

Abstract
Research on all stages of fibrin polymerization, using a variety of approaches including naturally occurring and recombinant variants of fibrinogen, x-ray crystallography, electron and light microscopy, and other biophysical approaches, has revealed aspects of the molecular mechanisms involved. The ordered sequence of fibrinopeptide release is essential for the knob-hole interactions that initiate oligomer formation and the subsequent formation of 2-stranded protofibrils. Calcium ions bound both strongly and weakly to fibrin(ogen) have been localized, and some aspects of their roles are beginning to be discovered. Much less is known about the mechanisms of the lateral aggregation of protofibrils and the subsequent branching to yield a 3-dimensional network, although the αC region and B:b knob-hole binding seem to enhance lateral aggregation. Much information now exists about variations in clot structure and properties because of genetic and acquired molecular variants, environmental factors, effects of various intravascular and extravascular cells, hydrodynamic flow, and some functional consequences. The mechanical and chemical stability of clots and thrombi are affected by both the structure of the fibrin network and cross-linking by plasma transglutaminase. There are important clinical consequences to all of these new findings that are relevant for the pathogenesis of diseases, prophylaxis, diagnosis, and treatment.
Introduction
Fibrin polymer is an end product of the enzymatic cascade of blood clotting. In vivo formation of the polymeric fibrin network, along with platelet adhesion and aggregation, are the key events in salutary stopping of bleeding at the site of injury (hemostasis) as well as in pathological vascular occlusion (thrombosis). Fibrin polymerization comprises a number of consecutive reactions, each affecting the ultimate structure and properties of the fibrin scaffold. These properties determine the development and outcomes of various diseases, such as heart attack, ischemic stroke, cancers, trauma, surgical and obstetrical complications, hereditary and acquired coagulopathies, and thrombocytopathies. In addition to providing a better understanding of the pathogenesis of such disorders, the knowledge of the molecular mechanisms of fibrin formation provides a foundation for new diagnostic tools and therapeutic approaches.
Fibrinopeptide release
Fibrinogen is 45 nm-long and made up of 6 paired polypeptide chains (Aα Bβ γ)2 held together by 29 disulfide bonds. There are now crystal structures of large parts of fibrinogen, including the γ- and β-nodules, the coiled coil, and the central nodule.1 Still missing in the crystal structures are the flexible NH2-terminal (N-terminal) ends of the Aα-chains and the carboxyl-terminal (C-terminal) portion of the Aα-chain, but modern simulation techniques make possible partial computational reconstructions of these parts of fibrin(ogen) (Figure 1).
Figure 1.
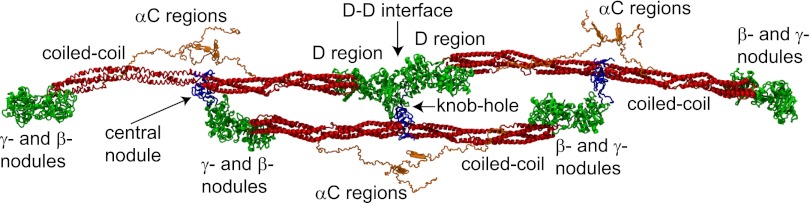
Schematic representation of a short oligomer formed by 3 fibrin monomers based on the x-ray crystallographic structure of fibrinogen (Protein Data Bank entry: 3GHG). Shown for each monomer are the central nodule (blue), coiled-coil connectors (red), the γ- and β-nodules (green), which make up the main part of the lateral D region, and the αC regions (orange). The molecules are shown with the addition of the missing parts of the crystal structure reconstructed from molecular dynamics simulations, namely the amino terminal ends of the α-chains in the central nodule and the αC regions. A:a knob-hole bonds that are the major basis of fibrin polymerization maintain the third (lower) monomer in a half-staggered arrangement. The intermolecular noncovalent coupling and the covalent cross-linking at the D-D interface hold the two (upper) monomers in a linear arrangement. Created by and published with permission from A. Zhmurov, O. Kononova, and V. Barsegov, University of Massachusetts–Lowell.
Fibrin polymerization is initiated by the thrombin cleavage of fibrinopeptides A (FpA) and B (FpB) from the N-termini of the Aα- and Bβ-chains of fibrinogen to produce fibrin monomer (α β γ)2. FpA is cleaved off more rapidly than FpB, but as polymerization proceeds, the rate of release of FpB increases, suggesting that it is preferentially released from polymers. Unlike in solution, in surface-attached fibrinogen, FpB can be cleaved at a faster rate than FpA,2 implying that the accessibility of fibrinopeptides for thrombin is dictated by fibrinogen’s conformation. Naturally occurring dysfibrinogenemias or recombinant fibrinogen variants with substitutions in AαArg16 or BβArg14 have impaired the cleavage of FpA or FpB, respectively, and defective fibrin polymerization.3,4
Knob-hole interactions
The release of FpAs exposes an N-terminal α-chain motif Gly-Pro-Arg (GPR), called knob ‘A,’1 which is complementary to pockets or holes ‘a’ located in the γ-nodules of another fibrin molecule, yielding A:a interactions (Figure 1 and Figure 2). The interactions between knobs ‘A’ and holes ‘a’ have been studied at the single-molecule level, and the A:a bonds have been found to be quite strong and stable.5
Figure 2.

Schematic representation of the consecutive steps of fibrin polymerization. The figure shows the following steps: (1) the enzymatic release of fibrinopeptides from fibrinogen, the formation of monomeric fibrin-containing exposed knobs, and the partial dissociation of the αC regions; (2) the self-assembly of monomeric fibrin via knob-hole interactions and the formation of half-staggered 2-stranded fibrin oligomers; (3) the lateral aggregation of protofibrils (fibrin oligomers made of 20 to 25 monomers) promoted by homophilic αC-αC-interactions within and between protofibrils, including the formation of αC-polymers; (4) the packing of protofibrils into a fiber with a 22.5-nm periodic cross-striation due to the half-staggered molecular structure and regular paracrystalline arrangement; and (5) the fibrin network formation due to the branching of fibers by either of 2 mechanisms.
Structures of the lateral D regions with the peptide GPRP representing the N-terminal sequence of fibrin’s α-chain (knob ‘A’) have shown this peptide in binding pockets in the γ-nodule,6,7 evidence that these are holes ‘a,’ but it is not yet known if the peptide represents the entire knob ‘A’ or only part of it. Cleavage of FpA and exposure of knobs ‘A’ are necessary and sufficient to form fibrin clots called desA-fibrin. Constitutively exposed fibrin(ogen) holes ‘a’ are also necessary for clot formation. If they are blocked by the GPRP peptide6 or impaired by a point mutation of the key residue γAsp364,8 fibrin polymerization is prevented. Together, these data suggest that A:a interactions are the driving force of fibrin polymerization.
