

Abstract
The natural target of the botulinum neurototoxin type A (BoNT-A) is the neuromuscular junction. When injected into a muscle, BoNT-A is internalized by motoneurone terminals where it functions as an endopeptidase, cleaving protein components of the synaptic machinery responsible for vesicle docking and exocytosis. As a result, BoNT-A induces a characteristic flaccid paralysis of the affected muscle. In animal models, BoNT-A applied in the periphery can also influence central activity via retrograde transport and transcytosis. An analogous direct central effect in humans is still debated. The present study was designed to address whether BoNT-A modifies the activity of the spinal recurrent inhibitory pathways, when injected at muscular level, in humans. To avoid methodological bias, the recurrent inhibition from an injected muscle (soleus) was investigated on an untreated muscle (quadriceps), and stimulation parameters (producing recurrent inhibition) were monitored on a third non-injected muscle but innervated by the same nerve as the soleus (flexor digitorum brevis). The experiments were performed on 14 post-stroke patients exhibiting spasticity in ankle plantarflexors, candidates for BoNT-A. One month after BoNT-A, the level of recurrent inhibition was depressed. It is suggested that the depression of recurrent inhibition was induced by BoNT-A, injected peripherally, through axonal transport and blockade of the cholinergic synapse between motoneurone recurrent collaterals and Renshaw cells.
Key points
Botulinum neurototoxin type A (BoNT-A) is known to block central synapses after muscular injection due to retrograde transport in animal models.
BoNT-A-induced changes in the human CNS activity have been attributed so far to indirect mechanisms involving peripheral afferent inputs modified after muscular injection.
The question of a possible direct central action of BoNT-A in humans was further addressed by investigating the modification of spinal recurrent inhibition in stroke patients after BoNT-A muscular injection.
Recurrent inhibition from soleus motor axons to motoneurones supplying quadriceps was depressed after BoNT-A injection in ankle plantarflexors.
BoNT-A, through retrograde transport, affects spinal synaptic transmission in humans.
Introduction
Botulinum neurotoxins produced by anaerobic bacteria of the genus Clostridium are the most poisonous proteins known, inducing flaccid paralysis by inhibiting synaptic transmission in cholinergic synapses. Once bound to the nerve cell membrane, botulinum neurotoxins are internalized into the cell and cleave a protein complex, preventing exocytosis and neurotransmitter release (Schiavo et al. 2000; Turton et al. 2002; Montal 2010). The extended action duration of botulinum neurotoxin type A (BoNT-A) at the neuromuscular junction has driven the widespread use of this serotype as a therapeutic agent in various neurological diseases with the aim of weakening the contraction of overactive muscles (Jankovic, 2004). Besides its well-known action at peripheral level, there are at least three possible mechanisms by which BoNT-A may affect central activity: (i) blockade of the gamma motor endings, reducing the spindle afferent inputs from the injected muscle (Filippi et al. 1993; Rosales et al. 1996); (ii) plastic changes following blockade of neuromuscular transmission (Abbruzzese & Berardelli, 2006; Caleo et al. 2009); and (iii) retrograde transport and transcytosis, i.e. release of the ligand in the synaptic cleft with possible uptake by second-order neurones (Antonucci et al. 2008; Torii et al. 2011).
Retrograde axonal transport was first detected with radiolabelled BoNT-A in animal intraspinal motor axons, after injection in gastrocnemius muscles (Habermann, 1974; Wiegand & Wellhoner, 1977; Black & Dolly, 1986). Later, catalytically active BoNT-A was visualized in the facial nucleus after injection of the toxin into rat whisker muscles and attributed to long-distance retrograde axonal transport of BoNT-A inside vesicles (Antonucci et al. 2008; Caleo et al. 2009). Using the visual pathway as a model system, it has been conclusively shown that at least a fraction of the injected BoNT-A undergoes retrograde and anterograde axonal transport in neurones and is then preferentially taken up in a catalytically active form by cholinergic afferent terminals (Antonucci et al. 2008; Restani et al. 2011). It has been speculated that transfer of BoNT-A in this way may reach the CNS and cause central effects in peripherally injected patients, although indications of this have never been observed in humans (Currà & Berardelli, 2009). Instead, alteration by BoNT-A of neurone excitability or synaptic transmission centrally is usually attributed to indirect consequences of its peripheral action (Priori et al. 1995; Girlanda et al. 1997; Modugno et al. 1998). In summary, current understanding is that BoNT-A has no direct effect on central synapses in humans (Curràet al. 2004; Rosales & Dressler, 2010).
Alpha motoneurones innervating the neuromuscular junction have cholinergic intraspinal recurrent collaterals projecting on Renshaw cells mediating recurrent inhibition to spinal motoneurones (Renshaw, 1941; Eccles et al. 1954; Alvarez et al. 1999; Fig. 1). Therefore, Renshaw cells are potential targets for a possible direct effect of BoNT-A on central synapses. Accordingly, animal studies have revealed that BoNT-A depressed recurrent inhibition (Hagenah et al. 1977; Wiegand & Wellhoner 1977) and interfered with the functional link between Renshaw cells and motoneurones (Sanna et al. 1993; Gonzalez-Forero et al. 2005; Clowry et al. 2006). Similarly, recurrent inhibition of soleus motoneurones was depressed after BoNT-A injection in the soleus muscle in patients with lower limb spasticity, but the results were difficult to interpret because low doses of BoNT-A were used to avoid peripheral changes in the soleus, and the effect at the neuromuscular junction could not be monitored (Mazzocchio et al. 2007).
Figure 1. Schematic diagram of the spinal connections.
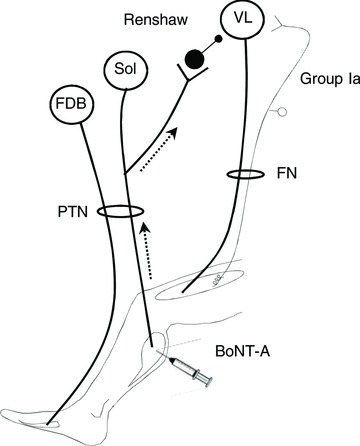
Open circles represent spinal motoneurones innervating FDB, soleus (Sol) and VL. Filled circles represent Renshaw cells activated by recurrent collaterals of soleus motoneurones. FDB and soleus motor axons run into the sciatic nerve and can be activated simultaneously by stimulating the PTN at the level of the plopliteal fossa. The grey line and open circle represent the muscle spindle group Ia afferents from VL, mediating H-reflex; VL sensory and motor axons run into the FN. BoNT-A was injected in soleus and leg muscles. The dotted line indicates the putative retrograde transport of BoNT-A.
The present study was designed to address whether muscular injection of BoNT-A directly influences the activity of central circuits, such as the spinal recurrent inhibitory pathways in humans. Given the widespread projections of motoneurone recurrent collaterals to Renshaw cells impinging on motor nuclei supplying muscles acting at the same joint or transjoint (Meunier et al. 1994), it is possible to test the effect of BoNT-A from one injected muscle (soleus) to motoneurones supplying another muscle, distant from the injection site (quadriceps; Fig. 1). This experimental design makes it possible to test BoNT-A at doses used routinely in post-stroke patients for spasticity of ankle plantarflexors and to monitor central effects of BoNT-A, assessed as a change in the recurrent inhibition at quadriceps level, without methodological bias due to the peripheral effects of BoNT-A at the soleus neuromuscular junction. This protocol implies a retrograde and possibly an anterograde transport of catalytically active BoNT-A in the CNS, both of which have been recently demonstrated in animals (Antonucci et al. 2008; Restani et al. 2011).
Methods
Ethical approval
The experiments were carried out on 14 stroke patients (four females), all of whom had given informed written consent to the experimental procedures, which had been approved by the ethics committee of Pitié-Salpêtrière Hospital (CPP Ile de France VI), and of the University of Siena (Italy). The study conformed to the standards set by the latest revision of the Declaration of Helsinki.
Patients
Inclusion criteria for study enrolment were spastic leg paresis, a marked increase in tone involving the ankle joint, consistent with a score of 4–5 on the 5-point Ashworth Scale, and a required interval since stroke of 6 weeks. Exclusion criteria included BoNT-A injection within the previous 4 months, previous alcohol or phenol blocks, surgical intervention, or casting of the lower limb, fixed contractures in the limb(s), or profound atrophy of the muscle(s) to be injected. The mean subject age was 61.5 ± 2.6 years (range: 49–75), and mean interval since stroke was 15.9 ± 3.2 months (range: 2–42; Table 1). Ongoing treatments (physical therapy and medication) remained unchanged throughout the study.
