

Abstract
Microphthalmia is an important developmental eye disorder. Although mutations in several genes have been linked to this condition, they only account for a minority of cases. We performed autozygome analysis and exome sequencing on a multiplex consanguineous family in which colobomatous microphthalmia is associated with profound global developmental delay, intractable seizures, and corpus callosum abnormalities, and we identified a homozygous truncating mutation in C12orf57 [c.1A>G; p.Met1?]. In a simplex case with a similar phenotype, we identified compound heterozygosity for the same mutation and another missense mutation [c.152T>A; p.Leu51Gln]. Little is known about C12orf57 but we show that it is expressed in several mouse tissues, including the eye and brain. Our data strongly implicate mutations in C12orf57 in the pathogenesis of a clinically distinct autosomal-recessive syndromic form of colobomatous microphthalmia.
Main Text
Microphthalmia is a condition in which partially arrested eye development results in a clinically recognizable small globe size unilaterally or bilaterally.1 Embryologically, the elongating optic stalk features a groove known as the choroid fissure, which closes as the optic stalk forms the optic cup, so a developmental arrest is expected to result not only in a small eye but also in failure of fissure closure and resultant coloboma.2–5 This is consistent with the clinical observation of nonrandom association between the two eye anomalies; one large epidemiological study on microphthalmia showed that around 70% of affected individuals have associated defects in fissure closure.6 Mechanisms that arrest eye development vary and include both environmental and genetic causes(see References7,8 and GeneReviews in Web Resources). Genetic forms of microphthalmia are particularly interesting because they provide insight into the molecular network that controls the development of this important sensory organ. For instance, the mapping of Pax6 as the mutated gene in the small eye mouse was a critical landmark in our understanding of eye development and revealed mechanisms that are remarkably conserved from mammals down to fruitflies.9
Both syndromic and nonsyndromic forms of microphthalmia are known to exist, although the latter account for the majority of cases (two-thirds). In humans, mutations in several genes have been identified in individuals with both forms of microphthalmia. Most of these mutations were described as autosomal dominant (usually de novo), but X-linked and autosomal-recessive forms have also been reported. Known causes of autosomal-recessive microphthalmia include mutations in FOXE3 (MIM 601094) (monoallelic mutations in this gene cause anterior-segment dysgenesis), CHX10 (MIM 142993), PITX3 (MIM 602669) (monoallelic mutations in this gene cause anterior-segment dysgenesis), RAX (MIM 601881), SIX6 (MIM 606326), and ODZ3 (MIM 610083).10–14
The high rate of consanguinity in the Saudi population has been shown to unmask numerous pathogenic recessive alleles that cause various birth defects, including those that cause microphthalmia, and these are typically amenable to homozygosity mapping approaches.13–15 We have previously shown the power of supplementing these approaches with next-generation sequencing to identify ODZ3 as a gene mutated in a family with autosomal-recessive microphthalmia.14 As part of our ongoing effort to explore the full spectrum of autosomal-recessive forms of microphthalmia in our population, we report here the identification of an autosomal-recessive form of colobomatous microphthalmia in which affected individuals have the additional features of profound global developmental delay, intractable seizures, and abnormal development of the corpus callosum. We show that affected members of this family harbor a homozygous truncating mutation in C12orf57. Reassuringly, we also identified an additional simplex case in an individual with a similar phenotype and show that she is compound heterozygous for that same truncating mutation and another allele in trans. Our findings strongly support the candidacy of C12orf57 as a gene that is mutated in a distinct syndromic form of colobomatous microphthalmia in humans.
Family 1 consists of second-cousin parents with two healthy and four affected children (the fourth was born during the study) (Figure 1 and Table 1). The older affected individual in this family is a 19-year-old male (VI:4). He was born after an uneventful pregnancy. Seizures developed at 3 weeks of age, and he still suffers from intractable generalized tonic clonic epilepsy. In addition, he has profound global developmental delay. Physical examination revealed no obvious dysmorphism but did reveal poor body build and hypertonia. Ophthalmological examination was significant for mild microphthalmia and bilateral iridal and inferior chorioretinal coloboma that extended to the optic disc and was associated with light perception only. His brain MRI revealed agenesis of the corpus callosum (Figure 1).