The release of FpBs exposes the new N-terminal β-chain motif Gly-His-Arg-Pro (GHRP), called knob ‘B,’ which is complementary to hole ‘b’ located in the globular β-nodule. The affinity of the knob ‘B’-mimetic GHRP peptide for fibrinogen (Kd = 140 μM) is relatively low compared with the knob ‘A’-mimetic GPRP (Kd = 25 μM).6 The desA-NDSK fibrin fragment corresponding to the central nodule with exposed knobs ‘A’ binds to surface-attached fibrinogen (bearing holes ‘a’) with Kd = 5.8 ± 1.1 μM.9 This suggests that in fibrin, the binding site corresponding to knob ‘A’ is not limited to the GPR sequence and therefore has substantially higher affinity.
Despite evidence for the physical existence of B:b interactions,10 their physiological role remains unclear and is based on indirect evidence. Cleavage of FpA without cleavage of FpB produces clots made up of thinner fibers than those initiated by cleavage of both fibrinopeptides. This suggests that B:b interactions are involved in the lateral aggregation of protofibrils, although lateral aggregation and fiber formation occurs without the cleavage of FpB. Evidence that B:b interactions can actually occur in fibrin comes from dysfibrinogenemias (Metz and Frankfurt XIII) in which all fibrinogen molecules are homodimers from which FpB, but not FpA, can be cleaved, yielding clots with thin fibers at low temperature.11 Furthermore, recombinant mutants of the γ364 residue that have no functional holes ‘a’ and cannot build A:a bonds form clots slowly with thrombin but not with reptilase.12 On the other hand, fibrinogen variant BβD432A with impaired hole ‘b’ displays normal polymerization.13 Therefore, it appears that B:b bonds can occur at least when A:a interactions are compromised, but their normal functional role is still mostly unknown. It is possible that B:b binding has an effect on the susceptibility of a clot to enzymatic destruction.14 In addition to the established A:a and B:b knob-hole complexes, there is some evidence for the physical existence of A:b interactions, as well as suggestions that the B:a interactions do not occur.5,10
Oligomer and protofibril formation
Two fibrin monomer molecules produced by the release of FpA interact with each other in a half-staggered manner, so that knob ‘A’ fits into hole ‘a,’ with 2 A:a knob-hole interactions holding the 2 monomers together. The addition of a third molecule to a half-staggered dimer produces an end-to-end junction where the ends of 2 molecules abut each other (Figure 1 and Figure 2). This molecular packing leads to a regular 22.5-nm repeat corresponding to one-half the length of the fibrin monomer, visualized by transmission electron microscopy and by atomic force microscopy as cross-striations in fibrin fibers.15,16 In addition to knob-hole interactions, the D-D interface comprising γ275-300 residues is an important feature of most of the known crystal forms6,17 and is likely to be similar to that at the end-to-end junction between monomers in fibrin. The D-D interactions are weak and yield first upon forced stretching of fibrin(ogen) oligomers.18 Studies on natural and recombinant fibrinogen variants revealed that the residues γ275, γ308, and γ309 are essential for D:D interactions and the elongation of fibrin strands.19–21
Additional fibrin monomers can add longitudinally to the dimer and trimer to form larger oligomers, which lengthen further to make protofibrils, a critically important intermediate product of fibrin polymerization, which is capable of lateral aggregation, leading to the formation of fibers (Figure 2). Protofibrils are defined as oligomers that are long enough to aggregate laterally, probably about 0.5 to 0.6 μm,22 which corresponds to 2-stranded soluble oligomeric structures made up of 20 to 25 half-staggered fibrin monomers. Protofibrils have been visualized using transmission electron microscopy22,23 and atomic force microscopy.24 Protofibril growth is partially impaired by the presence of the fibrinogen γ-chain splice variant named γ′, perhaps due to charge repulsion.24–26
Lateral aggregation of protofibrils
The precise mechanisms, particular structures, and driving forces supporting the lateral aggregation of protofibrils remain largely unknown. Protofibrils associate with each other laterally to make thicker or thinner fibers only when they reach a threshold length, which suggests that the interactions responsible for lateral aggregation are relatively weak and additive over the length of the protofibril. So far the following structures have been shown or hypothesized to contribute to interprotofibril lateral binding: knobs ‘B’ and holes ‘b,’ the C-terminal portions of the γ-chains and 2 adjacent β-nodules,27 αC regions (see “Role of the αC regions”), the coiled coils,28 and N-glycosaminoglycans at residues Bβ364Asn and γ52Asn.29
Since structural proteins that self-assemble tend to display similar binding interactions in vitro and in vivo,17 a plausible model for some aspects of lateral aggregation has been derived from analysis of the interactions between D regions in crystals, in which they interact, such that residues γ350-360 and γ370-380 are shielded from water.27 Yang et al27 proposed a mechanism of lateral aggregation via interactions between 2 β-nodules of adjoining protofibrils, specifically involving residues β330-375. There is also evidence for a particular importance in the lateral aggregation of protofibrils of the residues located in the N-terminal part of the Bβ-chain, namely, Bβ68Ala30 and Bβ15Gly, the end residue of knob ‘B,’ irrespective of whether FpB is cleaved.31
The packing in fibrin is paracrystalline, such that fibrin molecules are regularly arranged in the longitudinal direction but are less well ordered across the fibers. In addition, twisted protofibrils associate with each other in a specific manner that leads to twisted fibers.23 Because of the maintenance of the 22.5-nm repeat, as new protofibrils are added to the outside of a fiber, they must be stretched as their path length increases. This may provide a mechanism to limit the thickness of the fibers, as they stop growing laterally when the energy necessary to stretch an added protofibril exceeds the energy of bonding.