Table 1.
Clinical characteristics of the patients
| Lesion | Soleus Ashworth score | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients | BoNT | Time | Type | Site | Soleus | MG | LG | TP | PreBoNT | PostBoNT | Inh. |
| 1.M.70 | Y | 10 | Isch. | R | 3 | — | — | — | 4 | 3 | 8.6 |
| 2.M.56 | Y | 42 | Hem. | L | 3 | — | — | — | 5 | 5 | −6.1 |
| 3.M.71 | Y | 21 | Isch. | R | 3 | 1 | — | — | 5 | 4 | −11.0 |
| 4.M.51 | Y | 38 | HI+Hem. | L | 3 | — | — | — | 5 | 4 | −14.4 |
| 5.F.73 | — | 8 | Hem. | L | 3 | — | — | — | 5 | 4 | −26.7 |
| 6.M.61 | — | 15 | Hem. | L | 3 | 1 | 1 | 1 | 4 | 2 | −31.4 |
| 7.F.75 | Y | 21 | Isch. | R | 3 | 1 | — | — | 5 | 3 | −36.3 |
| 8.M.73 | Y | 20 | Isch. | R | 3 | — | 1 | — | 3 | 3 | −49.6 |
| 9.M.49 | — | 5 | Isch. | L | 3 | 1 | 1 | — | 4 | 2 | −58.9 |
| 10.F.51 | Y | 18 | Isch.+Hem. | R | 3 | — | — | 1 | 5 | 4 | −67.7 |
| 11.M.64 | — | 14 | Isch. | L | 3 | 1 | 1 | — | 4 | 3 | −71.0 |
| 12.M.63 | — | 3 | Isch. | R | 3 | 1 | 1 | — | 4 | 2 | −80.2 |
| 13.M.50 | — | 6 | Isch. | R | 3 | 1 | 1 | 1 | 4 | 2 | −96.4 |
| 14.F.54 | — | 2 | Isch. | R | 3 | 1 | 1 | — | 4 | 2 | −100 |
Patients: rank, gender (M, male; F, female), and age at the time of the investigation (years); BoNT indicates if the patients had received BoNT-A at least 3 months before the investigation (Y), or not (—); Lesion: Time = Time lapse between stroke and the first electrophysiological investigation before BoNT-A (months); Type = origin of the lesion (Isch., ischaemia; Hem., haemorrhage; HI, head injury); Site = cerebral hemisphere affected; Number of injection sites in muscles receiving BoNT-A: soleus, MG (medial gasctrocnemius), LG (lateral gasctrocnemius), and TP (tibialis posterior); soleus Ashworth score: estimation of muscle tone in soleus before (PreBoNT) and 1 month after toxin injection (PostBoNT); Inh. = change in inhibition 1 month after BoNT-A (Recurrent inhibition (RI) postBoNT-A – RI preBoNT-A/RI preBoNT-A)×100%.
BoNT-A was injected in spastic lower limb muscles with the exception of quadriceps (Table 1). Each subject was assessed by the same evaluator before and 1 month after BoNT-A injection. The target muscles for BoNT-A were determined according to the clinical features of the patients. The injection site was guided by EMG to localize motor end plates and muscle hyperactivity in soleus, medial head of the gastrocnemius (MG), lateral head of the gastrocnemius (LG) and tibialis posterior (TP; Table 1). Doses were established by the physicians based on their experience, muscle activity and degree of function, and the Worldwide Education and Awareness for Movement Disorders (WEMOVE, http://www.mdvu.org) guidelines.
Recordings
The patients were sitting in a comfortable reclining armchair, with head support. The hip was almost extended (110–135 deg), the knee semi flexed (30 deg) and the ankle in plantar flexion (120 deg). EMG activity was collected using bipolar surface electrodes (ZeroWire EMG, Aurion Srl, Milan, Italy) positioned on the lateral aspect of the thigh (10–15 cm above the knee) over vastus lateralis (VL, lateral head of quadriceps), on the posterior aspect of the leg (7–10 cm above the malleolus) over soleus, and on the plantar aspect of the foot, below the metatarsal eminence, over flexor digitorum brevis (FDB). EMG activity was filtered (10–1000 Hz), amplified (×1000) and digitalized for off-line analysis (Power 1401 and Signal Sofware, CED, Cambridge, UK). The patients were at rest, and EMG silence was monitored.
Electrical stimulations
Rectangular electrical pulses (1 ms duration) were delivered through surface electrodes placed along the nerve trajectory, and delivered by constant current stimulators (DS7A, Digitimer Ltd, Welwyn Garden City, UK). To stimulate the femoral nerve (FN), a 21 cm2 brass plaque was placed on the posterior aspect of the thigh (below the buttock) and a 7 cm2 brass hemisphere, in the femoral triangle. For the posterior tibial nerve (PTN), the plaque was above the patella and the hemisphere in the popliteal fossa. FN and PTN stimulation intensities were adjusted in relation to the muscular responses evoked in VL and FDB, respectively.
Experimental procedure
The protocol for the study of heteronymous recurrent inhibition from soleus to quadriceps motoneurones (Fig. 1) is based on the pattern of distribution of recurrent inhibition in human lower limbs, first described by studying the discharge of single motor units (Meunier et al. 1994). Inhibition with similar characteristics was then revealed by testing the influence of PTN stimulation on quadriceps Hoffmann reflex (H-reflex) amplitude (Iles & Pardoe, 1999; Iles et al. 2000). In these previous studies, PTN stimulation was adjusted according to the responses evoked in soleus EMG. The novelty of introducing FDB measures was necessary for the present study because soleus (and other calf muscles in some patients; Table 1) received BoNT-A, making it difficult to ensure that PTN-evoked EMG responses would be comparable before and after BoNT-A. Therefore, PTN stimulation was adjusted according to the EMG response of FDB, a distant muscle from the injection site (preventing a possible diffusive effect of BoNT-A; Carli et al. 2009), which is innervated by PTN, as soleus. Because the correlation between the EMG responses evoked by PTN stimulation in soleus and FDB was unknown, the protocol was first tested in four healthy subjects. The threshold intensity for an M response in soleus was lower than in FDB (on average, M in soleus was 5.7 ± 2.0% the soleus maximal motor response (Mmax) at the threshold intensity for M response in FDB) and the intensity to evoke Mmax in soleus and FDB was similar (cf. supplementary materials). These results supported the use of FDB EMG measures to monitor PTN stimulation for inhibiting the VL H-reflex as in previous studies (Iles & Pardoe, 1999; Iles et al. 2000).
At the beginning of the experiment, Mmax was evoked in VL, soleus and FDB by FN and PTN stimulation, respectively. FN stimulation intensity was then adjusted so as to evoke a sizeable H-reflex in VL, with or without a preceding M response (<10% Mmax; Fig. 2A–C). PTN stimulation intensity was adjusted so as to evoke an M response of 50% Mmax in FDB (FDB50), and its effect on VL H-reflex was investigated at inter-stimulus intervals (ISIs) between PTN and FN at 15, 20 and 25 ms (Iles & Pardoe 1999; Fig. 2A–D). The effect of varying the intensity of PTN stimulation was then investigated at the ISI at which the inhibition of VL H-reflex was the strongest. The intensity of PTN stimulation was adjusted at the threshold intensity for an M response in FDB (FDB0), or so as to evoke an M response of 25% (FDB25), 50% (FDB50), 75% (FDB75) or 100% Mmax (FDB100; Fig. 2E). For each investigation (changing ISI or PTN stimulation intensity), 20 test (isolated FN stimulation) and 20 conditioned (combined PTN + FN stimulations) VL H-reflexes were randomly alternated (0.3 Hz). The experiment was performed twice in each patient: before BoNT-A injection (preBoNT-A) and 1 month later (postBoNT-A), i.e. when clinical effects are seen in most patients (Kaji et al. 2010).
Figure 2. Characteristics of PTN-induced inhibition of VL H-reflex.
