synaptic plasticity. Recording electrodes were placed in the CA1 stratum radiatum of hippocampal slices from postnatal day 19–30 Clcn3−/− and WT mice. Field excitatory postsynaptic potentials (fEPSPs) were evoked by stimulating the Schaffer collaterals with a bipolar electrode. An input–output curve was measured to assay baseline neurotransmission, and the stimulus intensity was set to evoke field responses that were 30% of the maximal fEPSP amplitude (Fig. 2A). Although Clcn3−/− slices trended towards higher stimulation intensities (Fig. 2A left panel, WT, 74 ± 13 μA, n= 10; and KO, 124 ± 22 μA, n= 8; P= 0.06) and decreased maximal responses (Fig. 2A right panel, WT, 1.86 ± 0.18 mV, n= 10; and KO, 1.58 ± 0.13 mV, n= 8; P > 0.05), possibly a harbinger of impending neurodegeneration, neither parameter was significantly different between WT and Clcn3−/− slices.
Figure 2. Long-term potentiation (LTP) is increased in Clcn3−/− slices.
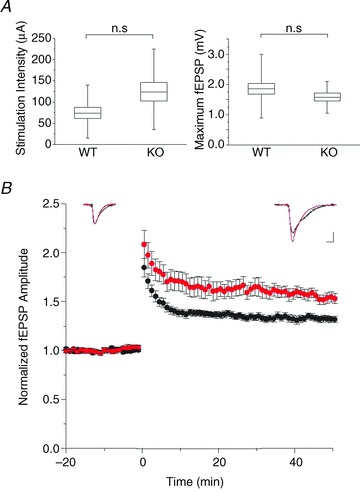
A, stimulation intensity (left panel) for WT (74 ± 13 μA, n= 10) and Clcn3−/− slices (124 ± 22 μA, n= 8, P= 0.06) required to produce a field excitatory postsynaptic potential (fEPSP) that is 30% of the maximal response (right panel; WT, 1.86 ± 0.18 mV, n= 10; and KO, 1.58 ± 0.13 mV, n= 8; P > 0.05). Box plots depict the mean (line across box), ± SEM (top and bottom of box) and range of the data. B, mean normalized fEPSP amplitude in response to theta burst stimulation at t= 0. Both WT (1.35 ± 4, n= 8) and Clcn3−/− slices (1.54±7, n= 10) show significant LTP at 50 min (P < 0.001, Student's paired t test). The Clcn3−/− (red traces and symbols) and WT slices (black traces and symbols) display significantly different magnitudes of LTP (P < 0.001, Student's unpaired t test). Each point is the mean ± SEM. Insets are averages of 10 fEPSPs (normalized to 1.0) for WT and Clcn3−/− slices before and after induction of LTP. Scale bar represents 0.5 and 10 ms.
Long-term potentiation was evoked using a theta burst stimulation protocol (Albensi et al. 2007) following a 20 min baseline period. After at least 50 min of stable LTP expression, we found that Clcn3−/− slices exhibited a 1.54 ± 7 (n= 8) fold-potentiation, significantly greater than the 1.35 ± 4 (n= 10) potentiation expressed by WT slices (P < 0.001; Fig. 2B). This result is consistent with prior studies showing that CLC-3 modulates synaptic events and evoked NMDAR currents in accordance with the Cl− gradient (Wang et al. 2006); removal of the inhibitory Cl− conductance allows for increased Ca2+ influx and, therefore, potentiation.
Paired-pulse facilitation is increased in Clcn3−/− slices
To control for changes in presynaptic function with LTP, we performed paired-pulse experiments before and after LTP. We measured fEPSPs 20 min before LTP induction in response to paired-pulse stimulation of the Schaffer collaterals at a range of interstimulus intervals (ISIs). Representative traces at ISI = 50 ms are shown in Fig. 3A, with data from all ISIs summarized in Fig. 3B. We observed robust paired-pulse facilitation (PPF) of the fEPSP in both WT and Clcn3−/− slices (PPF = amplitude of second/first peak; at 50 ms, WT, 1.60 ± 0.06, n= 14; and KO, 2.16 ± 0.09, n= 8), a well-characterized feature of the CA3–CA1 synapse (Zucker & Regehr, 2002; Ahmed & Siegelbaum, 2009). However, significantly greater PPF was measured in Clcn3−/− slices at all ISIs (Fig. 3B, *P < 0.01 and **P < 0.001, Student's unpaired t test).
Figure 3. Paired-pulse facilitation (PPF) is increased in Clcn3−/− slices.
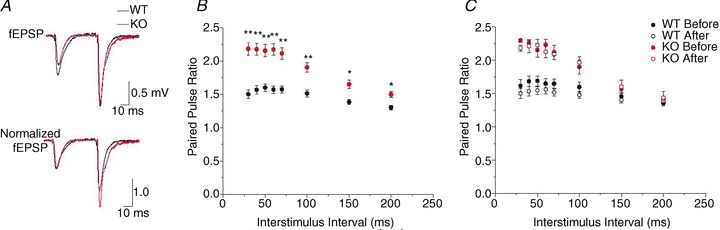
A, field response in CA1 stratum radiatum to paired-pulse stimulation of the Schaffer collaterals. Stimulation intensity was adjusted to 30% of the maximal evoked response. Traces shown are at a 50 ms interstimulus interval (ISI) in WT (black traces) and Clcn3−/− slices (red traces). Top traces, average fEPSPs from six consecutive sweeps. Bottom traces, average fEPSPs normalized to the peak of the first fEPSP. B, paired-pulse ratios for WT and Clcn3−/− slices, calculated as the amplitude of second/first peak (at 50 ms, WT, 1.60 ± 0.06, n= 14; and KO, 2.16 ± 0.09, n= 8). The Clcn3−/− slices display significantly stronger facilitation at all intervals between 30 and 200 ms (*P < 0.01, **P < 0.001, Student's unpaired t test). C, paired-pulse stimulation before and following 50 min of stable LTP expression in WT (n= 7) and Clcn3−/− slices (n= 4). At short ISIs, WT slices displayed a slight decrease in PPF (ISI = 30–100 ms, P < 0.05, Student's paired t test), but no significant difference appeared at longer intervals (ISI = 150–200 ms, P > 0.05, Student's paired t test). The Clcn3−/− slices showed small, inconsistent changes in PPF across ISIs, but these were significant at only one interval (ISI = 60 ms, P < 0.05; all other ISIs, P > 0.05; Student's paired t test), suggesting postsynaptic expression of LTP. Each point is the mean ± SEM.
