

Abstract
Mutations in several known or putative glycosyltransferases cause glycosylation defects in α-dystroglycan (α-DG), an integral component of the dystrophin glycoprotein complex. The hypoglycosylation reduces the ability of α-DG to bind laminin and other extracellular matrix ligands and is responsible for the pathogenesis of an inherited subset of muscular dystrophies known as the dystroglycanopathies. By exome and Sanger sequencing we identified two individuals affected by a dystroglycanopathy with mutations in β-1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2). B3GALNT2 transfers N-acetyl galactosamine (GalNAc) in a β-1,3 linkage to N-acetyl glucosamine (GlcNAc). A subsequent study of a separate cohort of individuals identified recessive mutations in four additional cases that were all affected by dystroglycanopathy with structural brain involvement. We show that functional dystroglycan glycosylation was reduced in the fibroblasts and muscle (when available) of these individuals via flow cytometry, immunoblotting, and immunocytochemistry. B3GALNT2 localized to the endoplasmic reticulum, and this localization was perturbed by some of the missense mutations identified. Moreover, knockdown of b3galnt2 in zebrafish recapitulated the human congenital muscular dystrophy phenotype with reduced motility, brain abnormalities, and disordered muscle fibers with evidence of damage to both the myosepta and the sarcolemma. Functional dystroglycan glycosylation was also reduced in the b3galnt2 knockdown zebrafish embryos. Together these results demonstrate a role for B3GALNT2 in the glycosylation of α-DG and show that B3GALNT2 mutations can cause dystroglycanopathy with muscle and brain involvement.
Introduction
Congenital muscular dystrophies (CMDs) are a heterogeneous group of inherited diseases characterized by the onset of muscle weakness at birth or within 2 years of life and variable degrees of dystrophic changes on muscle biopsy. A common subgroup within the CMDs are the dystroglycanopathies, characterized by reduced functional glycosylation of α-dystroglycan (α-DG). The dystroglycan gene DAG1 (MIM 128239) encodes for a protein that is posttranslationally glycosylated and cleaved into two noncovalently associated proteins, α-DG and β-dystroglycan (β-DG).1–3 α-DG is a peripheral membrane protein that binds to several extracellular matrix components, including laminin-α2 and other laminin-G domain containing ligands,2,4 and the transmembrane protein β-DG is anchored to the cytoskeleton by the dystrophin complex.1 This connection helps to maintain structural integrity and force transmission between the cytoskeleton and the extracellular matrix but also allows for efficient signal transduction. The correct glycosylation of α-DG is essential for its function as an extracellular matrix receptor.1 Dystroglycan is not restricted to skeletal muscle and is also present with varying degrees of glycosylation in many tissues such as the brain, peripheral nerve, and epithelia.5,6 In these tissues, dystroglycan has a multitude of functions including morphogenesis, early development, synaptogenesis, and signaling.7
Dystroglycanopathies have been linked to mutations in 12 genes: Protein O-mannosyltransferase 18 (POMT1 [MIM 607423]), Protein O-mannosyltransferase 29 (POMT2 [MIM 607439]), Protein O-mannose β-1, 2-N-acetylglucosaminyltransferase10 (POMGNT1 [MIM 606822]), Fukutin11 (FKTN [MIM 607440]), Fukutin-related protein12 (FKRP [MIM 606596]), like-acetylglucosaminyltransferase13 (LARGE [MIM 603590]), Dolichyl-phosphate mannosyltransferase 214 (DPM2 [MIM 603564]), Dolichyl-phosphate mannosyltransferase 315 (DPM3 [MIM 605951]), Dolichol Kinase16 (DOLK [MIM 610746]), Isoprenoid Synthase Domain Containing17–19 (ISPD [MIM 614631]), Glycosyltransferase-like domain containing 220 (GTDC2 [MIM 147730]), and Transmembrane protein 521 (TMEM5 [MIM 605862]). In these disorders, the reduced α-DG glycosylation1 results in lower-affinity binding to the extracellular matrix partners. Clinical severity in individuals with dystroglycanopathy varies markedly22–24 and is broadly related to the level of α-DG hypoglycosylation caused especially by mutations in some genes such as POMT1, POMT2, and POMGNT122 but less so with FKTN and FKRP,22 as assessed with antibodies that specifically recognize glycosylated epitopes on α-DG. Individuals at the severe end of the clinical spectrum have muscular dystrophy with brain and eye anomalies and the resulting clinical conditions known as Walker-Warburg syndrome (WWS [MIM 236670]), muscle-eye-brain disease (MEB [MIM 236670]), and Fukuyama congenital muscular dystrophy (FCMD [MIM 253800]).These conditions are often associated with a short life expectancy. Milder conditions often exclusively affect striated muscle and can have onset in childhood or later, as for example in limb-girdle muscular dystrophy type 2I (LGMD2I [MIM 607155]) and congenital muscular dystrophy type 1C with no mental retardation (MDC1C [MIM 606612]). Almost 50% of individuals with features of a dystroglycanopathy remain genetically uncharacterized.17,25,26
In this report we describe the identification of mutations in β1,3-N-acetylgalactosaminyl transferase (B3GALNT2 [MIM 610194]) in individuals with dystroglycanopathy. The clinical severity ranges from severe WWS to milder dystroglycanopathy variants, invariably associated with brain involvement. Further analysis suggests that B3GALNT2 is involved in the functional glycosylation of α-DG.