Figure 1.
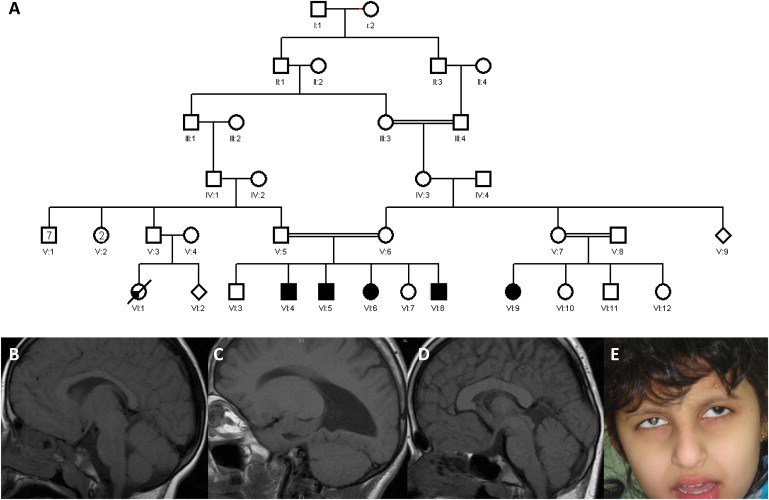
Identification of Individuals with a Syndromic form of Colobomatous Microphthalmia
(A) Pedigree of Family 1.
(B) An MRI of individual VI:6 shows a normal corpus callosum.
(C) The corpus callosum is absent in individual VI:4.
(D) Thickened corpus callosum in individual VI:5.
(E) Facial photos of the index individual in family 2 (iris coloboma is evident in both eyes, particularly the right because of flash reflection).
Table 1.
Summary of Clinical Features in Two Families Affected by a Syndromic Form of Colobomatous Microphthalmia
| Individual ID | Age | Gender | Colobomatous Microphthalmia | GDDa | Epilepsy | Corpus Callosal Abnormality | Others |
|---|---|---|---|---|---|---|---|
| VI:4 | 19 years | M | yes (bilateral) | yes | yes | yes (agenesis) | Hypertonia, self-mutilation and xylophagia |
| VI:5 | 18 years | M | yes (bilateral) | yes | yes | yes (hyperplasia) | |
| VI:6 | 12 years | F | no | yes | yes | no | Hypertonia |
| VI:8 | 2.5 years | M | no | yes | yes | yes (agenesis) | |
| Family 2 | 6 years | F | yes (bilateral) | yes | yes | no |
GDD: Global developmental delay.
The second affected child in this family (VI:5) is an 18-year-old male. Global developmental delay and poor vision were apparent in infancy. Seizures developed at 2.5 years of age and continue to be refractory to multiple antiepileptic medications. Physical examination revealed hypertonia and bilateral chorioretinal coloboma involving the optic disc, whereas the anterior segment appeared normal. Apart from thickening of the corpus callosum, his brain MRI was unremarkable.
The third affected child is a 12-year-old girl (VI:6). She has had refractory epilepsy since the age of 6 months, and she displays profound global developmental delay. Ophthalmological examination revealed a normal anterior segment and no clinical evidence of coloboma. Apart from mild hypertonia and slightly increased reflexes, her physical exam was unremarkable. A brain MRI was unremarkable.
During the course of the study, a fourth affected child (VI:8) was born and is now almost 2.5 years old. He developed seizures recently and showed only partial response to carbamazepine. A limited ophthalmological examination revealed no gross abnormalities. A brain MRI was significant for corpus callosum agenesis.
None of the four affected siblings shows any evidence of other system involvement. All underwent extensive medical evaluation that included metabolic screening; liver and renal function tests; a complete blood count; a bone profile; and test showing that blood gases, ammonia, and lactate levels were all within normal limits.