We repeated the paired-pulse protocol 1 h after induction of LTP to test whether presynaptic responses were altered (Fig. 3C). At short ISIs, WT slices displayed a slight decrease in PPF (n= 7; ISI = 30–100 ms, P < 0.05), but no significant difference appeared at longer intervals (ISI = 150–200 ms, P > 0.05). Minimal changes in PPF are reported as a consequence of postsynaptically expressed LTP at the CA3–CA1 synapse (Kerchner & Nicoll, 2008), consistent with our measurements. The Clcn3−/− slices showed small, inconsistent changes in PPF across ISIs, but these were significant at only one interval (n= 4; ISI = 60 ms, P < 0.05; all other ISIs, P > 0.05). Although loss of presynaptic CLC-3 has a striking effect on presynaptic function, presynaptic release is little changed by LTP induction in WT or Clcn3−/− slices.
CLC-3 is phosphorylated by CaMKII
Three putative CaMKII phosphorylation sites have been identified on CLC-3 (Kawasaki et al. 1994; Robinson et al. 2004). In order to examine phosphorylation of CLC-3 directly, we used a human embryonic kidney cell line derivative that was stably transfected with recombinant CLC-3 (tsA-CLC-3). Control experiments used tsA cells that were mock transfected with only a selection vector (tsA-mock). Figure 4A demonstrates strong expression of CLC-3 in tsA-CLC-3 cells and low basal expression in tsA-mock cells. Using an antibody directed against the intracellular C-terminal domain, we immunoprecipitated CLC-3 from tsA-CLC-3 lysate but not control lysate (Fig. 4B). We incubated the precipitated CLC-3 protein with autophosphorylated CaMKII. After phosphorylating to completion, the protein was eluted using sample buffer, and CLC-3 was resolved using SDS-PAGE. We assayed for phosphorylation using an antiphosphoserine antibody. With application of CaMKII, a distinct phosphorylation signal appeared at the molecular weight of CLC-3 that did not appear in the untreated immunoprecipitation lane or pre-immunoprecipitation lane (Fig. 4C).
Figure 4. CLC-3 is phosphorylated by Ca2+/calmodulin kinase II (CaMKII).
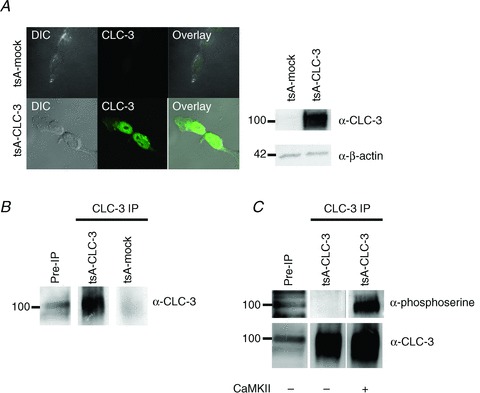
tsA cells were stably transfected with CLC-3 (tsA-CLC-3) or mock transfected with selection vector only (tsA-mock). A, tsA cells were fixed, permeablized, and labelled with an antibody to CLC-3. CLC-3 is expressed strongly in stably transfected tsA cells (bottom panels), while mock-transfected cells (top panels) have low endogenous expression. This is confirmed in a Western blot (right); tsA-mock cells do not show appreciable CLC-3 protein compared with tsA-CLC-3 cells. B, α-CLC-3 was incubated with protein A-conjugated magnetic Dynabeads and used to immunoprecipitate (IP) CLC-3 from tsA cells. We were able to immunoprecipitate CLC-3 from tsA-CLC-3 lysate but not from tsA-mock cells. C, CLC-3 is phosphorylated by CaMKII in vitro. Immunoprecipitated CLC-3 was incubated with activated CaMKII for 30 min, run out with SDS-PAGE, and probed for phosphorylation with an α-phosphoserine antibody. In the absence of kinase, there is negligible phosphorylation of the precipitated protein.
Robinson et al. (2004) showed that mutation of serine 109 to alanine (S109A) prevented the CaMKII-activated Cl− current in transiently transfected tsA cells, suggesting that plasma membrane CLC-3 is gated by CaMKII phosphorylation at S109. Thus, we hypothesized that by preventing CaMKII phosphorylation of CLC-3, we could mimic Clcn3−/− physiology in a WT preparation. Acute interference with CLC-3 function avoids developmental confounds of chronic genetic manipulation and secondary effects of the neurodegeneration associated with CLC-3 knockout.