Subjects and Methods
Subjects
This study covers two cohorts of affected individuals. The first cohort consisted of 47 dystroglycanopathy individuals25 recruited at the Dubowitz Neuromuscular Centre at Great Ormond Street Hospital (London, UK). The second cohort consisted of 55 dystroglycanopathy individuals that were enrolled at Boston Children’s Hospital (Boston, MA). All consents and local review board approvals were in accordance with the UK10K project ethical framework. Affected individuals P1 and P2 were identified from the UK cohort, and P3–P7 were identified from the USA cohort.
Exome and Sanger Sequencing
P1 was whole-exome sequenced as part of the UK10K project via the HiSeq platform (Illumina, San Diego, CA, USA) as paired-end 75 base pair (bp) reads. Sequencing, alignment of reads, and variant calling were performed as recently described.18 The mean coverage was 74×. P3 and P6 were whole-exome sequenced by Axeq Technologies (Seoul, Korea) with a SureSelect Human All Exon 50 Mb enrichment kit and the Illumina HiSeq platform. The mean coverages were 48.6× and 42×, respectively. P4 was whole-exome sequenced at the Broad Institute of Harvard and MIT with a SureSelect Human All Exon 37 Mb enrichment kit and the HiSeq platform (Illumina). It had a mean coverage of 110×. Additional B3GALNT2 mutations were identified in P2 and P7 by standard Sanger sequencing protocols.18 Primer sequences are available upon request.
For P1, functional annotation was added with Ensembl Variant Effect Predictor v2.2 against Ensembl 64 Variant filtering of the single-nucleotide variants, and indels for P1 were done at the B3GALNT2 locus as previously described18 assuming autosomal-recessive inheritance. The filtering for P4 was performed similarly as for P1 except that dbSNP132 variants were not excluded, and variants were also filtered against 821 exomes sequenced in the C.A.W. lab and 69 genome sequences available from Complete Genomics. We focused on variants in regions shared with the affected sibling. For P3, P4, and P6, functional annotation was performed with ANNOVAR.27
Skeletal Muscle Histology and Immunohistochemistry
Immunohistochemical studies were performed as described previously.28 Unfixed frozen serial sections (7 μm) were incubated with primary antibodies for 1 hr, and then with the appropriate biotinylated secondary antibodies for 30 min followed by streptavidin conjugated to Alexa Fluor 594 (Life Technologies, Paisley, UK) for 15 min. Primary antibodies used were mouse monoclonal: α-DG IIH6 (clone IIH6C4 from K.P.C.,3,29), β-DG (Leica, Milton Keynes, UK; clone 43DAG1/8D5), laminin-α2 (Merck Millipore, Nottingham, UK; catalog no. MAB1922 clone 5H2) to the 80 kDa C-terminal fragment, and goat polyclonal core α-dystroglycan GT20ADG.30 Sections were evaluated with a Leica DMR microscope interfaced to MetaMorph (Molecular Devices, Sunnyvale, CA, USA).
Plasmid Construction and Mutagenesis
Human B3GALNT2 cDNA (Image clone: IRATp970G0734D) was obtained from Source Bioscience (Nottingham, UK). The coding sequence of B3GALNT2 was amplified with the primers listed in Table S1 (available online). Amplified products were cloned into the pcDNA3.1/V5-His TOPO expression vector (Life Technologies) according to the manufacturer’s instructions. Missense mutations (c.740G>A [p.Gly247Glu], c.875G>C [p.Arg292Pro], c.802G>A [p.Val268Met]) were introduced via QuickChange II site-directed mutagenesis (Agilent Technologies, Cheshire, UK). Mutagenesis primers are listed in Table S2.
Flow Cytometry
Functional α-DG glycosylation was assessed in P1 and P2’s fibroblasts with the α-DG antibody IIH6 (Merck Millipore). Fibroblasts were grown in DMEM (Life Technologies) containing 20% fetal bovine serum (FBS, PAA, Yeovil, UK), 2% glutamine, and 1% penicillin-streptavidin (Life Technologies) at 37°C with 5% CO2. The method was modified from Rojek et al.31 Cells were detached with Accutase (Merck Millipore), centrifuged at 500 × g for 3 min, and resuspended to a final density of 200,000 cells/ml. They were subsequently fixed with 2% paraformaldehyde and washed with PBS 0.1% FBS. Subsequent centrifugations were at 3,000 × g for 3 min. Cells were then incubated with the following antibodies: anti-α-DG IIH6 (Merck Millipore), anti-mouse biotinylated IgM (Vector Labs, Peterborough, UK), and streptavidin-PE (BD PharMingen, Oxford, UK). Cells were resuspended in 500 μl of PBS and transferred to FACS tubes (BD Biosciences, Oxford, UK). Data were acquired with the Cyan ADP analyzer (Beckman Coulter, High Wycome, UK) and analyzed with the FlowJo software (Tree Star, Ashland, OR, USA). A separate control for each cell population was incubated only with the secondary antibody. Fluorescence staining observed in these cell populations was discounted as background and removed from the corresponding cell population stained with all three antibodies in order to select for IIH6-positive cells only (unpublished data). An unpaired two-tailed t test was utilized to interpret the flow cytometry data.