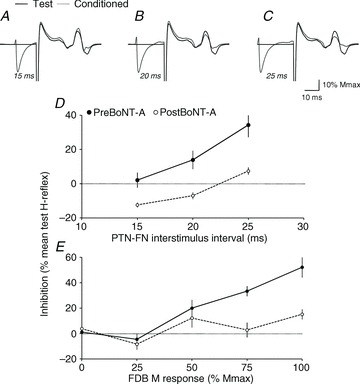
A–C, the dark line represents the VL test H-reflex in one patient after FN stimulation, and the grey line, the conditioned reflex by PTN stimulation evoking FDB50 at ISIs of 15 (A), 20 (B) and 25 ms (C); all reflex responses are normalized to Mmax in VL. D, the difference between test and conditioned reflex (% mean test reflex), illustrating the level of inhibition (horizontal dotted line above 0%) in another patient, is plotted against the ISI (ms) between PTN (evoking FDB50) and FN stimulations. The filled circle and dark line show the result before BoNT-A, and open circles and dashed line, the results after BoNT-A. Vertical bars are ± 1 SEM. E, the level of inhibition (as in D) in a third patient is plotted against the intensity of PTN stimulation (abscissa indicating the size of the M response in FDB expressed as a percentage of Mmax in FDB) when the ISI between PTN and FN stimulations was 20 ms. Filled circle, dark line, open circles, dashed line and vertical bars as in D.
Analysis
Peak-to-peak amplitudes of VL H-reflex and M response in VL, soleus and FDB were measured for quantitative analysis. The mean size of the test H-reflex was expressed as a percentage of Mmax in VL. The level of inhibition was evaluated using the difference (mean test H-reflex – conditioned H-reflexes), expressed as a percentage of the mean test H-reflex (Figs 2D and E and 3). The change in inhibition after BoNT-A was evaluated in each patient using the difference (inhibition postBoNT-A – inhibition preBoNT-A), expressed as a percentage of the level of inhibition preBoNT-A (Inh. in Table 1; Figs 4A and 6).
Figure 4. Loss of PTN-induced inhibition of VL H-reflex after BoNT-A.
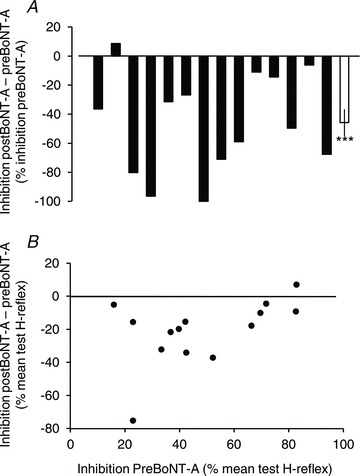
A, the change in inhibition after BoNT-A was estimated using the difference (inhibition postBoNT-A – inhibition preBoNT-A) expressed as a percentage of inhibition preBoNT-A. Each filled column represents one patient, and the last open column, the mean difference (14 patients). Vertical bar is ± 1 SEM. ***P < 0.001. B, relationship between the change in inhibition (inhibition postBoNT-A – inhibition preBoNT-A) and the level of inhibition preBoNT-A (both expressed as a percentage of the mean test H-reflex). Each dot represents one patient.
In each individual, paired t tests were performed to compare the test and conditioned H-reflexes, and to compare the inhibition before and after BoNT-A. Grouped data were analysed using two-way repeated-measures ANOVA to study the effects of PTN stimulation intensity on VL H-reflex before and after BoNT-A. If the tests provided significant P values, post hoc Fisher's least significant difference (LSD) tests were performed for comparison of two means. Maximal inhibition, Mmax in VL, soleus and FDB, PTN and FN stimulation intensity, and H-reflex test size before and after BoNT-A were compared using paired t tests. Pearson correlation analyses were performed to test the relationship between (i) the difference (inhibition postBoNT-A – inhibition preBoNT-A) and the level of inhibition before BoNT-A (both expressed as a percentage of mean test H-reflex; Fig. 4B), and (ii) the change in inhibition after BoNT-A and the date of stroke. Unpaired t tests were performed to compare the change in inhibition (i) in patients who had BoNT-A at least 3 months before the study and those for whom it was the first injection (BoNT in Table 1; Re-injection vs. First injection in Fig. 6A), and (ii) according to the number of injection sites. Because the cohort was too small for a valid statistical analysis, it was divided in two groups: patients with three to four injection sites (3 sites in soleus + 1 site in another calf muscle) vs. patients with five to six injection sites (3 in soleus and 2 or 3 in other calf muscles; Soleus, MG, LG and TP in Table 1; Fig. 6B). As the resulting subgroups of patients were too small to test for normality, non-parametric methods were used to compare the change in inhibition after BoNT-A according to the change in muscle tone (PreBoNT and PostBoNT in Table 1; Fig. 6C). A χ2 test was used to determine if the group of patients with five to six injection sites exhibited a better improvement of muscle tone than the group with three to four injection sites (Fig. 6D). Tests were performed using StatEL software (http://www.adscience.eu), and the significance level was set at P < 0.05. Mean data are indicated ± 1 standard error of the mean (SEM).
Figure 6. Correlation with clinical features.
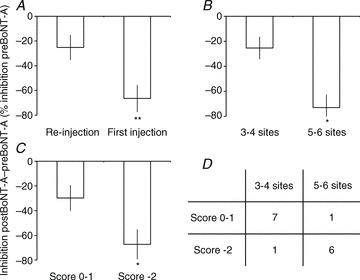
The change in inhibition after BoNT-A [(inhibition postBoNT-A – inhibition preBoNT-A)/(inhibition preBoNT-A)×100%]: A, in patients who had already received BoNT-A at least 3 months before the investigation (Re-injection) and patients who had never been treated (First injection); B, according to the number of injection sites (3 in soleus + 1 in another calf muscle = 3–4 sites vs. 3 in soleus + 2–3 in other calf muscles = 5–6 sites); and C, according to the difference in Ashworth score after BoNT-A patients with 0 or –1 point vs. patients with –2 points. Vertical bars are ± 1 SEM. *P < 0.05, **P < 0.01. D, distribution of the patients in the various subgroups.
Results
Characteristics of the PTN-induced inhibition of VL H-reflex
Figure 2A–C shows the mean test and mean conditioned (by PTN at FDB50) VL H-reflexes at ISIs of 15, 20 and 25 ms in one patient before BoNT-A, and Fig. 2D, the level of inhibition plotted against the same ISIs, in another patient. In both patients, PTN stimulation did not produce any change in VL H-reflex at an ISI of 15 ms, but depressed it significantly at 20 and 25 ms (paired t test, P < 0.05 and 0.0001, respectively). Figure 2E shows in a third patient that PTN stimulation produced inhibition of VL H-reflex at an ISI of 20 ms, before BoNT-A, only when it evoked FDB50 and above; inhibition was significant only at FDB75 and FDB100 (P < 0.001).
In all the patients, PTN stimulation did not produce any change in VL H-reflex at an ISI of 15 ms before BoNT-A. Statistically significant inhibition was observed at an ISI of 20 and/or 25 ms, and it was maximal at 20 ms in five patients, and at 25 ms in nine patients. The inhibition threshold intensity corresponded to PTN stimulation evoking FDB25 in five patients or FDB50 in seven patients; in the two remaining subjects, inhibition was only evoked at FDB100.
Changes in PTN-induced inhibition of VL H-reflex after BoNT-A
One month after BoNT-A in the patient recorded in Fig. 2D, PTN stimulation depressed VL H-reflex only at 25 ms, and to a significantly lesser extent than before BoNT-A (P < 0.0001). Figure 2E shows in the other patient no significant change when PTN stimulation produced FDB0, FDB25 and FDB50, but the inhibition was significantly less at FDB75 and FDB100 after BoNT-A (P < 0.001).
Figure 3A shows the grouped data. ANOVA revealed a significant increase of inhibition with PTN intensity (PTN factor, P < 0.00001), a significant difference between recordings pre- and postBoNT-A (BoNT-A factor, P < 0.00001), and a significant interaction between factors (PTNxBoNT-A, P < 0.00001), indicating that the effect of BoNT-A changed according to PTN stimulation intensity. Indeed, post hoc Fisher LSD tests indicated a significant difference between the results obtained at FDB100 as compared to FDB0, FDB25, FDB50 and FDB75 (0.01 < P < 0.00001), suggesting that the depression of PTN-induced VL H-reflex inhibition after BoNT-A was more marked at FDB100.
Figure 3. Changes in PTN-induced inhibition of VL H-reflex after BoNT-A.