There is no specific antagonist of CLC-3. We therefore used a strategy wherein we provided CaMKII with an alternative CLC-3-like target. We used the synthetic peptide CLC-3107−116 (INSKKKESAW), representing amino acids 107−116 of the CLC-3 N-terminus and including S109, to titrate CaMKII activity from CLC-3. Synthetic peptides have previously been used to interfere with phosphorylation of endogenous ion channels involved in synaptic transmission (Ahmadian et al. 2004; Lin et al. 2008). The CLC-3107−116 was made cell permeable by fusion to the human immunodeficiency virus type 1 Tat sequence (Tat-CLC-3107−117; YGRKKRRQRRR-INSKKKESAW; Green & Loewenstein, 1988; Schwarze et al. 1999, 2000). Control experiments used Tat-CLC-3scr, in which the Tat sequence remains but the subsequent residues are randomly arranged (Tat-CLC-3scr; YGRKKRRQRRR-KSIESANKWK). Two different scrambled peptides were tested, with similar results. The Tat sequence has been used previously to introduce a peptide that disrupted a specific protein–protein interaction required for long-term depression (Ronesi & Huber, 2008).
We measured the ability of neurons to take up the peptide by conjugating Tat-CLC-3scr to fluorescein. Cultured hippocampal neurons, used for improved visualization, displayed rapid uptake of the peptide, showing strong fluorescence after 15 min (Fig. 5A). We next tested whether the peptides could be phosphorylated by CaMKII in vitro (Fig. 5B). Tat-peptides were combined with 32P-radio-labelled ATP and autophosphorylated CaMKII. Control samples did not contain peptide and were tested with and without CaMKII. All conditions were tested in triplicate. The reaction was stopped after 30 min, and radioactivity was counted using a scintillation counter. When the peptide substrate was omitted, a baseline of <100 counts per minute (c.p.m.) was observed with and without CaMKII (85 ± 13 c.p.m. with CaMKII; 80±4 c.p.m. without CaMKII; P > 0.05, Student's unpaired t test). In contrast, Tat-CLC-3107−116 peptide was phosphorylated 12 times greater than baseline (1077 ± 51 c.p.m.; P < 0.001). Tat-CLC-3scr was also phosphorylated significantly over baseline (350 ± 33 c.p.m.; P < 0.01), but three times less than Tat-CLC-3107−116 (P < 0.001).
Figure 5. Application of the Tat-peptide Tat-CLC-3107−116 to a WT slice mimics increased LTP in adult Clcn3−/− slices.
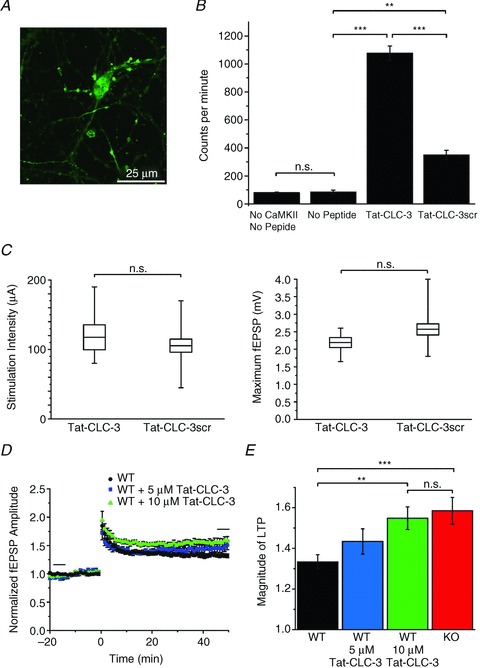
A, uptake of fluorescein-labelled Tat-CLC-3scr into cultured neurons. Cells displayed strong uptake within 15 min. B, in vitro phosphorylation of Tat-peptides by CaMKII using 32P-labelled ATP. When kinase or peptides are not included in the reaction mixture, there is no difference in baseline counts per minute (80 ± 4 c.p.m., n= 3; 85 ± 13 c.p.m., n= 3; P > 0.05, Student's unpaired t test), measured by a scintillation counter. Addition of Tat-CLC-3107−116 results in a 12-fold increase in 32P counts (1077 ± 51 c.p.m., n= 3; ***P < 0.001) compared with the no-peptide condition. Scrambled peptide also increases 32P counts from baseline (350 ± 33 c.p.m., n= 3; **P < 0.01) but significantly less than Tat-CLC-3107−116 (***P < 0.001). All bars are the average of three experiments ± SEM. C, stimulation intensity (left) for WT slices with 10 μm Tat-CLC-3107−116 (118 ± 18 μA, n= 6) and WT slices with 10 μm Tat-CLC-3scr (105 ± 9 μA, n= 13; P > 0.05) required to produce an fEPSP that is 30% of the maximal response (right; Tat-CLC-3107−116, 2.19 ± 0.14 mV, n= 6; and Tat-CLC-3scr, 2.57 ± 0.16 mV, n= 13; P > 0.05). Two different scrambled peptides were tested with the same result, so these data are combined. Box plots depict the mean (line across box), ± SEM (top and bottom of box) and range of the data. Time course (D) and average magnitude of LTP (E) in WT slices with 0, 5 and 10 μm Tat-CLC-3107−116 compared with Clcn3−/− slices (from left to right: 1.33 ± 0.04, n= 10; 1.43 ± 0.06, n= 4; 1.55 ± 0.06, n= 6; and 1.58 ± 0.07, n= 8; **P < 0.01, ***P < 0.001, Student's unpaired t test). At 10 μm, Tat-CLC-3107−116 produces LTP that is not significantly different from that in Clcn3−/− slices (P > 0.05).