Transfection of Wild-Type and Mutant B3GALNT2 on C2C12 Cells
C2C12 myoblast cells (ECCAC) were cultured in 20% FBS in DMEM supplemented with 2% L-glutamine. Transfection of cell lines was carried out with Gene Juice (Merck Millipore) in serum-free media. After 24 hr, cells were fixed in either 2% paraformaldehyde or methanol for immunohistochemistry. Immunolabeling was performed with V5-FITC (Life Technologies), ERp72 (Cell Signaling Technology for New England Biolabs, Ipswich, MA, USA), goat anti-rabbit Alexa Fluor 594 or goat anti-mouse Alexa Fluor 594 (Life Technologies), and Hoechst 33258 (Life Technologies).
Fluorescence images were taken with a Leica DMR microscope (Molecular Devices). Metamorph image capture software was used (Molecular Devices). Any further image processing was achieved with ImageJ software.
Immunoblot Analysis of Human Proteins
Immunoblotting of muscle protein lysate and fibroblast cell lysate was performed as previously described.32,33 The following antibodies were used: anti-α-DG IIH6 (Merck Millipore), anti-β-DG (Leica Microsystems, Milton Keynes, UK), biotinylated secondary antibody anti-IgM (Dako, Glostrup, Denmark) or anti-IgG (Dako), and HRP-streptavidin (Dako).
Knockdown of Zebrafish b3galnt2 and Characterization of the Phenotype
To determine the expression of zebrafish b3galnt2, we extracted RNA from 20–30 embryos with an RNeasy kit (QIAGEN, Crawley, UK), followed by reverse transcription with SuperScript III (Life Technologies). RT-PCR for b3galnt2 was done with RedTaq DNA polymerase kit (Sigma-Aldrich, Dorset, UK) with forward and reverse primers 5′-ACTCAGAGCTCCGCGATG-3′ and 5′-CAGAGCAGAGATCCCTCAAA-3′, respectively.
Translation blocking (TB) (5′-CGCCGCCGCTGCACTTCTCATGGAC-3′) and splice blocking (SB) (5′-GGTCTGTCTGTCAAGGAGAAATAAA-3′) morpholinos for b3galnt2 were obtained from Gene Tools, LLC (Philomath, OR, USA) and injected into 1- to 4-cell-stage Tuebingen Long Fin zebrafish embryos, which were reared as previously described.34 Sequences of p53 and dag1 morpholinos have been described.35,36
Zebrafish expression plasmids (pCS2+) containing b3galnt2 cDNA (Image clone: IRBOp991H0563D) and a C-terminal GFP tag were generated by Gateway Cloning Technology (Life Technologies) according to the manufacturer’s instructions. mRNA for injection was made with mMessage mMachine SP6 kit (Ambion, for Life Technologies). Immunofluorescence staining by primary antibodies against laminin (L-9393, Sigma-Aldrich) and β-DG (Leica Microsystems) was performed on 48 hr postfertilization (hpf) whole-mount embryos as previously described.35 For the Evans blue dye (EBD) assay, 48 hpf live embryos were immobilized in 1% low-melting-point agarose (Sigma-Aldrich) containing 0.016% tricaine (Sigma-Aldrich), and a solution of 0.1% EBD (Sigma-Aldrich) was injected into the pericardium. Two hours later, they were examined by confocal microscopy. Microsome preparation and immunoblot analysis of zebrafish proteins was performed as previously described.19
Results
B3GALNT2 Is Mutated in Individuals with Dystroglycanopathy
We diagnosed dystroglycanopathy in a single affected individual (P1). He presented at 4 months of age with developmental delay and a head circumference above 90th percentile. All motor milestones were delayed due to axial hypotonia and increased limb tone due to spasticity; he never acquired independent walking. From the age of 6 years he could take steps with hands held and used a standing device to stand. He is a sociable boy, but his communication skills are mostly confined to sign language. His vision was also severely impaired due to marked myopia complicated by bilateral partial retinal detachment. A brain MRI showed cerebellar cysts, a thin corpus callosum, diffusely abnormal white matter signal on T2-weighted images, and cortical dysplasia in the form of frontoparietal polymicrogyria. The diagnosis was a dystroglycanopathy with clinical features overlapping MEB and FCMD.25 Molecular genetic analysis of the genes previously associated with dystroglycanopathies was normal.
We performed whole-exome sequencing on P1 and variant filtering left three genes with two potentially deleterious variants each. These were alanyl-tRNAsynthetase (AARS [MIM 601065]), nesprin-2 (SYNE2 [MIM 608442]), and β-1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2). AARS mutations have been described to cause Charcot-Marie-Tooth disease, type 2N (MIM 613287), a phenotype not present in P1. SYNE2 variants have been associated with Emery-Dreifuss muscular dystrophy (MIM 612999), which is very distinct from P1’s phenotype. B3GALNT2 (RefSeq accession number NM_152490.2) encodes for a glycosyltransferase that synthesizes the structure GlcNac-β1, 3GalNac,37 a glycan that has previously been found on α-DG.38,39 Mutations in this gene could reduce the amount of functional α-DG glycosylation by preventing or impairing the formation of this particular glycan. P1 was compound heterozygous for the B3GALNT2 variants c.875G>C (p.Arg292Pro) and c.740G>A (p.Gly247Glu). We Sanger sequenced B3GALNT2 in the remaining UK cohort and found mutations in another individual of Turkish descent with a similar phenotype (Table 1). This individual (P2) had a history of delayed gross motor skills and was unable to walk. His communication was limited, but his vision was intact. His brain MRI showed features similar to those of P1 (Figure 1). For both P1 and P2, brain involvement was not detected in prenatal scans.