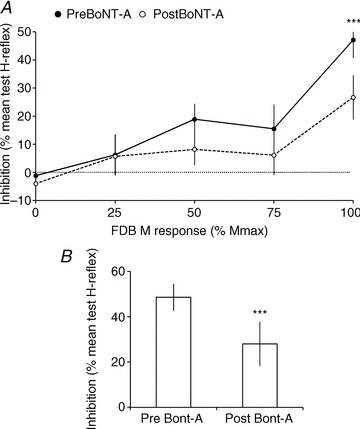
A, grouped data (14 patients) illustrating the level of inhibition (difference between test and conditioned reflex as a percentage of mean test reflex; horizontal dotted line above 0%) observed at the optimal ISI, according to the size of the M response in FDB produced by PTN stimulation (% Mmax in FDB), before (filled circle and dark line) and after BoNT-A (open circles and dashed line). B, mean maximal inhibition before (Pre BoNT-A) and after BoNT-A (Post BoNT-A). Vertical bars are ± 1 SEM. ***P < 0.001.
In 11/14 patients, maximal inhibition was observed at similar interval before and after BoNT-A, but the optimal ISI for inhibition was 25 ms before BoNT-A and 20 ms after BoNT-A in two patients, and the reverse in one patient. Moreover, maximal inhibition was observed at FDB100 in 24/28 experiments, and at FDB75 in four experiments. The maximal inhibition observed in each patient, before and after BoNT-A, was thus tested using a paired t test. Figure 3B shows a significant depression of the maximal inhibition after BoNT-A (48.8 ± 6.0 vs. 28.0 ± 9.8% mean test reflex; P < 0.001).
Figure 4A shows the change in inhibition after BoNT-A in each patient (see also Inh. in Table 1). In all patients but one, inhibition was depressed after BoNT-A. On average, inhibition was depressed by 45.8 ± 9.2% of its level recorded before BoNT-A (Fig. 4A, t test, P < 0.001). Because the inhibition before BoNT-A was different between patients, Pearson correlation analysis was performed to determine whether the level of inhibition before BoNT-A influenced the difference observed after BoNT-A, but no significant relationship was found (P= 0.07; Fig. 4B).
Stimulation parameters before and after BoNT-A
Figure 5A shows the mean Mmax in VL, soleus and FDB observed in the 14 patients before and after BoNT-A. As expected, only Mmax in soleus (injection site) was significantly depressed after BoNT-A (7.6 ± 1.2 vs. 5.1 ± 0.9 mV; paired t test, P < 0.05), Mmax in FDB and VL being comparable between the two recordings (FDB: 5.1 ± 0.7 vs. 5.3 ± 0.8 mV, P= 0.76; VL: 3.4 ± 0.5 vs. 2.6 ± 0.4 mV, P= 0.12). The PTN stimulation intensity at M threshold and Mmax in FDB was also compared before and after BoNT-A and no significant difference was found (P= 0.26 and 0.35 for PTN intensity at FDB0 and FDB100, respectively; Fig. 5B).
Figure 5. Characteristics of stimulation parameters.
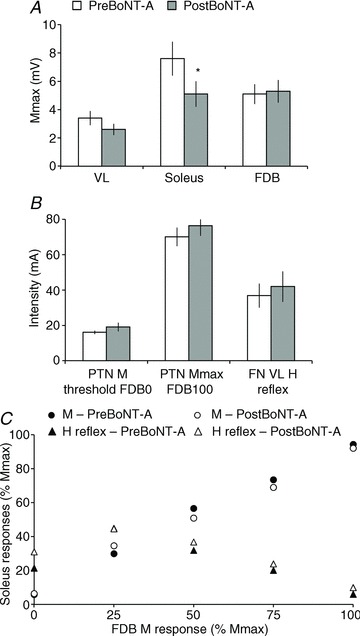
A, mean (14 patients) size (mV) of Mmax in VL, soleus and FDB before (open column) and after BoNT-A (shaded column). Vertical bars are ± 1 SEM. *P < 0.05. B, mean intensity (mA) of PTN stimulation at FDB0 (M threshold) and FDB100 (Mmax), and of FN stimulation evoking the test H-reflex in VL, before (open column) and after BoNT-A (shaded column). Vertical bars are ± 1 SEM. C, soleus M response (circles) and H-reflex (triangles) recruitment curves before (filled symbols) and after BoNT-A (open circles). Intensity of PTN stimulation was adjusted according to the M response in FDB.
FN stimulation intensity was adjusted so as to evoke H-reflex in VL of similar size before and after BoNT-A. Accordingly, no significant difference was found between recordings (21.4 ± 4.7 vs. 27.7 ± 5.0% Mmax in VL, during pre- and postBoNT-A experiments; P= 0.22). Moreover, FN stimulation intensity required to evoke the test H-reflex did not change significantly between recordings (36.4 ± 6.8 vs. 42.0 ± 8.6 mA, P= 0.18; Fig. 5B).
Lastly, the H-reflex and M response evoked in soleus at the different PTN stimulation intensities were compared before and after BoNT-A, and due to the peripheral changes after BoNT-A injection and for interindividual comparison, the amplitudes were normalized to the corresponding Mmax in soleus. Figure 5C shows that the mean soleus M response and H-reflex recruitment curves before and after BoNT-A are superimposed (two-way repeated-measures ANOVA; M response: BoNT-A P= 0.76, PTN intensity P < 0.00001, BoNT-AxPTN intensity P= 0.93; H-reflex: BoNT-A P= 0.31, PTN intensity P < 0.00001, BoNT-AxPTN intensity P= 0.96). On average: (i) the M threshold in soleus was lower than in FDB: at FDB0, M in soleus was 5.8 ± 1.6 vs. 6.4 ± 1.2% Mmax before and after BoNT, respectively; (ii) Mmax in soleus and FDB were evoked at similar PTN intensity; and (iii) at FDB25 and FDB50, i.e. about the threshold for the PTN-induced inhibition of VL H-reflex, the M response in soleus was about 40–60% Mmax.
Correlation with clinical features
The change in inhibition after BoNT-A was compared between patients according to the date of stroke (if the patients had already received BoNT-A before the investigation), the number of injection sites and the level of muscle tone. Other factor analyses (using crossing factors) could not be performed because the resulting subgroups of patients were too small for a valid statistical analysis (only one or two patients in some subgroups).
A significant negative relationship was found between the date of stroke and the loss of inhibition after BoNT-A: the older the lesion, the less the depression of the inhibition after BoNT-A (Pearson correlation analysis, P < 0.05, R2= 0.36). However, most of the patients with the oldest lesions had already received BoNT-A before the investigation (3 months for patients 1.M.70 and 4.M.51, more than 4 months for the other patients; Table 1). The change in inhibition after BoNT-A was thus compared in patients who had already received BoNT-A before the investigation and those who were not treated before (Re-injection vs. First injection in Fig. 6A; BoNT in Table 1). An unpaired t test revealed a loss of inhibition significantly stronger in patients who had not received BoNT-A before the investigation (−25.2 ± 10.2 vs.−66.4 ± 11.0% the level of inhibition preBoNT-A, P < 0.02). Correlation analysis was thus performed in each group, and no relationship was found between the date of stroke and the loss of inhibition (Re-injection group, P= 0.67; First injection group, P= 0.17).
All the patients received BoNT-A in soleus at three sites, and 10/14 patients were injected in other calf muscles (Table 1). Because this could have influenced the electrophysiological results, an unpaired t test was performed to compare the loss of inhibition according to the number of injection sites. Loss of inhibition was significantly stronger in patients who had 5–6 injection sites than in patients who had 3–4 injection sites (−73.0 ± 10.4 vs. −25.4 ± 8.8% the level of inhibition preBoNT-A, P < 0.05; Fig. 6B).
Lastly, we tested whether the change in inhibition after BoNT-A was related to the change in muscle tone in soleus. The cohort was first divided in three groups: patients with no change in Ashworth score (0), patients with –1 point and patients with –2 points 1 month after BoNT-A (difference between PreBoNT and PostBoNT in Table 1). A Kruskal–Wallis H test did not reveal any significant difference between the three groups (0 point: −27.9 ± 21.8 vs.−1 point: −30.4 ± 13.2 vs.−2 points: −67.2 ± 12.1% the level of inhibition preBoNT-A, P= 0.12). We therefore grouped the patients with 0 and −1 points to compare with the patients with −2 points (Fig. 6C). A Mann–Whitney U test revealed a loss of inhibition significantly stronger in the patients with −2 points than in the patients with 0–1 point (−67.2 ± 12.1 vs.−29.7 ± 10.5% the level of inhibition preBoNT-A, P < 0.05). A χ2 test revealed that the improvement of muscle tone was better in the subgroup of patients who had 5–6 injection sites (P < 0.05; Fig. 6D).