An alternative model of fibrin polymerization and structure has been proposed by the formation of ultrathin fibrin sheets, but the physiological relevance of this finding is unknown.32
Role of the αC regions
The C-terminal portion of fibrinogen’s Aα-chains (Aα392-610) in humans forms the relatively compact αC-domain tethered to the molecule with a flexible unstructured αC-connector (Aα221-391), together making up the αC region.1 During fibrin polymerization, the αC regions interact with each other intermolecularly (within and between protofibrils; Figure 2) and are cross-linked by a plasma transglutaminase (factor XIIIa), resulting in the formation of αC-polymers.33 The αC-domains adopt a physiologically active conformation upon self-association via their N-terminal subdomains that may involve β-hairpin swapping and C-terminal subdomain interaction with the αC-connector.34 Although the αC regions are not essential for lateral aggregation, they enhance it.15,33,35 Clots made from fibrinogen missing the αC regions are made up of thinner and denser fibers, with more branch points than fibers of control clots.36 A recombinant hybrid fibrinogen variant in which the human αC regions were replaced with the homologous but shorter chicken αC regions showed selectively impaired lateral aggregation of protofibrils.37
Role of calcium ions
Fibrin(ogen) has binding sites for calcium ions (Ca2+) that are important for its stability and that promote polymerization. Two calcium-binding sites are located in each of the γ-nodules (γ1 and γ2) and β-nodules (β1 and β2).6,7,38,39 The high-affinity γ1 site is located near hole ‘a’ and is composed of the side chains of residues γAsp318 and γAsp320 and the backbone carbonyls of γPhe322 and γGly324. The lower affinity site γ2 is composed of γAsp294 and γAsp301 and backbone carbonyls of residues γGly296 and γAsp298. The sites β1 and β2 both have a relatively low affinity for Ca2+, but the β2 site connects the β-nodule to the coiled coil via a Ca2+ bridge.7,38 Sialic acid provides additional low-affinity binding sites for Ca2+.40
There is a moderate effect of Ca2+on thrombin-catalyzed fibrinopeptide release,41 but it has strong effects on the subsequent steps of polymerization. Key high-affinity calcium-binding residues in the γ-nodule appear to be necessary for protofibril formation42 and fibrin properties,43 while substitutions of the low-affinity calcium-binding site do not cause significant changes in fibrin polymerization.39 Ca2+increases the rate and extent of lateral aggregation, such that thicker fibers are formed at higher Ca2+ concentrations. Impairments of the β2 calcium-binding site by mutation of 1 of the calcium-coordinating residues γGlu132 increase lateral aggregation,38 consistent with a role for B:b and/or β-nodule:β-nodule interactions in lateral aggregation, because this mutation makes the β-nodule more mobile. The (patho)physiological significance of the interaction of Ca2+ with fibrin(ogen) has not been clearly shown, but its importance may be deduced from the naturally occurring mutations in the vicinity of the calcium-binding sites, such as in γAla341, which affects Ca2+ binding and A:a knob-hole interactions.44
Fibrin branching and network formation
The elongation and the thickening of fibrin fibers are accompanied by branching, which is necessary to produce a 3-dimensional network. There is evidence from electron microscopy for 2 distinct molecular mechanisms by which branch points may form (Figure 2). One of them, called a “bilateral junction,” arises when 2 protofibrils undergo lateral aggregation to form a 4-stranded fibril, and then diverge again into 2 separate protofibrils.45 The second type of branch point, named a “trimolecular junction” or “equilateral junction,” arises when a molecule binds at the end of a protofibril via only 1 γ-nodule, such that both it and the molecule to which it is attached can elongate as 2-stranded protofibrils.46 In either case, most branch points consist of the junction of 3 fibers of about the same diameters,47 which suggests that additional protofibrils add approximately equally to the original structure, no matter what type of branch point. Finally, in general, as the number of branch points in a clot increase, the fiber diameters decrease.47 Together these observations suggest that branching and lateral aggregation compete; that is, conditions that favor lateral aggregation tend to produce clots with thick fibers and few branch points, while conditions that inhibit lateral aggregation tend to yield clots made up of thin fibers with many branch points.
During the course of fibrin polymerization, the population of fibrin structures initially consists of monomers and small 2-stranded structures, such as dimers, trimers, tetramers, and higher-order oligomers, whereas at the later stages the number of soluble, freely moving particles decreases, giving way to the branched fiber network at the gel point.22
Fibrin structure and the gel point
A fibrin clot or gel exists once the branching fibers form a space-filling, 3-dimensional network. The gel point or clotting time is used in clinical assays as an indication of altered coagulation. The gel point occurs relatively early in polymerization, when only about 15% to 20% of the fibrinogen has been incorporated into the gel.48 There are correlations between gel time and final clot structure, but the complete network is generally not established at the gel point, with new fibers and branch points still being established afterward.48 The structure of the clot can be characterized by the fiber diameter, density, number and nature of the branch points, distances between branch points, and size of the pores, all of which are strongly affected by variations in the course of the preceding steps. The fine nanostructure of fibrin clots at and after the gel point has been characterized using precise biophysical techniques that showed dynamic behavior and complex hierarchy at different scales.47,49–52 Although the diffusion of proteins is rarely affected by the fiber network because there are large spaces both within and between fibers, the perfusion of fluid53 or nanoparticles54 through the gel is a sensitive measure of pore size, and hence overall clot structure.
Factor XIIIa cross-linking and its consequences
During and after polymerization in blood, fibrin is covalently cross-linked by factor XIIIa, activated by thrombin. The C-terminal portion of each of fibrin(ogen)’s γ-chain contains 1 cross-linking site at which 2 adjacent molecules form an intermolecular γ-glutamyl-ε-lysyl covalent bond between the γ406Lys of one γ-chain and the γ398/399Gln of another γ-chain. There has been controversy as to whether the γ-γ-cross-linking occurs longitudinally within a strand of a protofibril or transversely between fibrin strands.55,56 Recent stretching experiments on individual fibrin fibers favor longitudinal cross-linking.57 The same intermolecular γ-glutamyl-ε-lysyl bonds form more slowly between C-terminal portions of fibrin α-chains (αC regions), thereby creating αC-polymers, but there are multiple donors and acceptors for the cross-linking reaction. Cross-linking also occurs between α- and γ-chains.58 The dense covalent cross-linking within and between protofibrils makes the polymerization process irreversible and stabilizes fibrin polymers, rendering them mechanically strong and resistant to lysis. A factor XIII genetic polymorphism, in which 34Val is replaced with 34Leu, alters fibrin structure by forming coarse, highly permeable clots or dense clots with reduced permeability, depending on the fibrinogen concentration.59
Variations in fibrin clot structure and properties
The complexity of fibrin polymerization, including variations in the rates of some steps, results in a staggering variety of clot structures and properties that is unequaled by any other biological polymer. The factors that affect fibrin formation and structure may be divided into 4 general categories.
(1). Hereditary and acquired variations in fibrinogen structure.
Different clot structures can arise from fibrinogen splice variants, single-nucleotide polymorphisms, and many possible posttranslational modifications to various parts of the molecule, including limited proteolysis, alterations of N-glycosaminoglycans, amino acid phosphorylation, tyrosine sulfation, glycation, nitration, proline hydroxylation, asparagine or glutamine deamidation, glutamine cyclization, acetylation, homocysteinylation, arginine citrullination, and oxidation. Some of these modifications affect the susceptibility of clots to fibrinolysis in vitro. In the case of homocysteinylation and glycation, evidence exists that fibrinogen modification affects clot stability in vivo.60 Nearly all dysfibrinogenemias have effects on fibrin polymerization.61
(2). Environmental conditions of polymerization.
Ionic conditions, including pH, ionic strength, and composition; numerous endogenous and exogenous compounds, such as polyphosphates, oligo- and polysaccharides, peptides, lipids; and medications, as well as albumin, fibronectin, lipoprotein(a), and other normal and pathological proteins present in plasma and injured tissue, influence clot structure. The concentration of active thrombin, which is determined by the relative rates of enzyme generation and inhibition/elimination, has a profound effect. High thrombin levels produce clots made up of many thin fibers, many branch points, and small pores, while clots made with low thrombin levels consist of fewer thick fibers with few branch points and large pores. Most of these observations are accounted for by the indirect effects on the kinetics of individual steps of fibrin polymerization62 and/or by directly interfering with particular polymerization reactions. A new example of the latter mechanism is the direct binding of factor XIIa (Hageman factor) to fibrinogen and fibrin, followed by modifications of fibrin structure independently from its enzymatic role in thrombin generation.63 The structure of fibrin networks is often characterized by the scanning electron microscopy of clots obtained from purified fibrinogen or platelet-poor plasma (Figure 3A).