Modulation of LTP by CLC-3 requires CaMKII phosphorylation
We next investigated the ability of the CLC-3 peptide to inhibit CLC-3 gating during LTP. We incubated slices with Tat-CLC-3107−116 or scrambled peptide for at least 2 h prior to recording. Baseline synaptic transmission was not different between slices treated with Tat-CLC-3107−116 or scrambled peptide, as indicated by the input–output relationship (Fig. 5C); neither the maximal response (WT + Tat-CLC-3107−116, 2.19 ± 0.14 mV, n= 6; and WT + Tat-CLC-3scr, 2.57 ± 0.16 mV, n= 13; P > 0.05) nor the stimulation intensity required to achieve 30% thereof (WT + 10 μm Tat-CLC-3107−116, 118 ± 18 μA, n= 6; and WT + 10 μm Tat-CLC-3scr, 105 ± 9 μA, n= 13; P > 0.05, Student's unpaired t test) differed between groups. The addition of Tat-peptides did not significantly change baseline synaptic transmission compared with WT slices alone (P > 0.05).
Treatment with Tat-CLC-3107−116 increased the magnitude of LTP expressed in WT slices in a dose-dependent manner (Fig. 5E, from left to right: 1.33 ± 0.04, n= 10; 1.43 ± 0.06, n= 4; 1.55 ± 0.06, n= 6; and 1.58 ± 0.07, n= 8; **P < 0.01, ***P < 0.001; Student's unpaired t test). At 10 μm, Tat-CLC-3107−116 resulted in the same magnitude of LTP as in Clcn3−/− slices (P > 0.05). Application of 10 μm Tat-CLC-3scr to a WT slice resulted in LTP that was not significantly different from WT slices alone (1.35 ± 0.05, n= 6; P > 0.05, Student's unpaired t test), suggesting that S109 and the surrounding residues are critical for recognition of CLC-3 by CaMKII (Fig. 6A). In addition, Tat-CLC-3107−116 did not affect LTP in Clcn3−/− slices (1.54 ± 0.10, n= 5; P > 0.05), providing further evidence of peptide specificity for disrupting primarily the CLC-3–CaMKII interaction (Fig. 6B, and summarized in Fig. 6C). These data show that there is no broad inhibition of CaMKII by the Tat-peptides; if this were the case, we would expect impaired LTP induction (Malenka et al. 1989; Lisman et al. 2002); however, the opposite response is evident in Fig. 5. Finally, equivalent baseline transmission between WT and Tat-peptide-treated slices further emphasizes that the acute effect of CLC-3 on LTP is dissociable from the long-term neurodegeneration phenotype.
Figure 6. Tat-CLC-3107−116 specifically blocks the postsynaptic interaction of CLC-3 and CaMKII.
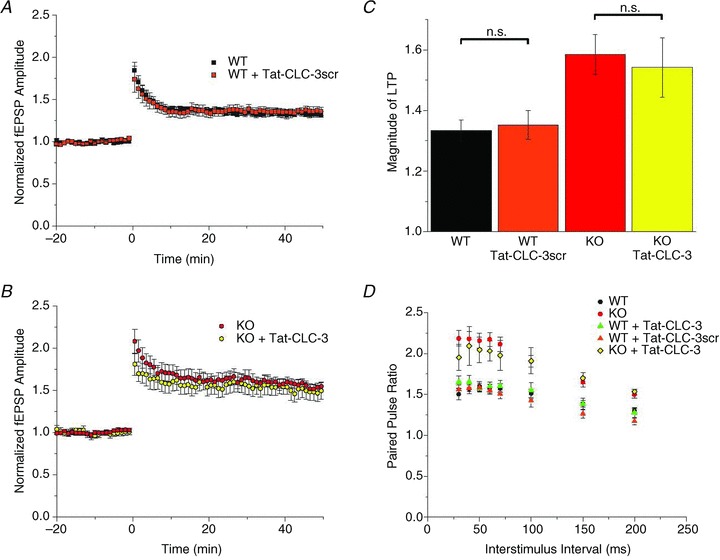
A, application of 10 μm Tat-CLC-3scr to a WT slice results in LTP that is not significantly different from that in WT slices alone (1.35 ± 0.05, n= 6; P > 0.05, Student's unpaired t test). B, 10 μm Tat-CLC-3107−116 does not alter Clcn3−/− LTP expression (1.54 ± 0.10, n= 5; P > 0.05), suggesting that it does not interfere with CaMKII phosphorylation of other substrates important for LTP. C, summary of LTP magnitudes in A and B, measured during the last 10 min of recording. D, presynaptic release is not significantly changed compared with WT slices alone (1.60 ± 0.06, n= 14) for WT slices with application of Tat-CLC-3107−116 (1.57 ± 0.04, n= 6; P > 0.05) or Tat-CLC-3scr (1.58 ± 0.06, n= 6; P > 0.05). In addition, facilitation measured in Clcn3−/− slices treated with Tat-CLC-3107−116 (2.05 ± 0.15, n= 5) is not significantly different from Clcn3−/− slices alone (2.16 ± 0.09, n= 8; P > 0.05), suggesting a purely postsynaptic action of Tat-CLC-3107−116 on LTP. Comparisons (Student's unpaired t test) were carried out for all ISIs, but reported for only ISI = 50 ms for brevity.
Presynaptic release is unaffected by Tat-CLC-3107−116
Both CaMKII and CLC-3 are expressed presynaptically as well as postsynaptically; CaMKII is involved in regulating presynaptic release (Wang, 2008; Pang et al. 2010), while vesicular CLC-3, at least in inhibitory vesicles, facilitates vesicle acidification and loading of neurotransmitter (Stobrawa et al. 2001; Schenck et al. 2009; Riazanski et al. 2011). It is possible that these proteins interact together presynaptically to increase vesicle release probability. We then would expect to see an increase in PPF with Tat-CLC-3107−116 application to WT slices to match the PPF measured in Clcn3−/− slices. We repeated the paired-pulse protocol in all slices treated with Tat-peptides, but found the opposite result; presynaptic release properties followed the genotype of the preparation, unaltered by application of Tat-CLC-3107−116 or Tat-CLC-3scr. The Clcn3−/− slices treated with Tat-CLC-3107−116 (50 ms ISI, 2.05 ± 0.15, n= 5) had increased PPF similar to Clcn3−/− slices without peptide (2.16 ± 0.09, n= 8; P > 0.05, Student's unpaired t test), while WT slices treated with Tat-CLC-3107−116 (1.57 ± 0.04, n= 6) or Tat-CLC-3scr (1.58 ± 0.06, n= 6) displayed WT PPF (1.60 ± 0.06, n= 14; P > 0.05 for both comparisons; Fig. 6D). The lack of peptide influence on presynaptic release suggests that the increase in PPF in Clcn3−/− slices is not due to a presynaptic interaction between CaMKII and CLC-3. Taken together, these data strongly suggest that CLC-3 is gated by CaMKII phosphorylation at the postsynaptic plasma membrane, not in presynaptic vesicles, and that this interaction is required for CLC-3 modulation of postsynaptically expressed LTP.