Discussion
This study has shown that the PTN-induced inhibition of VL H-reflex in stroke patients at rest was depressed after BoNT-A injection in the triceps surae muscle. The loss of inhibition was more marked in patients who received BoNT-A for the first time and/or through a larger number of injection sites.
Origin of H-reflex inhibition
In cats, heteronymous recurrent inhibition is distributed to motoneurones of synergistic muscles acting at the same joint or even at another joint, and there is a striking overlap between the distribution of recurrent inhibition and monosynaptic Ia excitation (Hultborn et al. 1971; Hultborn, 1989). Although the heteronymous connections are more largely distributed in the human lower limbs, the same rule has been observed. Accordingly, soleus group Ia afferents and Renshaw cells activated by soleus motoneurone recurrent collaterals project onto quadriceps motoneurones (Meunier et al. 1993, 1994; Katz & Pierrot-Deseilligny, 1998; Pierrot-Deseilligny & Burke, 2005).
In the present study, the PTN-induced inhibition of VL H-reflex was evoked at an ISI of >15 ms, and exhibited a threshold intensity above that for motor axons, as measured by the M threshold in FDB and soleus. Inhibition with similar characteristics has been observed in healthy subjects, first by testing the discharge of single motor units in post-stimulus time histograms: inhibition of the motor unit discharge was observed only when motor axons in the nerve stimulated for conditioning were activated (H-reflex response and/or direct M response). As a result, this inhibition was attributed to Renshaw cell activation (Meunier et al. 1994). Later, the inhibition from soleus motor axons to quadriceps motoneurones was further addressed using the H-reflex method, as in the present study, in healthy subjects at rest and during various contractions (Iles & Pardoe, 1999; Iles et al. 2000). The PTN-induced inhibition of quadriceps H-reflex had the same characteristics as those reported in the present study (ISI, threshold intensity); we simply added a monitoring of PTN stimulation parameters by recording FDB EMG activity to allow the comparison pre- vs. post-BoNT-A (cf. supplementary material). Interestingly, the levels of inhibition we found, both in healthy subjects (cf. supplementary material) and in stroke patients, were comparable to that reported in healthy subjects at rest by Iles and collaborators. Because recurrent inhibition has been found to be unchanged in resting stroke patients (Katz & Pierrot-Deseilligny, 1982), this further supports that the PTN-induced inhibition of VL H-reflex investigated here was probably mediated by Renshaw cells. This inhibition could also be due to presynaptic inhibition of group Ia terminals, but this would manifest at a lower stimulus intensity, below M threshold (Faist et al. 1994; Aymard et al. 2000). Moreover, BoNT-A did not affect presynaptic inhibition in the upper limbs of stroke patients (Girlanda et al. 1997) nor did it strengthen it in patients with dystonia or essential tremor (Priori et al. 1995; Modugno et al. 1998). Alternatively, group Ib inhibition can overlap with the beginning of recurrent inhibition, but such inhibition was depressed or even reversed to facilitation in stroke patients (Delwaide & Oliver, 1988).
Because VL H-reflex was similarly inhibited by activating selectively the nervous branches of soleus and medial gastrocnemius (Meunier et al. 1994), and because there are no intraspinal recurrent collaterals in motor axons supplying intrinsic foot muscles (Cullheim & Kellerth, 1978; Rossi & Mazzocchio, 1991), the level of VL H-reflex inhibition probably reflects the level of activation of Renshaw cells impinging on VL motoneurones, activated by motor axons from ankle plantarflexors (Fig. 1).
Influence of experimental conditions
Extrinsic (equipment, EMG sensor location) and intrinsic (tissue characteristics) factors influence the EMG signals (De Luca, 2008), and this hampers the comparison of the raw EMG activity collected in different subjects, and from one experiment to another. Normalization to a physiological constant recorded in similar conditions is thus required (Finsterer, 2001). Mmax, the maximal compound muscle action potential evoked when all motor axons in the nerve are activated, is commonly used in electrophyiological studies to monitor the size of EMG responses (Crone et al. 1990; Lackmy & Marchand-Pauvert, 2010). Although the size of Mmax has to be interpreted carefully, it was significantly decreased only in soleus receiving BoNT-A, but not in distant FDB and quadriceps muscles, which supports a low local diffusion of BoNT-A, restricted to the muscle near the injection site (Carli et al. 2009).
The superimposed soleus M and H-reflex recruitment curves before and after BoNT-A indicate that the H/M ratio did not change after BoNT-A in leg muscles, as observed in forearm muscles (Priori et al. 1995; Girlanda et al. 1997; Modugno et al. 1998), and that BoNT-A did not interfere with the gradual activation of motor and sensory axons, and with the recruitment of spinal motoneurones. In addition, the test size of VL H-reflex was similar between recordings, suggesting that recurrent inhibition was investigated on the same proportion of VL motoneurones (Crone et al. 1990). Lastly, the fact that PTN and FN stimulation intensities were comparable between recordings further validates the comparison of the results observed before and after BoNT-A, and that the depression of recurrent inhibition cannot be attributed to experimental bias.
Possible mechanisms underlying depression of recurrent inhibition after BoNT-A
As mentioned in the Introduction, there are three possible mechanisms by which BoNT-A could affect central activity (Caleo et al. 2009): (1) blockade of the gamma motor endings, (2) plastic changes following blockade of the neuromuscular transmission and (3) retrograde transport and transcytosis.
(1) BoNT-A is known to affect muscle spindle activity by blocking gamma motor endings in animals (Filippi et al. 1993; Rosales et al. 1996). In humans, changes in spinal (Priori et al. 1995; Modugno et al. 1998) and supraspinal excitability (Kim et al. 2006) after BoNT-A muscular injection have been attributed to modifications in muscle spindle afferent input (Curràet al. 2004). Although Renshaw cells receive supraspinal (Mazzocchio et al. 1994) and segmental afferent inputs (Wilson et al. 1964; Ryall & Piercey, 1971; Piercey & Goldfarb, 1974; Fromm et al. 1977; Rossi et al. 2003), it is unlikely that BoNT-A depressed recurrent inhibition through these indirect mechanisms because of the following.
Collaterals of corticospinal neurons make monosynaptic connections with reticulospinal neurons (Canedo & Lamas, 1993) which, in turn, contact Renshaw cells (Peterson, 1984). A lesion interrupting corticospinal but not reticulospinal projections, as in stroke patients, would cause a loss of supraspinal modulation of Renshaw cell activity (Katz & Pierrot-Deseilligny, 1982), but not a reduction in the level of excitability of these cells (Mazzocchio & Rossi, 1997).
Muscle spindle group Ia afferents produce disynaptic excitation in Renshaw cells only via the activation of motor axon collaterals by the reflex loop (Ross et al. 1972). If reduced group Ia afferent input after BoNT-A influenced Renshaw cell activity, recurrent inhibition would be less prominent at low PTN stimulus intensity, when evoking FDB0 or FDB25 (Fig. 3) corresponding to the highest reflex activity in the soleus (Fig. 5C).
Muscle spindle group II afferents have direct inhibitory control on Renshaw cells (Wilson et al. 1964; Fromm et al. 1977). Reduced group II inputs would lead to a disinhibition of Renshaw cells, and thus an increase in recurrent inhibition.
BoNT-A may also block group III–IV nociceptive free endings at muscular level and/or reduce the release of neuromodulators from central terminals of nociceptive afferents (Pavone & Luvisetto, 2010; Matak et al. 2011). Reduced sensitization at peripheral level and possibly at central level could lead to decreased activation of Renshaw cells (Piercey & Goldfarb, 1974; Rossi et al. 2003). However, it is not known whether BoNT-A may be operating on pain terminals in the absence of pain, as in our patients. Regardless, the net effect of conditions (c), i.e. increased recurrent inhibition, and (d), i.e. decreased recurrent inhibition, would be minimal on Renshaw cell activity. It is thus unlikely that a change in muscle spindle afferent activity is responsible for the consistent depression of recurrent inhibition after BoNT-A.
(2) Transitory loss of functional innervation at the neuromuscular junction may trigger rearrangements at the motoneuronal site and at the muscle itself (see Caleo et al. 2009). However, it is unlikely that these contributed to the depression of recurrent inhibition of VL motoneurones after BoNT-A because the toxin was injected in triceps surae muscle and had no distant effect on VL muscle and motoneurones. Moreover, any change at pre- and motoneuronal level would be revealed by a difference in the soleus H-reflex recruitment curve before and after BoNT-A. This was not so. Similarly, any change at muscle level would be offset by the reproducibility of stimulation and recording procedures after BoNT-A.