Figure 3.

Scanning electron microscope images of clots and thrombi. In vitro clots made from human (A) platelet-poor plasma and (B) platelet-rich plasma. (C) The fibrin formed on a surface coated with collagen-adherent platelets is made up of many fibers aligned along the direction of flow. (D) The image shows an ex vivo human coronary artery thrombus obtained by aspiration from a patient with ST-elevation myocardial infarction. Magnification bars represent 10 μm.
(3). Cellular effects.
Physiological clots and pathological thrombi contain or contact various cell types that have a profound effect on clot structure by directly interacting with fibrin and/or by releasing/expressing active compounds and microparticles.64 In vitro platelet-rich plasma clots contain platelet aggregates that are surrounded by a dense meshwork of fibers that are thinner than fibers elsewhere in the clot, and many of them radiate out from the activated platelets, perhaps along the gradient of thrombin activity (Figure 3B).65,66 Platelets can affect the clot structure by secreting polyphosphate67 and platelet factor 4,68 but the most dramatic platelet-mediated effect is the manifold densification of the fibrin network referred to as clot retraction.69 Other intravascular and extravascular cells (leukocytes, fibroblasts, endothelium, etc.) have effects on the fibrin formed in their vicinity, including those expressing tissue factor, and thus promote local thrombin generation.66,70,71 Red blood cells have effects on fibrin depending on the relative amounts incorporated into the clot, including thicker fibers, altered mechanical properties, and increased heterogeneity.72 The functional consequences of the cellular effects on fibrin clot formation and architecture include modulation in stiffness, stability, and resistance to fibrinolysis.73 Incorporation and infiltration of cells, mainly leukocytes, into the fibrin network promotes local pathophysiological reactions, such as inflammation and malignancy.74
(4). Hydrodynamic flow.
Hemostatic clots or obstructive thrombi are formed in the presence of blood flow that profoundly affects the formation of the fibrin network and its structure and properties. Clotting in static conditions ceases when all fibrinogen is converted to fibrin and polymerizes, such that in normal plasma containing about 2.5 g/L of fibrinogen, clots will contain only 0.25% protein. In contrast, flow continually brings more fibrinogen to the clot that is forming, which can therefore be much denser, with thicker and bundled fibers.75 Fluid flow can also cause the orientation of the fibrin fibers along the direction of flow (Figure 3C) both in vitro76,77 and in vivo,78 which has important consequences for clot mechanical properties and susceptibility to fibrinolysis.77,79 Importantly, based on in vitro microfluidics experiments, it has been proposed that shear forces of the bloodstream determine the likelihood of embolization.80
Clinical implications
An understanding of the mechanisms of fibrin polymerization is important for clinical medicine for a number of reasons. First, it has provided a basis for informative diagnostic tools, such as molecular markers of thrombin generation and/or intravascular fibrin deposition (D-dimer, soluble fibrin precursors, FpA). Many clinical assays in hospital coagulation laboratories involve the measurement of the gel time to diagnose a whole array of disorders. New and more-accurate instruments for the measurement of the gel time are being developed, for example, those based on the viscoelastic properties of incipient blood clots.51 Diagnostic methods employing peptide-derived fibrin-specific magnetic resonance imaging contrast agents to visualize in vivo fibrin deposits have emerged.81
Second, based on the mechanisms of fibrin clot polymerization and dissolution, it is possible to modulate fibrin formation and removal using pro- and anticoagulants, colloid fluids (dextran, hydroxyethyl-starch, gelatin) and thrombolytic therapy. Some other medications commonly used to prevent and treat cardiovascular diseases, such as aspirin,82 heparin,83 statins,84 angiotensin-converting enzyme inhibitors,85 and the hypoglycemic drug metformin,86 have turned out to have side effects that affect fibrin polymerization and clot structure, making fibrin more porous, permeable, and susceptible to lysis. Furthermore, a new generation of highly specific inhibitors of fibrin polymerization has been devised based on synthetic peptides that block knob-hole interactions.87,88
Finally, studies of fibrin formation and clot or thrombus structure in vitro and in vivo give insights into the mechanisms of many diseases associated with thrombotic complications. The architecture and properties of in vitro clots made from the plasma of patients have been shown to correlate with epidemiological and clinical data in cardiovascular diseases. Therefore, such studies have been widely used as a source of physiologically relevant information.89 The time course of in vivo clot formation has been studied by intravital confocal microscopy following laser-induced injury, especially in terms of the kinetics of the contributions of fibrin, tissue factor, and platelets.90 Spatial and temporal relationships between in vivo thrombin generation, the formation of a stable fibrin plug, and platelet deposits at the site of injury have been resolved.91 Genetic mouse models are being studied with this system to determine differences in the structure of the thrombi and the kinetics of formation as a consequence of disease.
The structures of human thrombi, including the fibrin scaffold, have also been studied by electron microscopy (Figure 3D). The dynamics of flow, the functional diversity of platelets, and spatial nonuniformity all result in the heterogeneity of clotting rates throughout the thrombus, so that the relative amounts of fibrin and cells vary considerably. Platelets are involved to a greater extent in the early stages of arterial thrombosis, with a platelet-rich head upstream and a fibrin-rich tail downstream. The traditional view has been that arterial thrombi are mainly composed of platelet aggregates held together by fibrin, while venous thrombi are composed of red cells and a larger amount of fibrin with relatively fewer platelets. More recently, coronary artery thrombi obtained by thromboaspiration in patients with ST-elevation myocardial infarction have been studied by scanning electron microscopy, showing that they were composed primarily of fibrin, with platelets being the second-most abundant component, as well as erythrocytes, cholesterol crystals, and leukocytes. Ischemic time affected the thrombus composition, resulting in a striking positive correlation with thrombus fibrin content and a negative correlation with platelet content.92 Studies have suggested that patients with thrombotic disorders tend to form plasma clots in vitro that are tight, rigid, and less permeable than those from control subjects.93,94 In addition, fibrin clot structure determines the rate of fibrinolysis,94 as well as the flow through the thrombus that is necessary for access to fibrinolytic agents introduced during treatment.
In many clinical studies, it has been shown that the altered structure of in vitro fibrin clots made from patients’ plasma is associated with the risk of thrombotic complications and the course of disease. Abnormal weak fibrin clots were formed from the plasma of people with hemophilia,95 while clots from the plasma of patients with abdominal aortic aneurisms were dense and fibrinolytically resistant.96 Altered fibrin clot structure associated with a resistance to fibrinolysis was observed in in vitro clots made from the plasma of patients with ischemic stroke,97 patients with venous thromboemolism,98 and in smokers.99 The importance of the structure and properties of fibrin clots formed at the sites of injury during wound healing and inflammation has been underestimated.100
In summary, research on the molecular mechanisms of fibrin polymerization and clot properties has increasingly provided a basis for understanding the fundamental mechanisms of the formation and dissolution of hemostatic clots and obstructive thrombi. Identifying structural details of fibrin formation provides novel targets and means for the modulation of blood clotting in vivo, underlying new approaches for the prophylaxis and treatment of thrombotic and bleeding complications in various diseases.