Discussion
Our data support a model of CLC-3 function presented in Fig. 7. Calcium ion influx through NMDARs activates CaMKII to initiate plasticity mechanisms, such as AMPA receptor insertion, to increase synaptic efficacy. Simultaneously, CaMKII enacts rapid measures to dampen Ca2+ influx by opening the Cl− shunt conductance CLC-3 via S109 phosphorylation. CLC-3 negatively regulates further Ca2+ influx through the NMDAR and moderates the amount of plasticity expressed, thereby protecting active synapses from excitotoxicity.
Figure 7. Model of CLC-3 regulation of LTP.
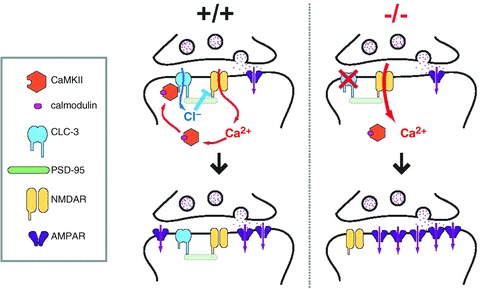
Left side, at a WT synapse in a mature animal, Ca2+ influx through the NMDAR activates CaMKII, which subsequently phosphorylates/gates CLC-3. The Cl− flux through CLC-3 results in shunting of membrane potential, reduces depolarization and promotes Mg2+ block of the NMDAR, thereby exerting negative feedback on the Ca2+ transient at the synapse. This moderates the amount of potentiation that the synapse undergoes. Right side, at a Clcn3−/− synapse, there is no regulation of Ca2+ influx through NMDARs by CLC-3. This leads to addition of excessive AMPA receptors (or changes in AMPA receptor conductance or distribution) and thus greater LTP for an equivalent stimulus.
In this study, we assayed the function of chloride channel CLC-3 at the postsynaptic membrane of excitatory synapses. Immunostaining in hippocampal slices demonstrated strong correlation of CLC-3 staining with postsynaptic markers NR1 and PSD-95, indicating spatial colocalization of the proteins (Fig. 1). Pixel-by-pixel comparison of the images revealed that CLC-3 staining overlapped spatially with the majority of both postsynaptic proteins. It has also been demonstrated that CLC-3 co-immunoprecipitates with NR1 and PSD-95 in isolated hippocampal neurons (Wang et al. 2006) and with CaMKII in human glioma cells (Cuddapah & Sontheimer, 2010). Plasma membrane CLC-3 is integrated into the postsynaptic density, where it cohabits in a microdomain with key structural and functional elements. These data advocate an important role for CLC-3 in determining the efficacy of synaptic transmission.
Prior work demonstrated tight functional coupling between CLC-3 and NMDARs, in which CLC-3 expression shifted the apparent NMDAR reversal potential negatively, from ENMDAR≍ 0 mV to ENMDAR=−20 mV. In addition, Ca2+ influx is required for CLC-3 gating, demonstrated by use of the fast Ca2+ buffer BAPTA in the intracellular solution (Wang et al. 2006). Long-term potentiation and its dependence upon NMDAR-mediated Ca2+ entry (Lynch et al. 1983; Kauer et al. 1988; Malenka et al. 1988) has been widely studied as a presumptive mechanism underlying learning and memory (Bliss & Collingridge, 1993; Reisel et al. 2002; Whitlock et al. 2006; Matsuo et al. 2008); however, despite the central role of Cl− in synaptic inhibition, relatively little attention has been paid to the role of anion channels at excitatory synapses or in synaptic plasticity. Here we extend the repertoire of players in LTP to include CLC-3, unique in its ability to confer the NMDAR with a functional Cl− sensitivity, which we propose is imperative for proper neuronal function and, ultimately, survival. Loss of CLC-3 expression or function (using Tat-CLC-3107−116) at CA3–CA1 synapses results in significantly increased expression of LTP (Figs 2 and 5). Furthermore, CLC-3 is activated directly by CaMKII, the quintessential molecule responsible for induction of plasticity at multiple types of synapses in the brain by directing the distribution or biophysical properties of AMPA receptors (Benke et al. 1998; Derkach et al. 1999; Shi et al. 1999; Bredt & Nicoll, 2003; Lee et al. 2003; Collingridge et al. 2004).