(3) In animal models, axonal transport and transcytosis of catalytically active BoNT-A from the periphery to the CNS has been demonstrated in the visual system and facial motoneurones (Antonucci et al. 2008; Restani et al. 2011), and central projections of nociceptive sensory neurones (see Pavone & Luvisetto, 2010). Specifically, axonally transported BoNT-A or its catalytically active fragments appeared in the trigeminal nucleus caudalis even at low peripheral anti-nociceptive doses, suggesting that BoNT-A, peripherally applied, can affect second order central sensory neurones, either presynaptically by cleaving its substrate in central terminals of primary afferent neurons or following transcytosis (Matak et al. 2011). Further, in a model of peripheral autonomic neurones, it has been demonstrated that BoNT-A, applied at distal neurites, moves retrogradely into cell bodies and then, anterogradely, passes through to neurites on the other side (Lawrence et al. 2012). Allowing for the much greater distance from injection sites to neuronal cell bodies in human recipients compared to the animal models, a similar migration of BoNT-A may still be replicated within the axons of our patients. Thus, extrapolation from animal model systems makes it possible to suggest that catalytically active BoNT-A may act presynaptically, reducing acetylcholine release in motoneurone recurrent terminals projecting on Renshaw cells impinging on VL motoneurones (Fig. 1). Transcytosis affecting presynaptic terminals of Renshaw cells is a further possible mechanism, as BoNT-A has the potential to interfere with release of several neurotransmitters other than acetylcholine from central neurones (Verderio et al. 2007). This, however, would be more likely to occur in Renshaw cells projecting to homonymous motoneurones (in our case, the soleus motoneurones) than in Renshaw cells projecting to heteronymous motoneurones (the quadriceps motoneurones), given the preferential uptake of BoNT-A by the cholinergic synapse.
Characteristics of BoNT-A-induced depression of recurrent inhibition
The extent of neuromuscular block depends on the dose of BoNT-A (Pascual-Pascual et al. 1997; Dressler & Rothwell, 2000). Assuming a similar mechanism of action of BoNT-A at central level, the higher the dose, the larger the number of central synapses blocked, the stronger the depression of recurrent inhibition. Dose-dependent effects could not be addressed in this study for ethical reasons; nevertheless, the depression of recurrent inhibition after BoNT-A was stronger in patients who had more injection sites.
Depression of recurrent inhibition was observed at FDB50 and reached significant values at FDB100. At FDB100, recurrent inhibition was mostly elicited by antidromic activation of all motor axons, including those of fast-type motoneurones, generating more recurrent axon collaterals and synaptic swellings and therefore a larger activation of the Renshaw cell pool (Windhorst, 1996). Similarly, in animals, depressed recurrent inhibition after BoNT-A muscle injection was observed when the global activity of Renshaw cells was tested (Wiegand & Wellhoner, 1977). It is possible that at intensity below FDB100, Renshaw cells were partly activated via the reflex loop (Ross et al. 1972), i.e. orthodromically, via recurrent collaterals not affected by BoNT-A, given that the engaged motoneurones were able to elicit a sizeable H-reflex in the soleus EMG (see Fig. 5C). This activation may partly offset the effect of BoNT-A on the Renshaw cell pool.
The depression of VL recurrent inhibition was greater in those patients receiving the first dose of BoNT-A than in the re-injected patients. This might be due to BoNT-A-induced plastic changes at soleus motoneurone level (Caleo et al. 2009), and the possible resulting change in the activation of Renshaw cells projecting onto VL motoneurones, and/or the sensitivity of the central synapse to BoNT-A that could have changed after the first injection. While this alternative is debated, the less central efficacy of the toxin in re-injected patients could also be related to an immune response (Atassi, 2004).
Lastly, the depression of recurrent inhibition was stronger in patients who had more injection sites and who exhibited the largest reduction in muscle tone. This might be due to a possible dose effect on recurrent inhibition, as observed for neuromuscular block (Pascual-Pascual et al. 1997; Dressler & Rothwell, 2000), given that no relationship between changes in muscle tone and in recurrent inhibition has been found so far.
Functional significance
Recurrent inhibition is normally depressed to assist the upright posture (Barbeau et al. 2000) and during the stance phase of walking (Lamy et al. 2008), partly via the inhibitory corticospinal control on Renshaw cells (Mazzocchio et al. 1994). The modulation of Renshaw cell activity may contribute to the shaping of muscle synergy through an increase in motoneurone synchronization (Mattei et al. 2003). After stroke, an altered descending drive may be responsible for the lack in task-dependent modulation of Renshaw cell activity (Katz & Pierrot-Deseilligny 1982). The loss of Renshaw cell adaptability to the motor task may make muscle synergies less flexible or abnormal (Mazzocchio & Rossi 1997). The BoNT-A-induced depression of recurrent inhibition could thus compensate for the lack of inhibitory corticospinal control after stroke and could contribute to the tuning of motor synergy (Dipietro et al. 2007) in the process of functional recovery of locomotion and upright posture.
Conclusion
Although the data were obtained with indirect electrophysiological methods, we present experimental evidence in support of a direct central effect of BoNT-A when injected at muscular level for the treatment of spasticity in humans. Specifically, 1 month after peripheral injection, the activity of spinal recurrent inhibitory pathways was depressed. We suggest that such modifications occur through axonal transport and blockade of the cholinergic synapse between motor axon recurrent collaterals and Renshaw cells. The method we have developed can be used to assess BoNT-A dose-dependent effects and to investigate the functional significance of BoNT-A central effects in humans.
Acknowledgments
The authors wish to express their gratitude to all the patients who participated in this study and to Alexandra Lackmy for her valuable technical assistance. L.S.G. was supported by a grant from UPMC Université Paris 6 (Ministère de l’Enseignement Supérieur et de la Recherche). The study was supported by UPMC Université Paris 6, Assistance Publique-Hôpitaux de Paris (AP-HP), Institut pour la Recherche sur la Moelle Epinière (IRME) and INSERM.
Glossary
- BoNT-A
botulinum neurotoxin type A
- FDB
flexor digitorum brevis
- FN
femoral nerve
- H-reflex
Hoffmann reflex
- ISI
interstimulus interval
- LG
lateral gastrocnemius
- MG
medial gastrocnemius
- Mmax
maximal motor response
- PTN
posterior tibial nerve
- TP
tibial posterior
- VL
vastus lateralis
Author contributions
V.M.P., C.A., A.R. & R.M. conceived the conceptual framework and designed the experiments. V.M.P., L.S.G., C.A., F.D. & R.M. performed the experiments, and participated in the data analysis and interpretation. V.M.P. & R.M. wrote the manuscript. All the authors have approved the final version of the manuscript.