Acknowledgments
We thank Artyom Zhmurov, Olga Kononova, and Valeri Barsegov for the image in Figure 1.
This work was supported by grants from the National Institutes of Health: National Heart, Lung and Blood Institute (HL090774 and HL030954).
Authorship
Contribution: J.W.W. and R.I.L. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John W. Weisel, Department of Cell and Developmental Biology, Perelman School of Medicine, University of Pennsylvania, 1054 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104-6058; e-mail: weisel@mail.med.upenn.edu.
References
- 1.Medved L, Weisel JW Fibrinogen and Factor XIII Subcommittee of Scientific Standardization Committee of International Society on Thrombosis and Haemostasis. Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost. 2009;7(2):355–359. doi: 10.1111/j.1538-7836.2008.03242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riedel T, Suttnar J, Brynda E, et al. Fibrinopeptides A and B release in the process of surface fibrin formation. Blood. 2011;117(5):1700–1706. doi: 10.1182/blood-2010-08-300301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galanakis DK, Henschen A, Peerschke EI, et al. Fibrinogen Stony Brook, a heterozygous A alpha 16Arg----Cys dysfibrinogenemia. Evaluation of diminished platelet aggregation support and of enhanced inhibition of fibrin assembly. J Clin Invest. 1989;84(1):295–304. doi: 10.1172/JCI114154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moen JL, Gorkun OV, Weisel JW, et al. Recombinant BbetaArg14His fibrinogen implies participation of N-terminus of Bbeta chain in desA fibrin polymerization. Blood. 2003;102(7):2466–2471. doi: 10.1182/blood-2003-01-0204. [DOI] [PubMed] [Google Scholar]
- 5.Litvinov RI, Gorkun OV, Owen SF, et al. Polymerization of fibrin: specificity, strength, and stability of knob-hole interactions studied at the single-molecule level. Blood. 2005;106(9):2944–2951. doi: 10.1182/blood-2005-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Everse SJ, Spraggon G, Veerapandian L, et al. Crystal structure of fragment double-D from human fibrin with two different bound ligands. Biochemistry. 1998;37(24):8637–8642. doi: 10.1021/bi9804129. [DOI] [PubMed] [Google Scholar]
- 7.Kostelansky MS, Betts L, Gorkun OV, et al. 2.8 A crystal structures of recombinant fibrinogen fragment D with and without two peptide ligands: GHRP binding to the “b” site disrupts its nearby calcium-binding site. Biochemistry. 2002;41(40):12124–12132. doi: 10.1021/bi0261894. [DOI] [PubMed] [Google Scholar]
- 8.Okumura N, Gorkun OV, Lord ST. Severely impaired polymerization of recombinant fibrinogen gamma-364 Asp —> His, the substitution discovered in a heterozygous individual. J Biol Chem. 1997;272(47):29596–29601. doi: 10.1074/jbc.272.47.29596. [DOI] [PubMed] [Google Scholar]
- 9.Geer CB, Tripathy A, Schoenfisch MH, et al. Role of ‘B-b’ knob-hole interactions in fibrin binding to adsorbed fibrinogen. J Thromb Haemost. 2007;5(12):2344–2351. doi: 10.1111/j.1538-7836.2007.02774.x. [DOI] [PubMed] [Google Scholar]
- 10.Litvinov RI, Gorkun OV, Galanakis DK, et al. Polymerization of fibrin: Direct observation and quantification of individual B:b knob-hole interactions. Blood. 2007;109(1):130–138. doi: 10.1182/blood-2006-07-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galanakis D, Spitzer S, Scharrer I. Unusual A alpha 16Arg—>Cys dysfibrinogenaemic family: absence of normal A alpha-chains in fibrinogen from two of four heterozygous siblings. Blood Coagul Fibrinolysis. 1993;4(1):67–71. [PubMed] [Google Scholar]
- 12.Okumura N, Terasawa F, Haneishi A, et al. B:b interactions are essential for polymerization of variant fibrinogens with impaired holes ‘a’. J Thromb Haemost. 2007;5(12):2352–2359. doi: 10.1111/j.1538-7836.2007.02793.x. [DOI] [PubMed] [Google Scholar]
- 13.Bowley SR, Lord ST. Fibrinogen variant BbetaD432A has normal polymerization but does not bind knob “B”. Blood. 2009;113(18):4425–4430. doi: 10.1182/blood-2008-09-178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doolittle RF, Pandi L. Binding of synthetic B knobs to fibrinogen changes the character of fibrin and inhibits its ability to activate tissue plasminogen activator and its destruction by plasmin. Biochemistry. 2006;45(8):2657–2667. doi: 10.1021/bi0524767. [DOI] [PubMed] [Google Scholar]
- 15.Weisel JW, Medved L. The structure and function of the alpha C domains of fibrinogen. Ann N Y Acad Sci. 2001;936:312–327. doi: 10.1111/j.1749-6632.2001.tb03517.x. [DOI] [PubMed] [Google Scholar]
- 16.Yermolenko IS, Lishko VK, Ugarova TP, et al. High-resolution visualization of fibrinogen molecules and fibrin fibers with atomic force microscopy. Biomacromolecules. 2011;12(2):370–379. doi: 10.1021/bm101122g. [DOI] [PubMed] [Google Scholar]
- 17.Weisel JW, Warren SG, Cohen C. Crystals of modified fibrinogen: size, shape and packing of molecules. J Mol Biol. 1978;126(2):159–183. doi: 10.1016/0022-2836(78)90357-1. [DOI] [PubMed] [Google Scholar]
- 18.Zhmurov A, Brown AE, Litvinov RI, et al. Mechanism of fibrin(ogen) forced unfolding. Structure. 2011;19(11):1615–1624. doi: 10.1016/j.str.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullin JL, Brennan SO, Ganly PS, et al. Fibrinogen Hillsborough: a novel gammaGly309Asp dysfibrinogen with impaired clotting. Blood. 2002;99(10):3597–3601. doi: 10.1182/blood.v99.10.3597. [DOI] [PubMed] [Google Scholar]
- 20.Marchi RC, Carvajal Z, Boyer-Neumann C, et al. Functional characterization of fibrinogen Bicêtre II: a gamma 308 Asn—>Lys mutation located near the fibrin D:D interaction sites. Blood Coagul Fibrinolysis. 2006;17(3):193–201. doi: 10.1097/01.mbc.0000220241.22714.68. [DOI] [PubMed] [Google Scholar]
- 21.Bowley SR, Okumura N, Lord ST. Impaired protofibril formation in fibrinogen gamma N308K is due to altered D:D and “A:a” interactions. Biochemistry. 2009;48(36):8656–8663. doi: 10.1021/bi900239b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chernysh IN, Nagaswami C, Weisel JW. Visualization and identification of the structures formed during early stages of fibrin polymerization. Blood. 2011;117(17):4609–4614. doi: 10.1182/blood-2010-07-297671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medved’ L, Ugarova T, Veklich Y, et al. Electron microscope investigation of the early stages of fibrin assembly. Twisted protofibrils and fibers. J Mol Biol. 1990;216(3):503–509. doi: 10.1016/0022-2836(90)90376-W. [DOI] [PubMed] [Google Scholar]