Table 1.
Mutations and Predicted Protein Changes for P1–P4, P6, and P7
| Affected Persona | Mutationsb | Predicted Protein Effect | Mutation Detection Technique | Parental Carrier Status |
|---|---|---|---|---|
| P1 (M) | exon 6: c.740G>A | p.Gly247Glu | exome | mother: carrier |
| exon 8: c.875G>C | p.Arg292Pro | father: carrier | ||
| P2 (M) | exon 1: c.51_73dup (homozygous) | p.Ser25Cysfs∗38 | Sanger sequencing | father: carrier |
| mother: carrier | ||||
| P3 (F) | exon 6: c.726_727del | p.Val243Glufs∗2 | exome | mother: carrier |
| exon 7: c.822_823dup | p.Ile276Leufs∗26 | no paternal DNA | ||
| P4 (M) | exon 3: c.308_309del | p.Val103Glyfs∗10 | exome | mother: carrier |
| exon 6: c.755T>G | p.Val252Gly | father: carrier | ||
| P6 (F) | exon 7: c.802G>A (homozygous) | p.Val268Met | exome | father: carrier |
| mother: carrier | ||||
| P7 (M) | exon 12: c.1423C>T (homozygous) | p.Gln475∗ | Sanger sequencing | no parental DNA |
Affected person’s sex in brackets: M, male; F, female. DNA was not available for P5.
Mutation nomenclature follows the recommended guidelines of the Human Genome Variation Society with the nucleotide numbering based on GenBank reference sequence NM_152490.2 and verified with Mutalyzer 2.0.beta-21. Nucleotide numbering denotes the adenosine of the annotated translation start codon as nucleotide position +1.
Figure 1.
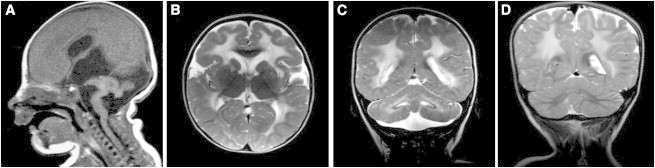
Individuals with B3GALNT2 Mutations Have Structural Brain Abnormalities
(A) Sagittal T1-weighted MRI of P3 demonstrating severe hydrocephalus, ponto-cerebellar hypoplasia, and anterior concavity of the brainstem.
(B) Axial T2-weighted MRI of P2 demonstrating abnormal cerebral white matter.
(C) Coronal T2-weighted MRI of P2 demonstrating cerebellar dysplasia and cysts.
(D) Coronal T2-weighted MRI of P6 demonstrating abnormal cerebral white matter and cerebellar dysplasia.
Six additional mutations in B3GALNT2 were subsequently identified in four individuals by exome sequencing (P3, P4, and P6) or by Sanger sequencing (P7) from a cohort with dystroglycanopathy and severe brain involvement.40,41 P3, P4, and P7 have at least one truncating mutation in B3GALNT2 and more severe clinical features than do P1 and P2 with evidence of severe hydrocephalus and cobblestone lissencephaly on brain MRI (Figure 1), leading to a diagnosis of WWS, the most severe form of dystroglycanopathy. P4 had two affected siblings: an affected female diagnosed with WWS (P5, for whom no DNA was available) and another affected sibling who was diagnosed prenatally with severe hydrocephalus and ocular malformations based on fetal ultrasound, leading to a pregnancy termination. The fetal DNA confirmed the presence of the compound heterozygous B3GALNT2 mutations identified in P4 and carried by the parents. P6 had a less severe presentation, with clinical and radiological features of dystroglycanopathy associated with abnormal white matter signal on T2-weighted MRI images. All affected individuals had severe hypotonia and did not attain any motor milestones. P3, P4, and P5 also had epilepsy. See Table 2 for further clinical details and Figure S1 for a summary of the mutations found in P1–P4, P6, and P7.
Table 2.