Supplementary material
Supplementry Material
References
- Abbruzzese G, Berardelli A. Neurophysiological effects of botulinum toxin type A. Neurotox Res. 2006;9:109–114. doi: 10.1007/BF03033927. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Dewey DE, McMillin P, Fyffe RE. Distri-bution of cholinergic contacts on Renshaw cells in the rat spinal cord: a light microscopic study. J Physiol. 1999;515:787–797. doi: 10.1111/j.1469-7793.1999.787ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonucci F, Rossi C, Gianfranceschi L, Rossetto O, Caleo M. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28:3689–3696. doi: 10.1523/JNEUROSCI.0375-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atassi MZ. Basic immunological aspects of botulinum toxin therapy. Mov Disord. 2004;9:S68–84. doi: 10.1002/mds.20020. [DOI] [PubMed] [Google Scholar]
- Aymard C, Katz R, Lafitte C, Lo E, Pénicaud A, Pradat-Diehl P, Raoul S. Presynaptic inhibition and homosynaptic depression: a comparison between lower and upper limbs in normal human subjects and patients with hemiplegia. Brain. 2000;123:1688–1702. doi: 10.1093/brain/123.8.1688. [DOI] [PubMed] [Google Scholar]
- Barbeau H, Marchand-Pauvert V, Meunier S, Nicolas G, Pierrot-Deseilligny E. Posture-related changes in heteronymous recurrent inhibition from quadriceps to ankle muscles in humans. Exp Brain Res. 2000;130:345–61. doi: 10.1007/s002219900260. [DOI] [PubMed] [Google Scholar]
- Black JD, Dolly JO. Interaction of 125I-labeled botulinum neurotoxins with nerve terminals. II. Autoradiographic evidence for its uptake into motor nerves by acceptor-mediated endocytosis. J Cell Biol. 1986;103:535–544. doi: 10.1083/jcb.103.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleo M, Antonucci F, Restani L, Mazzocchio R. A reappraisal of the central effects of botulinum neurotoxin type A: by what mechanism. J Neurochem. 2009;109:15–24. doi: 10.1111/j.1471-4159.2009.05887.x. [DOI] [PubMed] [Google Scholar]
- Canedo A, Lamas JA. Pyramidal and corticospinal synaptic effects over reticulospinal neurones in the cat. J Physiol. 1993;463:475–489. doi: 10.1113/jphysiol.1993.sp019606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carli L, Montecucco C, Rossetto O. Assay of diffusion of different botulinum neurotoxin type A formulations injected in the mouse leg. Muscle Nerve. 2009;40:374–830. doi: 10.1002/mus.21343. [DOI] [PubMed] [Google Scholar]
- Clowry GJ, Walker L, Davies P. The effects of botulinum neurotoxin A induced muscle paresis during a critical period upon muscle and spinal cord development in the rat. Exp Neurol. 2006;202:456–469. doi: 10.1016/j.expneurol.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Crone C, Hultborn H, Mazières L, Morin C, Nielsen J, Pierrot-Deseilligny E. Sensitivity of monosynaptic test reflexes to facilitation and inhibition as a function of the test reflex size: a study in man and the cat. Exp Brain Res. 1990;81:35–45. doi: 10.1007/BF00230098. [DOI] [PubMed] [Google Scholar]
- Cullheim S, Kellerth JO. A morphological study of the axons and recurrent axon collaterals of cat α-motoneurones supplying different hind-limb muscles. J Physiol. 1978;281:285–299. doi: 10.1113/jphysiol.1978.sp012422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currà A, Berardelli A. Do the unintended actions of botulinum toxin at distant sites have clinical implications. Neurology. 2009;72:1095–1099. doi: 10.1212/01.wnl.0000345010.98495.fc. [DOI] [PubMed] [Google Scholar]
- Currà A, Trompetto C, Abbruzzese G, Berardelli A. Central effects of botulinum toxin type A: evidence and supposition. Mov Disord. 2004;19:S60–S64. doi: 10.1002/mds.20011. [DOI] [PubMed] [Google Scholar]
- De Luca CJ. A practicum on the use of sEMG signals in movement sciences. Boston, MA: Delsys Inc; 2008. http://www.delsys.com/KnowledgeCenter/Practicum.html. [Google Scholar]
- Delwaide PJ, Oliver E. Short-latency autogenic inhibition (IB inhibition) in human spasticity. J Neurol Neurosurg Psychiatry. 1988;51:1546–1550. doi: 10.1136/jnnp.51.12.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipietro L, Krebs HI, Fasoli SE, Volpe BT, Stein J, Bever C, Hogan N. Changing motor synergies in chronic stroke. J Neurophysiol. 2007;98:757–768. doi: 10.1152/jn.01295.2006. [DOI] [PubMed] [Google Scholar]
- Dressler D, Rothwell JC. Electromyographic quantification of the paralysing effect of botulinum toxin in the sternocleidomastoid muscle. Eur Neurol. 2000;43:13–16. doi: 10.1159/000008122. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Fatt P, Koketsu K. Cholinergic and inhibitory synapses in a pathway from motor-axon collaterals to motoneurones. J Physiol. 1954;126:524–562. doi: 10.1113/jphysiol.1954.sp005226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faist M, Mazevet D, Dietz V, Pierrot-Deseilligny E. A quantitative assessment of presynaptic inhibition of Ia afferents in spastics. Differences in hemiplegics and paraplegics. Brain. 1994;117:1449–1455. doi: 10.1093/brain/117.6.1449. [DOI] [PubMed] [Google Scholar]
- Filippi GM, Errico P, Santarelli R, Bagolini B, Manni E. Botulinum A toxin effects on rat jaw muscle spindles. Acta Otolaryngol. 1993;113:400–404. doi: 10.3109/00016489309135834. [DOI] [PubMed] [Google Scholar]
- Finsterer J. EMG-interference pattern analysis. J Electromyogr Kinesiol. 2001;4:231–246. doi: 10.1016/s1050-6411(01)00006-2. [DOI] [PubMed] [Google Scholar]
- Fromm C, Haase J, Wolf E. Depression of the recurrent inhibition of extensor motoneurons by the action of group II afferents. Brain Res. 1977;120:459–468. doi: 10.1016/0006-8993(77)90399-7. [DOI] [PubMed] [Google Scholar]
- Girlanda P, Quartarone A, Sinicropi S, Nicolosi C, Roberto ML, Picciolo G, Macaione V, Battaglia F, Ruggeri M, Messina C. Botulinum toxin in upper limb spasticity: study of reciprocal inhibition between forearm muscles. Neuroreport. 1997;8:3039–3044. doi: 10.1097/00001756-199709290-00008. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Forero D, Pastor AM, Geiman EJ, Benitez-Temino B, Alvarez FJ. Regulation of gephyrin cluster size and inhibitory synaptic currents on Renshaw cells by motor axon excitatory inputs. J Neurosci. 2005;25:417–429. doi: 10.1523/JNEUROSCI.3725-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermann E. 125I-labeled neurotoxin from Clostridium botulinum A: preparation, binding to synaptosomes and ascent to the spinal cord. Naunyn Schmiedebergs Arch Pharmacol. 1974;281:47–56. doi: 10.1007/BF00500611. [DOI] [PubMed] [Google Scholar]
- Hagenah R, Benecke R, Wiegand H. Effects of type A botulinum toxin on the cholinergic transmission at spinal Renshaw cells and on the inhibitory action at Ia inhibitory interneurones. Naunyn Schmiedebergs Arch Pharmacol. 1977;299:267–272. doi: 10.1007/BF00500319. [DOI] [PubMed] [Google Scholar]
- Hultborn H. Overview and critiques of Chapters 22 and 23: Recurrent inhibition – in search of a function. In: Allum JHJ, Hulliger M, editors. Progress in Brain Research. Vol. 80. Amsterdam: Elsevier; 1989. pp. 269–271. [Google Scholar]
- Hultborn H, Jankowska E, Lindström S. Relative contribution from different nerves to recurrent depression of Ia IPSPs in motoneurones. J Physiol. 1971;215:637–664. doi: 10.1113/jphysiol.1971.sp009489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles JF, Pardoe J. Changes in transmission in the pathway of heteronymous spinal recurrent inhibition from soleus to quadriceps motor neurons during movement in man. Brain. 1999;122:1757–1764. doi: 10.1093/brain/122.9.1757. [DOI] [PubMed] [Google Scholar]
- Iles JF, Ali A, Pardoe J. Task-related changes of transmission in the pathway of heteronymous spinal recurrent inhibition from soleus to quadriceps motor neurones in man. Brain. 2000;123:2264–2272. doi: 10.1093/brain/123.11.2264. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75:951–957. doi: 10.1136/jnnp.2003.034702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji R, Osako Y, Suyama K, Maeda T, Uechi Y, Iwasaki M GSK1358820 Spasticity Study Group. Botulinum toxin type A in post-stroke lower limb spasticity: a multicenter, double-blind, placebo-controlled trial. J Neurol. 2010;257:1330–1337. doi: 10.1007/s00415-010-5526-3. Erratum in: J Neurol (2010) 257, 1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz R, Pierrot-Deseilligny E. Recurrent inhibition of α-motoneurons in patients with upper motor neuron lesions. Brain. 1982;105:103–124. doi: 10.1093/brain/105.1.103. [DOI] [PubMed] [Google Scholar]