- 24.Allan P, Uitte de Willige S, Abou-Saleh RH, et al. Evidence that fibrinogen γ’ directly interferes with protofibril growth: implications for fibrin structure and clot stiffness. J Thromb Haemost. 2012;10(6):1072–1080. doi: 10.1111/j.1538-7836.2012.04717.x. [DOI] [PubMed] [Google Scholar]
- 25.Cooper AV, Standeven KF, Ariëns RA. Fibrinogen gamma-chain splice variant gamma’ alters fibrin formation and structure. Blood. 2003;102(2):535–540. doi: 10.1182/blood-2002-10-3150. [DOI] [PubMed] [Google Scholar]
- 26.Gersh KC, Nagaswami C, Weisel JW, et al. The presence of gamma’ chain impairs fibrin polymerization. Thromb Res. 2009;124(3):356–363. doi: 10.1016/j.thromres.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Z, Mochalkin I, Doolittle RF. A model of fibrin formation based on crystal structures of fibrinogen and fibrin fragments complexed with synthetic peptides. Proc Natl Acad Sci USA. 2000;97(26):14156–14161. doi: 10.1073/pnas.97.26.14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okumura N, Terasawa F, Hirota-Kawadobora M, et al. A novel variant fibrinogen, deletion of Bbeta111Ser in coiled-coil region, affecting fibrin lateral aggregation. Clin Chim Acta. 2006;365(1-2):160–167. doi: 10.1016/j.cca.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Langer BG, Weisel JW, Dinauer PA, et al. Deglycosylation of fibrinogen accelerates polymerization and increases lateral aggregation of fibrin fibers. J Biol Chem. 1988;263(29):15056–15063. [PubMed] [Google Scholar]
- 30.Mullin JL, Gorkun OV, Lord ST. Decreased lateral aggregation of a variant recombinant fibrinogen provides insight into the polymerization mechanism. Biochemistry. 2000;39(32):9843–9849. doi: 10.1021/bi000045c. [DOI] [PubMed] [Google Scholar]
- 31.Hirota-Kawadobora M, Terasawa F, Yonekawa O, et al. Fibrinogens Kosai and Ogasa: Bbeta15Gly—>Cys (GGT—>TGT) substitution associated with impairment of fibrinopeptide B release and lateral aggregation. J Thromb Haemost. 2003;1(2):275–283. doi: 10.1046/j.1538-7836.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien ET, III, Falvo MR, Millard D, et al. Ultrathin self-assembled fibrin sheets. Proc Natl Acad Sci USA. 2008;105(49):19438–19443. doi: 10.1073/pnas.0804865105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsurupa G, Mahid A, Veklich Y, et al. Structure, stability, and interaction of fibrin αC-domain polymers. Biochemistry. 2011;50(37):8028–8037. doi: 10.1021/bi2008189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsurupa G, Pechik I, Litvinov RI, et al. On the mechanism of αC polymer formation in fibrin. Biochemistry. 2012;51(12):2526–2538. doi: 10.1021/bi2017848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Litvinov RI, Yakovlev S, Tsurupa G, et al. Direct evidence for specific interactions of the fibrinogen alphaC-domains with the central E region and with each other. Biochemistry. 2007;46(31):9133–9142. doi: 10.1021/bi700944j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collet JP, Moen JL, Veklich YI, et al. The alphaC domains of fibrinogen affect the structure of the fibrin clot, its physical properties, and its susceptibility to fibrinolysis. Blood. 2005;106(12):3824–3830. doi: 10.1182/blood-2005-05-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ping L, Huang L, Cardinali B, et al. Substitution of the human αC region with the analogous chicken domain generates a fibrinogen with severely impaired lateral aggregation: fibrin monomers assemble into protofibrils but protofibrils do not assemble into fibers. Biochemistry. 2011;50(42):9066–9075. doi: 10.1021/bi201094v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kostelansky MS, Lounes KC, Ping LF, et al. Calcium-binding site beta 2, adjacent to the “b” polymerization site, modulates lateral aggregation of protofibrils during fibrin polymerization. Biochemistry. 2004;43(9):2475–2483. doi: 10.1021/bi0359978. [DOI] [PubMed] [Google Scholar]
- 39.Kostelansky MS, Lounes KC, Ping LF, et al. Probing the gamma2 calcium-binding site: studies with gammaD298,301A fibrinogen reveal changes in the gamma294-301 loop that alter the integrity of the “a” polymerization site. Biochemistry. 2007;46(17):5114–5123. doi: 10.1021/bi602607a. [DOI] [PubMed] [Google Scholar]
- 40.Dang CV, Shin CK, Bell WR, et al. Fibrinogen sialic acid residues are low affinity calcium-binding sites that influence fibrin assembly. J Biol Chem. 1989;264(25):15104–15108. [PubMed] [Google Scholar]
- 41.Profumo A, Turci M, Damonte G, et al. Kinetics of fibrinopeptide release by thrombin as a function of CaCl2 concentration: different susceptibility of FPA and FPB and evidence for a fibrinogen isoform-specific effect at physiological Ca2+ concentration. Biochemistry. 2003;42(42):12335–12348. doi: 10.1021/bi034411e. [DOI] [PubMed] [Google Scholar]
- 42.Lounes KC, Ping L, Gorkun OV, et al. Analysis of engineered fibrinogen variants suggests that an additional site mediates platelet aggregation and that “B-b” interactions have a role in protofibril formation. Biochemistry. 2002;41(16):5291–5299. doi: 10.1021/bi011988s. [DOI] [PubMed] [Google Scholar]
- 43.Averett LE, Akhremitchev BB, Schoenfisch MH, et al. Calcium dependence of fibrin nanomechanics: the γ1 calcium mediates the unfolding of fibrinogen induced by force applied to the “A-a” bond. Langmuir. 2010;26(18):14716–14722. doi: 10.1021/la1017664. [DOI] [PubMed] [Google Scholar]
- 44.Park R, Ping L, Song J, et al. Fibrinogen residue γAla341 is necessary for calcium binding and ‘A-a’ interactions. Thromb Haemost. 2012;107(5):875–883. doi: 10.1160/TH11-10-0731. [DOI] [PubMed] [Google Scholar]
- 45.Mosesson MW, DiOrio JP, Siebenlist KR, et al. Evidence for a second type of fibril branch point in fibrin polymer networks, the trimolecular junction. Blood. 1993;82(5):1517–1521. [PubMed] [Google Scholar]