Summary of Clinical Findings
| Affected Person | Sex | Origin | Age at Presentation | Presenting Symptom | Maximal Motor Ability | Main Neurologic Features | Ophthalmologic Findings | Maximum CK (U/L) | Brain MRI Findings | Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | Europe | 5 weeks | borderline macrocephaly | few steps with support | cognitive delay | optic nerve hypoplasia | 1,132 | polymicrogyria, fronto-temporal leukoencephalopathy, cerebellar cysts | MEB/FCMD-like |
| P2 | M | Turkey | 17 months | motor and cognitive delays | few steps with support | cognitive impairment | none | 894 | polymicrogyria, fronto-temporal leukoencephalopathy, brainstem and pontine hypoplasia, cerebellar dysplasia, cerebellar cysts | MEB/FCMD-like |
| P3 | F | USA (white) | birth | hypotonia, severe hydrocephalus | roll over (halfway) | epilepsy and absent cognitive development | optic nerve hypoplasia, bilateral lens opacities | 21,000 | severe hydrocephalus, cobblestone lissencephaly, ponto-cerebellar hyoplasia | WWS/WWS-like |
| P4 | M | South Africa | birth | hypotonia, severe hydrocephalus | no motor milestones attained | epilepsy and absent cognitive development | right-sided congenital glaucoma, left-sided microphthalmia and cataract, buphthalmia | 6,964 | severe hydrocephalus, cobblestone lissencephaly | WWS/WWS-like |
| P5 | F | South Africa | birth | hypotonia, hydrocephalus | no motor milestones attained | epilepsy and absent cognitive development | blindness | NA | hydrocephalus, cobblestone lissencephaly, profound hypomyelination | WWS/WWS-like |
| P6 | F | Saudi Arabia | 15 months | motor and cognitive delays | bottom shuffling | cognitive impairment | none (no formal ophthalmologic exam) | 1,740 | fronto-temporal leukoencephalopathy, cerebellar dysplasia | MEB/FCMD-like |
| P7 | M | USA (Hispanic) | birth | hypotonia and respiratory distress | no motor milestones attained | absent cognitive development | bilateral microphthalmia | 1,086 | severe hydrocephalus, cobblestone lissencephaly | WWS/WWS-like |
Abbreviations are as follows: CK, creatine kinase; M, male; F, female; FCMD, Fukuyama congenital muscular dystrophy; MEB, muscle-eye-brain disease; NA, not available; and WWS, Walker-Warburg syndrome. DNA was not available for P5.
Skeletal Muscle Biopsies of Individuals with B3GALNT2 Mutations Reveal Dystrophic Processes and Reduced α-DG Glycosylation
A muscle biopsy of the quadriceps performed in P1 at 11 months of age showed features consistent with muscular dystrophy, including abnormal variation in fiber size, regenerating fibers, endomysial connective tissue, and internal nuclei. A muscle biopsy performed in P2 at 2 years of age showed less severe changes, with no obvious necrosis. Immunolabeling of laminin α2 with antibodies to the 80 and 300 kDa fragments showed a slight reduction of labeling of some fibers in P1 but was normal in P2 (data not shown). Labeling of α-DG with IIH6 was variably reduced whereas labeling of β-DG, of core α-DG (Figure 2), and of laminin chains α5, β1, and γ1 was normal (data not shown).
Figure 2.
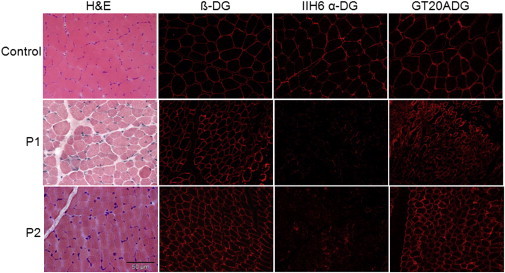
Functional α-DG Glycosylation Is Reduced in Individuals with B3GALNT2 Mutations
An examination of skeletal muscle cryosections from control, P1, and P2 revealed that glycosylated α-DG is reduced in individuals with B3GALNT2 mutations (P1 and P2) whereas β-DG was well preserved. Staining with the GT20ADG antibody against the core α-DG protein showed that the core was better preserved in P2 than in P1 as some variability was seen among the fibers.
Immunoblotting for α-DG with the IIH6 antibody in muscle protein lysates showed a profound reduction in α-DG functional glycosylation for P1 and a mild reduction with a lower molecular weight for P2. There was no change in the amount of β-DG (Figure 3). Immunoblotting for IIH6 in fibroblasts of both P1 and P2 showed a severe reduction in the IIH6 epitope (Figure S2).
Figure 3.
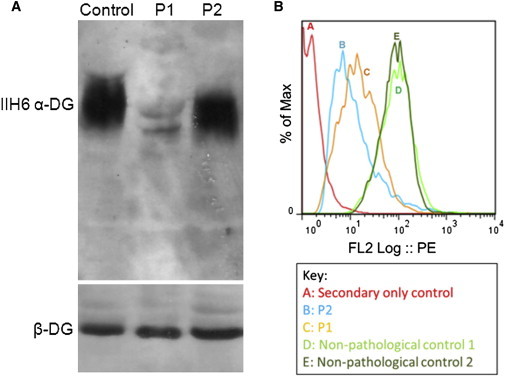
Immunoblotting of Muscle Protein Lysate and Flow Cytometry of P1 and P2 Fibroblasts Reveals Aberrant α-DG Glycosylation in P1 and P2
(A) The membrane was incubated with α-DG IIH6 and β-DG antibodies. P1 showed a profound reduction in α-DG IIH6 accumulation compared to the control whereas in P2 the level of IIH6 was similar to the control, but a slight shift in molecular weight was seen in both individuals. There was no change in β-DG levels.