- Katz R, Pierrot-Deseilligny E. Recurrent inhibition in humans. Prog Neurobiol. 1998;57:325–355. doi: 10.1016/s0301-0082(98)00056-2. [DOI] [PubMed] [Google Scholar]
- Kim DY, Oh BM, Paik NJ. Central effect of botulinum toxin type A in humans. Int J Neurosci. 2006;116:667–680. doi: 10.1080/00207450600674525. [DOI] [PubMed] [Google Scholar]
- Lackmy A, Marchand-Pauvert V. The estimation of short intra-cortical inhibition depends on the proportion of spinal motoneurones activated by corticospinal inputs. Clin Neurophysiol. 2010;121:612–621. doi: 10.1016/j.clinph.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Lamy JC, Iglesias C, Lackmy A, Nielsen JB, Katz R, Marchand-Pauvert V. Modulation of recurrent inhibition from knee extensors to ankle motoneurones during human walking. J Physiol. 2008;586:5931–5946. doi: 10.1113/jphysiol.2008.160630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence GW, Ovsepian SV, Wang J, Aoki KR, Dolly JO. Extravesicular intraneuronal migration of internalized botulinum neurotoxins without detectable inhibition of distal neurotransmission. Biochem J. 2012;441:443–452. doi: 10.1042/BJ20111117. [DOI] [PubMed] [Google Scholar]
- Matak I, Bach-Rojecky L, Filipovic B, Lackovic Z. Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience. 2011;186:201–207. doi: 10.1016/j.neuroscience.2011.04.026. [DOI] [PubMed] [Google Scholar]
- Mattei B, Schmied A, Mazzocchio R, Decchi B, Rossi A, Vedel JP. Pharmacologically induced enhancement of recurrent inhibition in humans: effects on motoneurone discharge patterns. J Physiol. 2003;548:615–29. doi: 10.1113/jphysiol.2002.033126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzocchio R, Rossi A. Involvement of spinal recurrent inhibition in spasticity. Further insight into the regulation of Renshaw cell activity. Brain. 1997;120:991–1003. doi: 10.1093/brain/120.6.991. [DOI] [PubMed] [Google Scholar]
- Mazzocchio R, Rossi A, Rothwell JC. Depression of Renshaw recurrent inhibition by activation of corticospinal fibres in human upper and lower limb. J Physiol. 1994;481:487–498. doi: 10.1113/jphysiol.1994.sp020457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzocchio R, Spidalieri R, Dominici F, Popa T, Hallett M, Rossi A. Putative central effects of botulinum toxin, possibly mediated by changes in Renshaw cell activity, following intramuscular injection in humans. Movement Disorders. 2007;22:S122. [Google Scholar]
- Meunier S, Pierrot-Deseilligny E, Simonetta M. Pattern of monosynaptic heteronymous Ia connections in the human lower limb. Exp Brain Res. 1993;96:533–544. doi: 10.1007/BF00234121. [DOI] [PubMed] [Google Scholar]
- Meunier S, Pierrot-Deseilligny E, Simonetta-Moreau M. Pattern of heteronymous recurrent inhibition in the human lower limb. Exp Brain Res. 1994;102:149–159. doi: 10.1007/BF00232447. [DOI] [PubMed] [Google Scholar]
- Modugno N, Priori A, Berardelli A, Vacca L, Mercuri B, Manfredi M. Botulinum toxin restores presynaptic inhibition of group Ia afferents in patients with essential tremor. Muscle Nerve. 1998;21:1701–1705. doi: 10.1002/(sici)1097-4598(199812)21:12<1701::aid-mus12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Montal M. Botulinum neurotoxin: a marvel of protein design. Annu Rev Biochem. 2010;79:591–617. doi: 10.1146/annurev.biochem.051908.125345. [DOI] [PubMed] [Google Scholar]
- Pascual-Pascual SI, Sánchez de Muniain P, Roche MC, Pascual-Castroviejo I. Botulinum toxin as a treatment for infantile cerebral palsy. Rev Neurol. 1997;25:1369–1375. [PubMed] [Google Scholar]
- Pavone F, Luvisetto S. Botulinum neurotoxin for pain management: insights from animal models. Toxins. 2010;2:2890–2913. doi: 10.3390/toxins2122890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BW. The reticulospinal system and its role in the control of movement. In: Barnes CD, editor. Brain Stem Control of Spinal Cord Function. Orlando: Academic Press; 1984. pp. 141–214. [Google Scholar]
- Piercey MF, Goldfarb J. Discharge patterns of Renshaw cells evoked by volleys in ipsilateral cutaneous and high-threshold muscle afferents and their relationship to reflexes recorded in ventral roots. J Neurophysiol. 1974;37:294–302. doi: 10.1152/jn.1974.37.2.294. [DOI] [PubMed] [Google Scholar]
- Pierrot-Deseilligny E, Burke D. The Circuitry of the Human Spinal Cord. New York: Cambridge University Press; 2005. Recurrent inhibition; pp. 151–196. [Google Scholar]
- Priori A, Berardelli A, Mercuri B, Manfredi M. Physiological effects produced by botulinum toxin treatment of upper limb dystonia. Changes in reciprocal inhibition between forearm muscles. Brain. 1995;118:801–807. doi: 10.1093/brain/118.3.801. [DOI] [PubMed] [Google Scholar]
- Renshaw B. Influence of discharge of motoneurons upon excitation of neighboring motoneurons. J Neurophysiol. 1941;4:67–183. [Google Scholar]
- Restani L, Antonucci F, Gianfranceschi L, Rossi C, Rossetto O, Caleo M. Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A) J Neurosci. 2011;31:15,650–15,659. doi: 10.1523/JNEUROSCI.2618-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales RL, Dressler D. On muscle spindles, dystonia and botulinum toxin. Eur J Neurol. 2010;17:71–80. doi: 10.1111/j.1468-1331.2010.03056.x. [DOI] [PubMed] [Google Scholar]
- Rosales RL, Arimura K, Takenaga S, Osame M. Extrafusal and intrafusal muscle effects in experimental botulinum toxin-A injection. Muscle Nerve. 1996;19:488–496. doi: 10.1002/(SICI)1097-4598(199604)19:4<488::AID-MUS9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Ross HG, Cleveland S, Haase J. Quantitative relation of Renshaw cell discharges to monosynaptic reflex height. Pflugers Arch. 1972;332:73–79. doi: 10.1007/BF00603815. [DOI] [PubMed] [Google Scholar]
- Rossi A, Mazzocchio R. Presence of homonymous recurrent inhibition in motoneurones supplying different lower limb muscles in humans. Exp Brain Res. 1991;84:367–373. doi: 10.1007/BF00231458. [DOI] [PubMed] [Google Scholar]
- Rossi A, Mazzocchio R, Decchi B. Effect of chemically activated fine muscle afferents on spinal recurrent inhibition in humans. Clin Neurophysiol. 2003;114:279–287. doi: 10.1016/s1388-2457(02)00334-6. [DOI] [PubMed] [Google Scholar]
- Ryall RW, Piercey MF. Excitation and inhibition of Renshaw cells by impulses in peripheral afferent nerve fibers. J Neurophysiol. 1971;34:242–51. doi: 10.1152/jn.1971.34.2.242. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Celio MR, Bloom FE, Rende M. Presumptive Renshaw cells contain decreased calbindin during recovery from sciatic nerve lesions. Proc Natl Acad Sci U S A. 1993;90:3048–3052. doi: 10.1073/pnas.90.7.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- Torii Y, Akaike N, Harakawa T, Kato K, Sugimoto N, Goto Y, Nakahira S, Kohda T, Kozaki S, Kaji R, Ginnaga A. Type A1 but not type A2 botulinum toxin decreases the grip strength of the contralateral foreleg through axonal transport from the toxin-treated foreleg of rats. J Pharmacol Sci. 2011;117:275–285. doi: 10.1254/jphs.11121fp. [DOI] [PubMed] [Google Scholar]
- Turton K, Chaddock JA, Acharya KR. Botulinum and tetanus neurotoxins: structure, function and therapeutic utility. Trends Biochem Sci. 2002;27:552–558. doi: 10.1016/s0968-0004(02)02177-1. [DOI] [PubMed] [Google Scholar]
- Verderio C, Grumelli C, Raiteri L, Coco S, Paluzzi S, Caccin P, Rossetto O, Bonanno G, Montecucco C, Matteoli M. Traffic of botulinum toxins A and E in excitatory and inhibitory neurons. Traffic. 2007;8:142–153. doi: 10.1111/j.1600-0854.2006.00520.x. [DOI] [PubMed] [Google Scholar]
- Wiegand H, Wellhoner HH. The action of botulinum A neurotoxin on the inhibition by antidromic stimulation of the lumbar monosynaptic reflex. Naunyn Schmiedebergs Arch Pharmacol. 1977;298:235–238. doi: 10.1007/BF00500893. [DOI] [PubMed] [Google Scholar]
- Wilson VJ, Talbot WH, Kato M. Inhibitory convergence upon Renshaw cells. J Neurophysiol. 1964;27:1063–1079. doi: 10.1152/jn.1964.27.6.1063. [DOI] [PubMed] [Google Scholar]
- Windhorst U. On the role of recurrent inhibitory feedback in motor control. Prog Neurobiol. 1996;49:517–587. doi: 10.1016/0301-0082(96)00023-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.