Calcium transients last hundreds of milliseconds, but CaMKII activation extends for several seconds after the transient is complete (Lee et al. 2009). Consequently, CLC-3 gating is likely to extend beyond the NMDAR Ca2+ signal itself in order to influence subsequent synaptic events. A similar Ca2+-mediated feedback loop at the synapse has been described involving small-conductance Ca2+-activated K+ channels (SK channels; Ngo-Anh et al. 2005; Lin et al. 2008; Lee et al. 2011). The SK2 channels are removed from the plasma membrane by protein kinase A-dependent endocytosis following LTP (Ngo-Anh et al. 2005; Lin et al. 2008). Lin et al. (2008) suggest that elimination of the SK2 conductance would remove inhibition of Ca2+ influx after induction of LTP, but this may be compensated through other homeostatic mechanisms. Given that CLC-3 is regulated in an activity-dependent fashion (increased Ca2+ influx is communicated to CLC-3 via CaMKII activity), CLC-3 is a candidate for such a homeostatic role. Removal of SK2 channels was estimated to be responsible for 13% of LTP induced (Lin et al. 2008). We found that loss of CLC-3 expression or gating nearly doubles LTP from 33% above baseline to 58%; therefore, we estimate that the contribution of CLC-3 reduces synaptic potentiation on average by ∼43%. The addition of CLC-3 Cl− inhibition more than compensates for the loss of SK channels, and probably also diminishes the LTP-induced changes in AMPA receptor distribution (as illustrated in Fig. 7) or biophysical properties. Further experiments will be required to identify the exact effects of CLC-3 gating on AMPA receptors.
Long-term potentiation in young animals is dependent on protein kinase A (Yasuda et al. 2003), not CaMKII as extensively documented in adult preparations (Malenka et al. 1989; Lisman et al. 2002). In fact, CaMKII is expressed at somewhat reduced levels until the third postnatal week (Kelly & Vernon, 1985), but this does not appear to lessen CLC-3 function. Wang et al. (2006) measured a significant effect of CLC-3 expression on miniature EPSPs in immature cultured neurons consistent with a depolarizing Cl− gradient at that age. As a result of this developmental switch in kinase dependence of LTP, pinpointing the role of CLC-3 in LTP in immature preparations is complex. However, in combination with previous results, our data predict an opposite effect of CLC-3 in the early postnatal brain in comparison to juvenile and adult brains.
In addition to its postsynaptic function in plasticity, CaMKII is involved in the regulation of presynaptic release (Wang, 2008; Pang et al. 2010); application of a CaMKII inhibitor reduces presynaptic function (Waxham et al. 1993; Sanhueza et al. 2007). Vesicular CLC-3 has also been shown to influence presynaptic function. In inhibitory vesicles particularly, CLC-3 serves as the primary charge shunt pathway to facilitate vesicle acidification and transmitter loading, manifested in the Clcn3−/− as a significant decrease in miniature IPSC amplitude (Riazanski et al. 2011). In excitatory vesicles, CLC-3 appears to play a lesser role in acidification due to an apparent Cl− flux through the glutamate transporter VGLUT (Schenck et al. 2009). Further confounding the role of CLC-3, the original characterization of the Clcn3−/− mouse showed that loading of glutamate is severely compromised in Clcn3−/− vesicles, but this is accompanied by an increase in miniature EPSC amplitudes (Stobrawa et al. 2001). These inconsistent observations could be explained by an increase in postsynaptic responsiveness, resulting from loss of plasma membrane CLC-3, that conceals reduced glutamate loading. The consequences of plasma membrane chloride channel loss are further obscured by the common convention of setting ECl= 0 mV by equalizing [Cl−] in the bath and pipette solutions. Although this convention simplifies recordings, it alters the endogenous Cl− gradient and results in inconsistencies between whole-cell and extracellular recordings which maintain endogenous [Cl−]i.
Generally, the amount of PPF is inversely related to initial vesicle release probability (Dobrunz & Stevens, 1997; Zucker & Regehr, 2002; Sun & Dobrunz, 2006). Considering this, it is possible that CaMKII acts through CLC-3 to promote vesicle release, raising initial release probability and dampening the amount of PPF. If this were the case, we would expect application of the decoy Tat-CLC-3107−116 peptide to increase WT PPF to that of Clcn3−/− levels, but we did not observe this; presynaptic release properties tracked with the genotype of the preparation, unaffected by Tat-peptide application (Fig. 6B). These results may be explained by a secondary structural function of CLC-3, not requiring N-terminal phosphorylation or ion channel activity (Riazanski et al. 2011). Given that only a subset of synaptic vesicles contain CLC-3 (Stobrawa et al. 2001; Riazanski et al. 2011), we consider it improbable that CLC-3 is structurally necessary for adept fusion.
An alternative means of CLC-3 gating from Matsuda et al. (2008a) showed that CLC-3 can be activated by low extracellular pH, corresponding to intravesicular pH. This supplementary gating mechanism would be particularly practical for a protein involved in shunting charge build-up in acidifying compartments. Our results do not directly address the gating mechanism of vesicular CLC-3 beyond the observation that CaMKII interaction with residues 107–116 of the N-terminus is unlikely to be involved. A more plausible explanation is that the decreased release probability is a global side-effect of chronic CLC-3 loss and Ca2+ dysregulation. For example, the increase in postsynaptic efficacy accompanying CLC-3 loss may induce compensatory mechanisms on the presynaptic side in an attempt to equilibrate baseline signalling and avert excitotoxicity.
We propose that Tat-CLC-3107−116 titrates kinase activity from the true CLC-3 N-terminus, as suggested by studies using exogenous peptides in an analogous manner (Ahmadian et al. 2004; Lin et al. 2008), but it may alternatively interfere with the structural changes necessary for gating. We selected residues 107–116 of the N-terminus as the decoy peptide because phosphorylation of S109 has been implicated in CaMKII-dependent gating of the channel; however, the mechanism by which phosphorylation is translated into channel gating is unknown. Future studies into the gating mechanism of CLC-3 will reveal opportunities for more precise control of ion channel activity.