(B) Flow cytometry was utilized to assess the level of functional α-DG glycosylation in P1 and P2 compared to nonpathological controls. The level of functional α-DG glycosylation was evaluated by the fluorescence intensity of the IIH6 epitope conjugated to streptavidin-PE. This histogram shows the mean fluorescence intensity (MFI) of the IIH6 epitope. The MFI of the controls depicted in the histogram were 75.28 (line D) and 79.98 (line E), whereas P1 (line C) and P2 (line B) had MFI values of 33.53 and 28.5, respectively. There is a statistically significant (p < 0.05) reduction in MFI in both P1 and P2 compared to controls (each fibroblast sample was analyzed three times). The secondary antibody-only control (line A) was used to remove background from the populations incubated with all three antibodies.
Affected Fibroblasts with Mutations in B3GALNT2 Have Reduced α-DG Glycosylation
Flow cytometry was used to assess the level of functional α-DG glycosylation in P1 and P2 fibroblasts compared to the fibroblasts of two nonpathological controls (previously found to have normal α-DG glycosylation by immunohistochemistry and immunoblot [not shown]). The level of functional α-DG glycosylation was evaluated by the fluorescence intensity of the IIH6 epitope conjugated to streptavidin-PE.
Flow cytometry showed that P1 and P2 fibroblasts were reduced in functionally glycosylated α-DG compared to the two controls (p < 0.05). The controls had 75.28 average and 79.98 mean fluorescence intensity (MFI), whereas B3GALNT2 fibroblasts have 33.53 average MFI (P1) and 28.5 average MFI (P2), suggesting a profound reduction of the IIH6 epitope (Figure 3; Table S3).
To further validate this technique, in addition to the nonpathological fibroblast controls, we also tested fibroblasts from an individual with a ryanodine receptor mutation (RYR1 [MIM 180901]) and from an individual with an α-glucosidase mutation (GAA [MIM 232300]). These fibroblasts all had MFI values that were not statistically different than the controls listed above (Table S3). Additionally, two individuals carrying mutations in two genes previously associated with dystroglycanopathy (POMT1 and FKRP) had significantly (p < 0.05) reduced MFI values compared to both the nonpathological and the pathological controls described here (Table S3).
B3GALNT2 Localizes to the Endoplasmic Reticulum and This Localization Is Disrupted by Dystroglycanopathy Mutations
In order to assess the functional effect of the B3GALNT2 mutations, we examined the localization of the wild-type and mutant proteins. Wild-type B3GALNT2 colocalized with the ER-specific marker ERp72 (Figure 4), suggesting B3GALNT2 localization to the ER despite the absence of an obvious ER retention signal. Like wild-type, the c.875G>C (p.Arg292Pro) mutant colocalized with the ER marker, indicating that this mutation did not alter the protein localization (Figure 4). However, both c.740G>A (p.Gly247Glu) and c.802G>A (p.Val268Met) mutants had diffuse localization compared to wild-type and the ER marker (Figure 4), suggesting that these mutations disrupt the ER localization of B3GALNT2.
Figure 4.
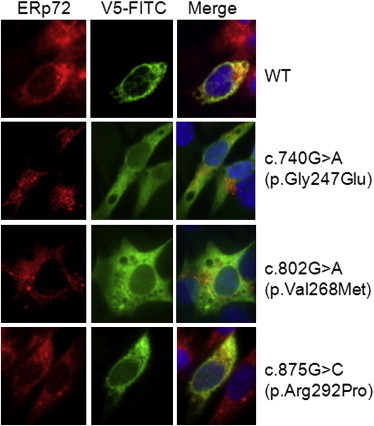
Transfection of C212 Myoblasts with Wild-Type and Mutant B3GALNT2
C2C12 myoblasts were transfected with B3GALNT2 pcDNA 3.1 V5/HIS-TOPO (either wild-type or with the missense mutation c.740G>A [p.Gly247Glu], c.875G>C [p.Arg292Pro], c.802G>A [p.Val268Met]). Wild-type B3GALNT2 colocalized with the ER-specific marker ERp72. The c.740G>A (p.Gly247Glu) and c.802G>A (p.Val268M) mutations caused the enzyme to localize differently than the wild-type enzyme, but the c.875G>C (p.Arg292Pro) mutation did not alter the subcellular localization compared to wild-type.
Knockdown of Zebrafish b3galnt2 Recapitulates Several Aspects of the Human Phenotypes Including Reduced α-DG Glycosylation
To analyze the role of B3GALNT2 in vivo, we used zebrafish as a vertebrate model. Human B3GALNT2 has a single ortholog in zebrafish (Ensembl ID ENSDARG00000046133). Zebrafish B3galnt2 is 53% identical to human B3GALNT2 and the amino acid sequence of the galactosyltransferase domain alone is conserved with 68.5% identity. Publically available in situ hybridization data indicate that b3galnt2 is ubiquitously expressed at early stages of zebrafish development, becoming more anteriorly localized as development progresses (ZFIN database). By using RT-PCR, we confirmed that b3galnt2 expression was detectable throughout early development (Figure S3).
To knock down b3galnt2 in zebrafish embryos, we injected a translation-blocking (TB) morpholino (4 ng) together with a p53 morpholino (2 ng) to reduce p53-mediated off-target effects.42,43 Under confocal microscopy, we showed that the expression of GFP-tagged B3galnt2 was effectively suppressed, confirming the morpholino specificity (Figure 5). Zebrafish b3galnt2 knockdown embryos (morphants) showed curved body, mild retinal degeneration, and severely impaired motility (Figure 5). Similar to fktn, fkrp, and ispd morphants,19 b3galnt2 morphants recapitulated the hydrocephalus phenotype present in P3, P4, and P7. To further confirm the morpholino specificity, we injected a b3galnt2 splice-blocking morpholino, which also caused very similar phenotypes (data not shown).