Parents signed a written consent to participate in an IRB-approved research protocol (King Faisal Specialist Hospital and Research Center RAC# 2070023) before blood was drawn in EDTA tubes for DNA extraction. Genotyping was performed on an Axiom platform according to the manufacturer’s protocol (Affymetrix, Santa Clara, CA, USA), and the full set of autozygosity blocks per individual (autozygome) was determined by autoSNPa.16 One member of family 1 (VI:5) underwent exome sequencing. Exome capture was performed with the TruSeq Exome Enrichment kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target with the Illumina Exome Enrichment protocol. The captured libraries were sequenced with Illumina HiSeq 2000 Sequencer. The reads were mapped against UCSC hg19 with the Burrows-Wheeler Aligner (see Web Resources). The SNPs and Indels were detected with SAMTOOLS.
Although we did expect smaller or fewer runs of homozygsity (ROH) in family 1 because parents were second cousins, we could not identify any ROH that were exclusively shared by the affected children even after we lowered our threshold to just 1Mb (Figure S1). In view of the apparent lack of an obvious lead from autozygome analysis, we were unable to use it to filter the exome results and instead used the following filters: total variants → homozygous variants → variants that are absent in dbSNP → variants that are coding or splicing → variants that are predicted to be pathogenic → variants that are absent in 200 in-house Saudi exomes. Nine variants survived this filtration scheme, but only one passed the segregation test (Figure S2 in the Supplemental Data available online). This variant in C12orf57 (RefSeq accession number NM_138425.2: c.1A>G) abolishes the normal initiation codon and is predicted to completely abolish the protein because the next ATG is out of frame in all three known protein-coding transcripts (Figure 2). When we re-examined the autozygome data in this family, we identified a very small (500 kb) run of homozygosity encompassing this gene in the three genotyped siblings (Figure S1). This represents a highly unusual sequence of recombination events given that the affected individuals are only four generations removed from the nearest common ancestor.15,17 Indeed, the centromeric recombination was <70 kb away from the gene locus (Figure S1).
Figure 2.
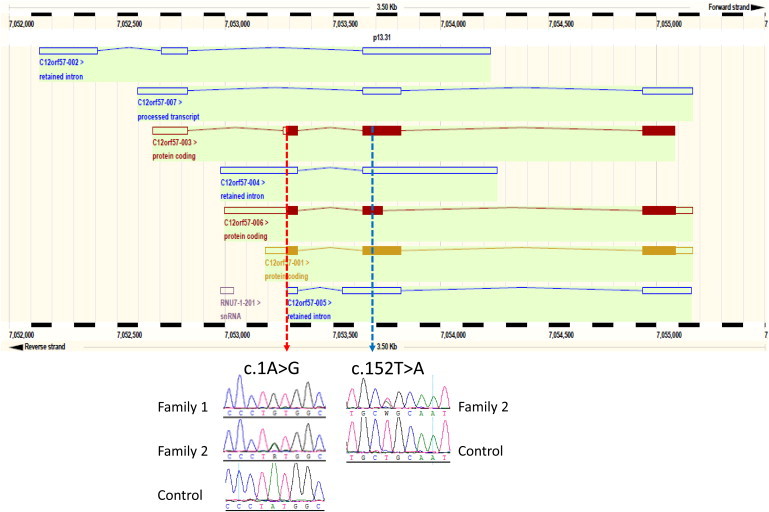
Ensembl View Showing All Known C12orf57 Transcripts Drawn to Scale
The location of the p.Met1? mutation is indicated by a red dotted line showing that it affects the same start codon in all three coding transcripts. The blue dotted line denotes the location of the p.Leu51Gln mutation. Sequence chromatograms are shown for both mutations.
We then searched our database of >30 microphthalmia families for potential clinical overlap and identified family 2, which consists of two young Saudi parents who belong to the same tribe but deny identifiable consanguinity otherwise. Their only child is a 6-year-old girl with profound global developmental delay and epilepsy that began at 6 months of age and is currently under control after numerous trials of antiepilepsy medications. The girl also has bilateral microphthalmia and coloboma involving the iris, choroid, and retina and extending to the optic cup (Figure 1 and Table 1). Her brain MRI was unremarkable.