- 46.Fogelson AL, Keener JP. Toward an understanding of fibrin branching structure. Phys Rev E Stat Nonlin Soft Matter Phys. 2010;81(5):051922. doi: 10.1103/PhysRevE.81.051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryan EA, Mockros LF, Weisel JW, et al. Structural origins of fibrin clot rheology. Biophys J. 1999;77(5):2813–2826. doi: 10.1016/S0006-3495(99)77113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chernysh IN, Weisel JW. Dynamic imaging of fibrin network formation correlated with other measures of polymerization. Blood. 2008;111(10):4854–4861. doi: 10.1182/blood-2007-08-105247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferri F, Greco M, Arcòvito G, et al. Structure of fibrin gels studied by elastic light scattering techniques: dependence of fractal dimension, gel crossover length, fiber diameter, and fiber density on monomer concentration. Phys Rev E Stat Nonlin Soft Matter Phys. 2002;66(1):011913. doi: 10.1103/PhysRevE.66.011913. [DOI] [PubMed] [Google Scholar]
- 50.Guthold M, Liu W, Stephens B, et al. Visualization and mechanical manipulations of individual fibrin fibers suggest that fiber cross section has fractal dimension 1.3. Biophys J. 2004;87(6):4226–4236. doi: 10.1529/biophysj.104.042333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans PA, Hawkins K, Morris RH, et al. Gel point and fractal microstructure of incipient blood clots are significant new markers of hemostasis for healthy and anticoagulated blood. Blood. 2010;116(17):3341–3346. doi: 10.1182/blood-2010-02-269324. [DOI] [PubMed] [Google Scholar]
- 52.Yeromonahos C, Polack B, Caton F. Nanostructure of the fibrin clot. Biophys J. 2010;99(7):2018–2027. doi: 10.1016/j.bpj.2010.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okada M, Blombäck B. Factors influencing fibrin gel structure studied by flow measurement. Ann N Y Acad Sci. 1983;408:233–253. doi: 10.1111/j.1749-6632.1983.tb23248.x. [DOI] [PubMed] [Google Scholar]
- 54.Spero RC, Sircar RK, Schubert R, et al. Nanoparticle diffusion measures bulk clot permeability. Biophys J. 2011;101(4):943–950. doi: 10.1016/j.bpj.2011.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weisel JW. Cross-linked gamma-chains in fibrin fibrils bridge transversely between strands: no. J Thromb Haemost. 2004;2(3):394–399. doi: 10.1111/j.1538-7933.2003.00621.x. [DOI] [PubMed] [Google Scholar]
- 56.Mosesson MW. Cross-linked gamma-chains in fibrin fibrils bridge ‘transversely’ between strands: yes. J Thromb Haemost. 2004;2(3):388–393. doi: 10.1111/j.1538-7933.2004.00613.x. [DOI] [PubMed] [Google Scholar]
- 57.Guthold M, Carlisle C. Single fibrin fiber experiments suggest longitudinal rather than transverse cross-linking: reply to a rebuttal. J Thromb Haemost. 2010;8(9):2090–2091. [Google Scholar]
- 58.Standeven KF, Carter AM, Grant PJ, et al. Functional analysis of fibrin gamma-chain cross-linking by activated factor XIII: determination of a cross-linking pattern that maximizes clot stiffness. Blood. 2007;110(3):902–907. doi: 10.1182/blood-2007-01-066837. [DOI] [PubMed] [Google Scholar]
- 59.Lim BC, Ariëns RA, Carter AM, et al. Genetic regulation of fibrin structure and function: complex gene-environment interactions may modulate vascular risk. Lancet. 2003;361(9367):1424–1431. doi: 10.1016/S0140-6736(03)13135-2. [DOI] [PubMed] [Google Scholar]
- 60.Hoffman M. Alterations of fibrinogen structure in human disease. Cardiovasc Hematol Agents Med Chem. 2008;6(3):206–211. doi: 10.2174/187152508784871981. [DOI] [PubMed] [Google Scholar]
- 61.de Moerloose P, Neerman-Arbez M. Congenital fibrinogen disorders. Semin Thromb Hemost. 2009;35(4):356–366. doi: 10.1055/s-0029-1225758. [DOI] [PubMed] [Google Scholar]
- 62.Weisel JW, Nagaswami C. Computer modeling of fibrin polymerization kinetics correlated with electron microscope and turbidity observations: clot structure and assembly are kinetically controlled. Biophys J. 1992;63(1):111–128. doi: 10.1016/S0006-3495(92)81594-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Konings J, Govers-Riemslag JW, Philippou H, et al. Factor XIIa regulates the structure of the fibrin clot independently of thrombin generation through direct interaction with fibrin. Blood. 2011;118(14):3942–3951. doi: 10.1182/blood-2011-03-339572. [DOI] [PubMed] [Google Scholar]
- 64.Aleman MM, Gardiner C, Harrison P, et al. Differential contributions of monocyte- and platelet-derived microparticles towards thrombin generation and fibrin formation and stability. J Thromb Haemost. 2011;9(11):2251–2261. doi: 10.1111/j.1538-7836.2011.04488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collet JP, Montalescot G, Lesty C, et al. A structural and dynamic investigation of the facilitating effect of glycoprotein IIb/IIIa inhibitors in dissolving platelet-rich clots. Circ Res. 2002;90(4):428–434. doi: 10.1161/hh0402.105095. [DOI] [PubMed] [Google Scholar]
- 66.Campbell RA, Overmyer KA, Bagnell CR, et al. Cellular procoagulant activity dictates clot structure and stability as a function of distance from the cell surface. Arterioscler Thromb Vasc Biol. 2008;28(12):2247–2254. doi: 10.1161/ATVBAHA.108.176008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mutch NJ, Engel R, Uitte de Willige S, et al. Polyphosphate modifies the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plasminogen to fibrin. Blood. 2010;115(19):3980–3988. doi: 10.1182/blood-2009-11-254029. [DOI] [PubMed] [Google Scholar]
- 68.Amelot AA, Tagzirt M, Ducouret G, et al. Platelet factor 4 (CXCL4) seals blood clots by altering the structure of fibrin. J Biol Chem. 2007;282(1):710–720. doi: 10.1074/jbc.M606650200. [DOI] [PubMed] [Google Scholar]
- 69.Feghhi S, Sniadecki NJ. Mechanobiology of platelets: techniques to study the role of fluid flow and platelet retraction forces at the micro- and nano-scale. Int J Mol Sci. 2011;12(12):9009–9030. doi: 10.3390/ijms12129009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ovanesov MV, Ananyeva NM, Panteleev MA, et al. Initiation and propagation of coagulation from tissue factor-bearing cell monolayers to plasma: initiator cells do not regulate spatial growth rate. J Thromb Haemost. 2005;3(2):321–331. doi: 10.1111/j.1538-7836.2005.01128.x. [DOI] [PubMed] [Google Scholar]
- 71.Campbell RA, Overmyer KA, Selzman CH, et al. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114(23):4886–4896. doi: 10.1182/blood-2009-06-228940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gersh KC, Nagaswami C, Weisel JW. Fibrin network structure and clot mechanical properties are altered by incorporation of erythrocytes. Thromb Haemost. 2009;102(6):1169–1175. doi: 10.1160/TH09-03-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wolberg AS. Plasma and cellular contributions to fibrin network formation, structure and stability. Haemophilia. 2010;16(suppl): 37-12. [DOI] [PubMed]