Figure 5.
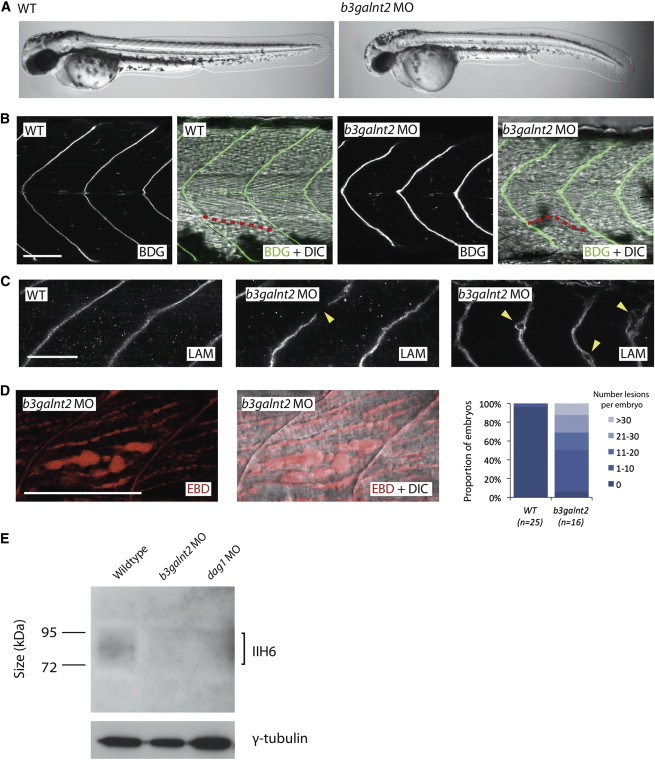
b3galnt2 Knockdown Zebrafish Embryos Have Muscle Defects and Hypoglycosylated α-DG at 48 hpf
(A) Whole-mount pictures of live embryos show gross morphological defects.
(B) Immunofluorescence staining by an antibody against β-DG and differential interference contrast (DIC) microscopy showed that the muscle fibers are disordered. One sample fiber is highlighted in red.
(C) Immunofluorescence staining by an antibody against laminin (LAM) shows gaps in the myosepta of knockdown embryos and degeneration of the extracellular matrix.
(D) Evans blue dye assay (EBD) highlights frequent lesions between muscle fibers in b3galnt2 morphants, which are very rarely seen in wild-type embryos (the same image is shown with and without DIC microscopy).
(A–D) b3galnt2 MO: b3galnt2 TB morpholino (4 ng) coinjected with p53 TB morpholino (2 ng). Scale bars represent 50 μm.
(E) Immunoblotting by isolated microsome protein from 48 hpf embryos and IIH6 antibody showed a reduction in glycosylated α-DG in the b3galnt2 knockdown embryos. b3galnt2 MO: b3galnt2 TB morpholino (5 ng) coinjected with p53 TB morpholino (2.5 ng). dag1 MO: dag1 TB morpholino (5 ng).
To characterize the muscle phenotype, we performed immunofluorescence staining. Compared to the V-shaped somite boundaries flanking straight muscle fibers in wild-type embryos, b3galnt2 morphants consistently showed slightly U-shaped somites and disordered muscle fibers (Figure 5). Laminin staining revealed occasional gaps in the myosepta (the connective tissues where the muscle fibers anchor), suggesting disruption of the extracellular matrix (Figure 5). Next, we quantified the severity of muscle damage by Evans blue dye (EBD) assay.44 EBD penetrates compromised sarcolemma or accumulates at lesions in muscle. Compared to wild-type embryos, b3galnt2 morphants showed more severely damaged muscle with increased number of lesions, ranging from less than 10 to more than 30 lesions per embryo (Figure 5).
Immunoblot analysis with the IIH6 antibody on protein extracts from wild-type embryos and b3galnt2 morphants showed a reduction of the IIH6 signal in b3galnt2 morphants, indicating that knockdown of b3galnt2 led to reduced functional glycosylation of α-DG. This is consistent with the human data and strongly suggests that this may be the molecular mechanism behind the phenotypes described.
Discussion
In this study we describe the use of exome and Sanger sequencing for the identification of mutations in B3GALNT2 in six dystroglycanopathy-affected families characterized by the invariable presence of structural brain involvement and, in some severe cases, ocular involvement. The spectrum of severity in individuals with B3GALNT2 mutations ranged from severe WWS to mild MEB-like or FCMD-like conditions45 (Tables 1 and 2). In terms of correlation between genotype and phenotype, biallelic loss-of-function mutations were associated with the most severe phenotype, as demonstrated in P3 and P7, affected by severe WWS. P1, with compound heterozygosity for two missense mutations in exons 6 and 8 of B3GALNT2, had a milder CMD phenotype, associated with less severe structural brain involvement and better motor and cognitive achievements. Surprisingly, P2, with a biallelic duplication in exon 1, also presented with a milder CMD phenotype with brain involvement.