Subsequent direct sequencing of C12orf57 in family 2 revealed that the affected individual was compound heterozygous for the same mutation as well as a previously undescribed missense variant, c.152T>A [p.Leu51Gln]. Sequencing of the parents confirmed that the two mutations are in trans in their affected daughter (Figure 2). The p.Leu51Gln variant is predicted to be pathogenic in silico (PolyPhen score of 0.997, SIFT score of 0.01, and MutationTaster score of 1) and is highly conserved (Figure S3). Like the p.Met1? substitution, p.Leu51Gln was absent in EVS (exome variant server), 200 in-house Saudi exomes, and 576 Saudi individuals (1552 Saudi chromosomes) who underwent direct sequencing. In order to address the possibility that C12orf57 is a gene that accumulates pathogenic variants in the general population, we fully sequenced the gene in 96 Saudi controls and found no pathogenic variants.
Colobomatous microphthalmia affects around 10% of blind children and is associated with significant morbidity that is proportionate to the degree of severity.1,18 Most children with this condition lack a definite molecular diagnosis because the clinical presentation usually predicts the likely genetic defect only in a subset of distinct syndromic associations, as in the case of CHD7 (MIM 608892)- and BMP4 (MIM 112262)-related microphthalmia. In fact, the true contribution of Mendelian mutations to the causation of this condition is very difficult to determine, especially in simplex cases, which account for the majority of cases. Indeed, colobomatous microphthalmia in family 2 could have been due to many causes, and we were only able to identify the biallelic mutation in C12orf57 through the study of family 1. Thus, although the syndrome we describe here clearly has important additional features, we believe that colobomatous microphthalmia is the most helpful defining feature. However, as is typical of many other syndromes, even this feature is not fully penetrant, and its absence does not exclude the possibility of C12orf57 mutation.
Virtually nothing is known about C12orf57 other than that it has recently been reported to be mutated in a family described as having “nonsyndromic mental retardation.”19 However, we show that affected members of that family clearly display a distinct phenotype that consists of profound delay in all aspects of development, early-onset intractable seizures, an abnormal corpus callosum, and 50% penetrance of colobomatous microphthalmia. The penetrance of colobomatous microphthalmia is increased to 60% if we include family 2. Thus, C12orf57 must be playing an important developmental role in both the brain and eye, although the exact role remains unknown. Our expression profiling in mice revealed ubiquitous expression, including in the brain and eye (Figure 3).
Figure 3.
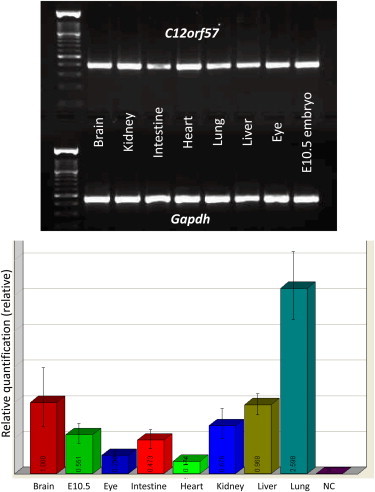
Expression Profiling of C12orf57 in the Mouse
Upper panel: Gel picture of RTPCR using RNA from various mouse tissues (Gapdh is shown below as an internal control for each reaction).
Lower panel: Bar chart summarizing the results of quantitative RTPCR for the same tissues (error bars for the experimental triplicates are shown).
In summary, we add to the literature an autosomal-recessive cause of colobomatous microphthalmia that seems to be invariably associated with profound global developmental delay and intractable early-onset seizures, as well as occasional defects of the corpus callosum. We believe that individuals with this syndromic from of colobomatous microphthalmia should be screened for mutations in C12orf57. Future studies are needed to understand the role of this gene in a developmental context and to determine the minimal diagnostic criteria for this syndrome.