Our data suggest a fundamental difference in pre- and postsynaptic CLC-3 function. In support of this, there are reports of differential localization of CLC-3A and CLC-3B to intracellular compartments and plasma membrane, respectively (Gentzsch et al. 2002). CLC-3B contains an extended C-terminus with a PDZ-binding domain that may be involved in the trafficking of CLC-3 to the plasma membrane and/or the recruitment of CaMKII to CLC-3. In support of this, studies in primary cell culture have corroborated interactions between CLC-3B and PDZ-binding proteins and kinases, including CaMKII, although whether these interactions promote cell surface trafficking remains unclear (Gentzsch et al. 2002; Denton et al. 2005; Cuddapah & Sontheimer, 2010). Similar to CLC-3, Allen et al. (2011) reported functional and spatial partitioning of the short and long isoforms of SK2 channels. Our results contribute a functional segregation between vesicular and membrane CLC-3 that we predict may be a reflection of the underlying difference in isoform structure.
Chloride channels have been associated with neurodegeneration previously. Loss of CLC-7 leads to CA3 neuron degeneration, in addition to lysosomal storage defects and osteopetrosis (Kasper et al. 2005). Neurodegeneration, alterations in tonic GABAergic signalling and upregulation of CLC-2 currents have been recorded in CA1 pyramidal cells in an animal model of temporal lobe epilepsy (Ge et al. 2011). Our data suggest a mechanism whereby CLC-3 channels are required for Ca2+ regulation during induction of plasticity; long-term dysregulation of Ca2+ and excessive expression of LTP eventually leads to excitotoxicity and the degeneration of vulnerable tissues. Therefore, CLC-3 Cl− channel gating during high activity endows the excitatory synapse with a novel form of Cl−-dependent regulation and may provide a protective limit on Ca2+ influx.
Acknowledgments
The authors wish to express their appreciation to Dr Aaron Fox for many helpful conversations during the course of the work and to Dr Fred Lamb at the University of Iowa who provided the Clcn3−/− strain. We thank Dr Radmila Sarac and Erwin Gomez for their careful help with the manuscript and Dr Ivan Goussikov and Erwin Gomez for technical advice. Much gratitude is due to Stephanie Klenotich for her help in making cryosections and to Dr Christine Labno for her imaging expertise. L.M.F. was supported by T32 GM07839-30 and NIH F31 NS076226. The study was supported by NIH R01 GM36823 and NIH R01 DK080364 to D.J.N.
Glossary
- CaM
calmodulin
- CaMKII
Ca2+/calmodulin kinase II
- DIC
differential interference contrast
- fEPSP
field excitatory postsynaptic potential
- ISI
interstimulus interval
- KCC2
K+–Cl− cotransporter
- KO
knockout
- LTP
long-term potentiation
- NMDAR
NMDA receptor
- NR1
NMDA receptor subunit 1
- PFA
paraformaldehyde
- PPF
paired-pulse facilitation
- PSD
postsynaptic density
- SK channel
small-conductance Ca2+-activated K+ channel
- VGLUT1
vesicular glutamate transporter 1
- WT
wild-type
Author contributions
L.M.F. and D.J.N. designed the project. L.M.F. interpreted the results and wrote the manuscript. L.M.F. and B.N.L. collected and analysed the data. All authors approved the final version for publication.
References
- Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, Taghibiglou C, Wang Y, Lu J, Wong TP, Sheng M, Wang YT. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 2004;23:1040–1050. doi: 10.1038/sj.emboj.7600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MS, Siegelbaum SA. Recruitment of N-type Ca2+ channels during LTP enhances low release efficacy of hippocampal CA1 perforant path synapses. Neuron. 2009;63:372–385. doi: 10.1016/j.neuron.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albensi BC, Oliver DR, Toupin J, Odero G. Electrical stimulation protocols for hippocampal synaptic plasticity and neuronal hyper-excitability: are they effective or relevant. Exp Neurol. 2007;204:1–13. doi: 10.1016/j.expneurol.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Allen D, Bond CT, Lujan R, Ballesteros-Merino C, Lin MT, Wang K, Klett N, Watanabe M, Shigemoto R, Stackman RW, Jr, Maylie J, Adelman JP. The SK2-long isoform directs synaptic localization and function of SK2-containing channels. Nat Neurosci. 2011;14:744–749. doi: 10.1038/nn.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C, Groc L, Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated ‘ménage à trois’. Trends Neurosci. 1997;20:523–529. doi: 10.1016/s0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- Benke TA, Luthi A, Isaac JT, Collingridge GL. Modulation of AMPA receptor unitary conductance by synaptic activity. Nature. 1998;393:793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. Journal of Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Cuddapah VA, Sontheimer H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J Biol Chem. 2010;285:11188–11196. doi: 10.1074/jbc.M109.097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton J, Nehrke K, Yin X, Morrison R, Strange K. GCK-3, a newly identified Ste20 kinase, binds to and regulates the activity of a cell cycle-dependent ClC anion channel. J Gen Physiol. 2005;125:113–125. doi: 10.1085/jgp.200409215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson LW, Bonthius DJ, Schutte BC, Yang B, Barna TJ, Bailey MC, Nehrke K, Williamson RA, Lamb FS. Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC-3 voltage-gated chloride channels. Brain Res. 2002;958:227–250. doi: 10.1016/s0006-8993(02)03519-9. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Ge YX, Liu Y, Tang HY, Liu XG, Wang X. ClC-2 contributes to tonic inhibition mediated by α5 subunit-containing GABAA receptor in experimental temporal lobe epilepsy. Neuroscience. 2011;186:120–127. doi: 10.1016/j.neuroscience.2011.04.029. [DOI] [PubMed] [Google Scholar]
- Gentzsch M, Cui L, Mengos A, Chang X, Chen J, Riordan J. The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with CFTR-interacting PDZ proteins. J Biol Chem. 2002;278:6440–6449. doi: 10.1074/jbc.M211050200. [DOI] [PubMed] [Google Scholar]
- Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- Huang P, Liu J, Di A, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ. Regulation of human CLC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 2001;276:20093–20100. doi: 10.1074/jbc.M009376200. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl− channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poet M, Steinfeld R, Schweizer M, Kornak U, Jentsch TJ. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005;24:1079–1091. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. NMDA application potentiates synaptic transmission in the hippocampus. Nature. 1988;334:250–252. doi: 10.1038/334250a0. [DOI] [PubMed] [Google Scholar]
- Kawasaki M, Uchida S, Monkawa T, Miyawaki A, Mikoshiba K, Marumo F, Sasaki S. Cloning and expression of a protein kinase C-regulated chloride channel abundantly expressed in rat brain neuronal cells. Neuron. 1994;12:597–604. doi: 10.1016/0896-6273(94)90215-1. [DOI] [PubMed] [Google Scholar]
- Kelly PT, Vernon P. Changes in the subcellular distribution of calmodulin-kinase II during brain development. Brain Res. 1985;350:211–224. doi: 10.1016/0165-3806(85)90265-2. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Luján R, Watanabe M, Adelman JP, Maylie J. SK2 channel plasticity contributes to LTP at Schaffer collateral–CA1 synapses. Nat Neurosci. 2008;11:170–177. doi: 10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxam MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Matsuda JJ, Filali MS, Collins MM, Volk KA, Lamb FS. The ClC-3 Cl−/H+ antiporter becomes uncoupled at low extracellular pH. J Biol Chem. 2008a;285:2569–2579. doi: 10.1074/jbc.M109.018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JJ, Filali MS, Volk KA, Collins MM, Moreland JG, Lamb FS. Overexpression of CLC-3 in HEK293T cells yields novel currents that are pH dependent. Am J Physiol Cell Physiol. 2008b;294:C251–C262. doi: 10.1152/ajpcell.00338.2007. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Reijmers L, Mayford M. Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science. 2008;319:1104–1107. doi: 10.1126/science.1149967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Ogura T, Furukawa T, Toyozaki T, Yamada K, Zheng Y, Katayama Y, Nakaya H, Inagaki N. ClC-3B, a novel ClC-3 splicing variant that interacts with EBP50 and facilitates expression of CFTR-regulated ORCC. FASEB J. 2002;16:863–865. doi: 10.1096/fj.01-0845fje. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of Stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Cao P, Xu W, Sudhof TC. Calmodulin controls synaptic strength via presynaptic activation of calmodulin kinase II. J Neurosci. 2010;30:4132–4142. doi: 10.1523/JNEUROSCI.3129-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–689. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, Seeburg PH, Rawlins JN. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci. 2002;5:868–873. doi: 10.1038/nn910. [DOI] [PubMed] [Google Scholar]
- Riazanski V, Deriy LV, Shevchenko PD, Le B, Gomez EA, Nelson DJ. Presynaptic CLC-3 determines quantal size of inhibitory transmission in the hippocampus. Nat Neurosci. 2011;14:487–494. doi: 10.1038/nn.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NC, Huang P, Kaetzel MA, Lamb FS, Nelson DJ. Identification of an N-terminal amino acid of the CLC-3 chloride channel critical in phosphorylation-dependent activation of a CaMKII-activated chloride current. J Physiol. 2004;556:353–368. doi: 10.1113/jphysiol.2003.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci. 2008;28:543–547. doi: 10.1523/JNEUROSCI.5019-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, McIntyre CC, Lisman JE. Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor. J Neurosci. 2007;27:5190–5199. doi: 10.1523/JNEUROSCI.5049-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck S, Wojcik SM, Brose N, Takamori S. A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat Neurosci. 2009;12:156–162. doi: 10.1038/nn.2248. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Hruska KA, Dowdy SF. Protein transduction: unrestricted delivery into all cells. Trends Cell Biol. 2000;10:290–295. doi: 10.1016/s0962-8924(00)01771-2. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–196. doi: 10.1016/s0896-6273(01)00189-1. [DOI] [PubMed] [Google Scholar]
- Sun HY, Dobrunz LE. Presynaptic kainate receptor activation is a novel mechanism for target cell-specific short-term facilitation at Schaffer collateral synapses. J Neurosci. 2006;26:10796–10807. doi: 10.1523/JNEUROSCI.2746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Deriy LV, Foss S, Huang P, Lamb FS, Kaetzel MA, Bindokas V, Marks JD, Nelson DJ. CLC-3 channels modulate excitatory synaptic transmission in hippocampal neurons. Neuron. 2006;52:321–333. doi: 10.1016/j.neuron.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Wang ZW. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol Neurobiol. 2008;38:153–166. doi: 10.1007/s12035-008-8039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxham MN, Malenka RC, Kelly PT, Mauk MD. Calcium/calmodulin-dependent protein kinase II regulates hippocampal synaptic transmission. Brain Res. 1993;609:1–8. doi: 10.1016/0006-8993(93)90847-g. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signalling cascades for LTP induction. Nat Neurosci. 2003;6:15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- Yoshikawa M, Uchida S, Ezaki J, Rai T, Hayama A, Kobayashi K, Kida Y, Noda M, Koike M, Uchiyama Y, Marumo F, Kominami E, Sasaki S. CLC-3 deficiency leads to phenotypes similar to human neuronal ceroid lipofuscinosis. Genes Cells. 2002;7:597–605. doi: 10.1046/j.1365-2443.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Li X, Hao J, Winston JH, Weinman SA. The ClC-3 chloride transport protein traffics through the plasma membrane via interaction of an N-terminal dileucine cluster with clathrin. J Biol Chem. 2007;282:29022–29031. doi: 10.1074/jbc.M703506200. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]