- 74.Wolberg AS. Determinants of fibrin formation, structure, and function. Curr Opin Hematol. 2012;19(5):349–356. doi: 10.1097/MOH.0b013e32835673c2. [DOI] [PubMed] [Google Scholar]
- 75.Neeves KB, Illing DA, Diamond SL. Thrombin flux and wall shear rate regulate fibrin fiber deposition state during polymerization under flow. Biophys J. 2010;98(7):1344–1352. doi: 10.1016/j.bpj.2009.12.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gersh KC, Edmondson KE, Weisel JW. Flow rate and fibrin fiber alignment. J Thromb Haemost. 2010;8(12):2826–2828. doi: 10.1111/j.1538-7836.2010.04118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Campbell RA, Aleman M, Gray LD, et al. Flow profoundly influences fibrin network structure: implications for fibrin formation and clot stability in haemostasis. Thromb Haemost. 2010;104(6):1281–1284. doi: 10.1160/TH10-07-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Whittaker P, Przyklenk K. Fibrin architecture in clots: a quantitative polarized light microscopy analysis. Blood Cells Mol Dis. 2009;42(1):51–56. doi: 10.1016/j.bcmd.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Varjú I, Sótonyi P, Machovich R, et al. Hindered dissolution of fibrin formed under mechanical stress. J Thromb Haemost. 2011;9(5):979–986. doi: 10.1111/j.1538-7836.2011.04203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Colace TV, Muthard RW, Diamond SL. Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: role of thrombin with and without fibrin. Arterioscler Thromb Vasc Biol. 2012;32(6):1466–1476. doi: 10.1161/ATVBAHA.112.249789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Katoh M, Haage P, Wiethoff AJ, et al. Molecular magnetic resonance imaging of deep vein thrombosis using a fibrin-targeted contrast agent: a feasibility study. Invest Radiol. 2009;44(3):146–150. doi: 10.1097/RLI.0b013e318195886d. [DOI] [PubMed] [Google Scholar]
- 82.He S, Bark N, Wang H, et al. Effects of acetylsalicylic acid on increase of fibrin network porosity and the consequent upregulation of fibrinolysis. J Cardiovasc Pharmacol. 2009;53(1):24–29. doi: 10.1097/FJC.0b013e3181953e0f. [DOI] [PubMed] [Google Scholar]
- 83.Yeromonahos C, Marlu R, Polack B, et al. Antithrombin-independent effects of heparins on fibrin clot nanostructure. Arterioscler Thromb Vasc Biol. 2012;32(5):1320–1324. doi: 10.1161/ATVBAHA.112.245308. [DOI] [PubMed] [Google Scholar]
- 84.Tehrani S, Mobarrez F, Antovic A, et al. Atorvastatin has antithrombotic effects in patients with type 1 diabetes and dyslipidemia. Thromb Res. 2010;126(3):e225–e231. doi: 10.1016/j.thromres.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 85.Undas A, Celinska-Löwenhoff M, Löwenhoff T, et al. Statins, fenofibrate, and quinapril increase clot permeability and enhance fibrinolysis in patients with coronary artery disease. J Thromb Haemost. 2006;4(5):1029–1036. doi: 10.1111/j.1538-7836.2006.01882.x. [DOI] [PubMed] [Google Scholar]
- 86.Standeven KF, Ariëns RA, Whitaker P, et al. The effect of dimethylbiguanide on thrombin activity, FXIII activation, fibrin polymerization, and fibrin clot formation. Diabetes. 2002;51(1):189–197. doi: 10.2337/diabetes.51.1.189. [DOI] [PubMed] [Google Scholar]
- 87.Stabenfeldt SE, Gourley M, Krishnan L, et al. Engineering fibrin polymers through engagement of alternative polymerization mechanisms. Biomaterials. 2012;33(2):535–544. doi: 10.1016/j.biomaterials.2011.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watson JW, Doolittle RF. Peptide-derivatized albumins that inhibit fibrin polymerization. Biochemistry. 2011;50(45):9923–9927. doi: 10.1021/bi201406c. [DOI] [PubMed] [Google Scholar]
- 89.Undas A, Ariëns RA. Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2011;31(12):e88–e99. doi: 10.1161/ATVBAHA.111.230631. [DOI] [PubMed] [Google Scholar]
- 90.Falati S, Gross P, Merrill-Skoloff G, et al. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8(10):1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 91.Welsh JD, Colace TV, Muthard RW, et al. Platelet-targeting sensor reveals thrombin gradients within blood clots forming in microfluidic assays and in mouse. J Thromb Haemost. 2012;10(11):2344–2353. doi: 10.1111/j.1538-7836.2012.04928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Silvain J, Collet JP, Nagaswami C, et al. Composition of coronary thrombus in acute myocardial infarction. J Am Coll Cardiol. 2011;57(12):1359–1367. doi: 10.1016/j.jacc.2010.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Collet JP, Allali Y, Lesty C, et al. Altered fibrin architecture is associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol. 2006;26(11):2567–2573. doi: 10.1161/01.ATV.0000241589.52950.4c. [DOI] [PubMed] [Google Scholar]
- 94. Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost. 2007;5(suppl 1):116-124. [DOI] [PubMed]
- 95.Brummel-Ziedins KE, Branda RF, Butenas S, et al. Discordant fibrin formation in hemophilia. J Thromb Haemost. 2009;7(5):825–832. doi: 10.1111/j.1538-7836.2009.03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scott DJ, Prasad P, Philippou H, et al. Clot architecture is altered in abdominal aortic aneurysms and correlates with aneurysm size. Arterioscler Thromb Vasc Biol. 2011;31(12):3004–3010. doi: 10.1161/ATVBAHA.111.236786. [DOI] [PubMed] [Google Scholar]
- 97.Undas A, Podolec P, Zawilska K, et al. Altered fibrin clot structure/function in patients with cryptogenic ischemic stroke. Stroke. 2009;40(4):1499–1501. doi: 10.1161/STROKEAHA.108.532812. [DOI] [PubMed] [Google Scholar]
- 98.Undas A, Zawilska K, Ciesla-Dul M, et al. Altered fibrin clot structure/function in patients with idiopathic venous thromboembolism and in their relatives. Blood. 2009;114(19):4272–4278. doi: 10.1182/blood-2009-05-222380. [DOI] [PubMed] [Google Scholar]
- 99.Parastatidis I, Thomson L, Burke A, et al. Fibrinogen beta-chain tyrosine nitration is a prothrombotic risk factor. J Biol Chem. 2008;283(49):33846–33853. doi: 10.1074/jbc.M805522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost. 2006;4(5):932–939. doi: 10.1111/j.1538-7836.2006.01861.x. [DOI] [PubMed] [Google Scholar]