Acknowledgments
We thank the families for enthusiastic participation. Genotyping and sequencing were performed by the Genotyping and Sequencing Core Facilities at King Faisal Specialist Hospital and Research Center. This study was supported by King Abdulaziz City for Science and Technology grant 08-MED497-20 (F.S.A.) and a Dubai-Harvard Foundation for Medical Research collaborative grant (F.S.A.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
Ensembl Genome Browser, http://www.ensembl.org/index.html
GeneReviews, Bardakjian, T., Weiss, A., and Schneider, A.S. (1993). Anophthalmia/Microphthalmia Overview, http://www.ncbi.nlm.nih.gov/books/NBK1378/
MutationTaster, www.mutationtaster.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen, www.genetics.bwh.harvard.edu/pph2/
SAMtools, http://samtools.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Traboulsi E.I. Oxford University Press; Oxford: 2011. Genetic Diseases of the Eye. [Google Scholar]
- 2.Donner A.L., Maas R.L. Conservation and non-conservation of genetic pathways in eye specification. Int. J. Dev. Biol. 2004;48:743–753. doi: 10.1387/ijdb.041877ad. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert S.F. Sinauer Associates; Sunderland. MA: 2010. Developmental biology. [Google Scholar]
- 4.Lachke S.A., Maas R.L. Building the developmental oculome: systems biology in vertebrate eye development and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010;2:305–323. doi: 10.1002/wsbm.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wawersik S., Maas R.L. Vertebrate eye development as modeled in Drosophila. Hum. Mol. Genet. 2000;9:917–925. doi: 10.1093/hmg/9.6.917. [DOI] [PubMed] [Google Scholar]
- 6.Morrison D., FitzPatrick D., Hanson I., Williamson K., van Heyningen V., Fleck B., Jones I., Chalmers J., Campbell H. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J. Med. Genet. 2002;39:16–22. doi: 10.1136/jmg.39.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bardakjian T.M., Schneider A. The genetics of anophthalmia and microphthalmia. Curr. Opin. Ophthalmol. 2011;22:309–313. doi: 10.1097/ICU.0b013e328349b004. [DOI] [PubMed] [Google Scholar]
- 8.Verma A.S., Fitzpatrick D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007;2:47. doi: 10.1186/1750-1172-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill R.E., Favor J., Hogan B.L., Ton C.C., Saunders G.F., Hanson I.M., Prosser J., Jordan T., Hastie N.D., van Heyningen V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991;354:522–525. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- 10.Valleix S., Niel F., Nedelec B., Algros M.P., Schwartz C., Delbosc B., Delpech M., Kantelip B. Homozygous nonsense mutation in the FOXE3 gene as a cause of congenital primary aphakia in humans. Am. J. Hum. Genet. 2006;79:358–364. doi: 10.1086/505654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voronina V.A., Kozhemyakina E.A., O’Kernick C.M., Kahn N.D., Wenger S.L., Linberg J.V., Schneider A.S., Mathers P.H. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum. Mol. Genet. 2004;13:315–322. doi: 10.1093/hmg/ddh025. [DOI] [PubMed] [Google Scholar]
- 12.Ferda Percin E., Ploder L.A., Yu J.J., Arici K., Horsford D.J., Rutherford A., Bapat B., Cox D.W., Duncan A.M., Kalnins V.I. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat. Genet. 2000;25:397–401. doi: 10.1038/78071. [DOI] [PubMed] [Google Scholar]
- 13.Aldahmesh M.A., Khan A.O., Mohamed J., Alkuraya F.S. Novel recessive BFSP2 and PITX3 mutations: Insights into mutational mechanisms from consanguineous populations. Genet. Med. 2011;13:978–981. doi: 10.1097/GIM.0b013e31822623d5. [DOI] [PubMed] [Google Scholar]
- 14.Aldahmesh M.A., Mohammed J.Y., Al-Hazzaa S., Alkuraya F.S. Homozygous null mutation in ODZ3 causes microphthalmia in humans. Genet. Med. 2012;14:900–904. doi: 10.1038/gim.2012.71. [DOI] [PubMed] [Google Scholar]
- 15.Alkuraya F.S. Autozygome decoded. Genet. Med. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 16.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 17.Alkuraya F.S. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protoc. Hum. Genet. 2012;6 doi: 10.1002/0471142905.hg0612s75. 6.12. [DOI] [PubMed] [Google Scholar]
- 18.Parker S.E., Mai C.T., Canfield M.A., Rickard R., Wang Y., Meyer R.E., Anderson P., Mason C.A., Collins J.S., Kirby R.S., Correa A., National Birth Defects Prevention Network Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res. A Clin. Mol. Teratol. 2010;88:1008–1016. doi: 10.1002/bdra.20735. [DOI] [PubMed] [Google Scholar]
- 19.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.