In order to assess the pathogenicity of the missense mutations found in P1 (c.740G>A [p.Gly247Glu], c.875G>C [p.Arg292Pro]) and P6 (c.802G>A [p.Val268Met]), which do not affect a known functional domain (Figure 6), we transfected mutated V5-tagged constructs into C2C12 cells and observed that two of them (c.740G>A [p.Gly247Glu] and c.802G>A [p.Val268Met]) altered the ER localization, potentially preventing normal enzymatic function (Figure 4). The missense mutation (c.875G>C [p.Arg292Pro]) showed no obvious changes in localization compared to wild-type (Figure 4), but this amino acid is highly conserved in vertebrates (Figure 6), so it is likely to be of functional importance. To determine whether mutations in B3GALNT2 led to a reduction in the level of functional α-DG glycosylation, we assessed the muscle and fibroblasts of P1 and P2 by muscle immunohistochemistry (Figure 2), flow cytometry, and immunoblotting (Figures 3 and S2). Although each technique revealed some degree of hypoglycosylation, the fact that flow cytometry and immunoblotting of the fibroblasts indicated a more severe depletion compared to the immunoblot of muscle protein lysate may be related to tissue-specific glycosylation of α-DG.5,6,46
Figure 6.
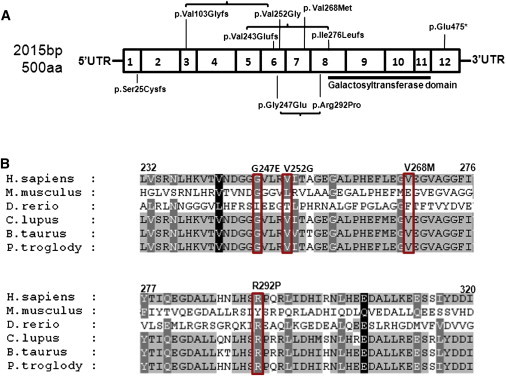
Schematic Diagram of B3GALNT2 Exons, Mutations, and Alignment
(A) B3GALNT2 is 57,249 bp in length and contains 12 exons that are transcribed into a 4,720 bp RNA transcript (NM_152490.2). The coding sequence of B3GALNT2 is 2,015 bp long and contains 500 amino acids. This schematic diagram indicates the location of the known galactosyltransferase domain (black line indicating position in the protein, amino acids 307–457) as well as the location of the mutations found in affected individuals. These mutations are listed in Table 2.
(B) Alignment of a section of the protein sequence of B3GALNT2 in six species (Homo sapiens, Pan troglodytes, Mus musculus, Danio rerio, Canis lupus, Bos taurus) shows that the missense mutations identified in affected individuals (c.740G>A [p.Gly247Glu] and c.875G>C [p.Arg292Pro] in P1, c.755T>G [p.Val252Gly] in P4, c.802G>A [p.Val268Met] in P6) are well conserved across these species. This suggests structural or functional importance for these residues. Protein alignments were performed with NCBI Homologene software.
Knockdown of zebrafish b3galnt2 recapitulated aspects of the human phenotype including dystrophic muscle, hydrocephalus, and reduced functional α-DG glycosylation. The myotomal lesions revealed by EBD (Figure 5) are very similar to the phenotype caused by knockdown of other known dystroglycanopathy genes including FKTN and FKRP.43
α-DG is heavily O-mannosylated in its mucin-like domain.1 These mannose residues can themselves be modified. For example, a β1,2 linked N-acetyl glucosamine (GlcNAc) group can be added by POMGNT138 or a β1,4 linked GlcNAc group can be added by an unknown GlcNAc transferase.38 We speculate that after the addition of β1,4 linked GlcNAc, B3GALNT2 completes the trisaccharide GalNAc-b1,3-GlcNAc-b1,4-Man.38,39 This trisaccharide is believed to contain a LARGE-dependent 6-O-phosphoryl modification on the mannose residue38 and is required for ligand binding glycan of α-DG.37 There may be other protein targets of B3GALNT2, which are currently unknown.37 We propose that the phosphorylated trisaccharide is produced in the endoplasmic reticulum, as suggested by the subcellular localization of B3GALNT2. Although O-mannosylation occurs in the ER and O-GalNAcylation in the Golgi, it has been recently suggested that certain GalNAc glycosyltransferases can be located in the ER,47 which appears to be the case for B3GALNT2.
Although further study is needed to establish the precise role of B3GALNT2 in the glycosylation of α-DG, our data show that mutations in B3GALNT2 can cause dystroglycanopathy. Furthermore, this study provides insight into the complexity of α-DG glycoslyation and illustrates the utility of whole-exome sequencing with functional analysis for the identification of mutations that cause dystroglycanopathies. Additional genetic discoveries will help to complete the understanding of α-DG glycosylation as well as elucidate the pathological mechanisms underlying the muscle and brain phenotypes associated with these diseases.
Acknowledgments
We are grateful to the UK10K consortium for making this study possible. We wish to thank the following funding bodies: the UK National Specialised Commissioned Team funding for the Congenital Muscular Dystrophies and Congenital Myopathy service, the Great Ormond Street Children’s Charity and the GOSH Biomedical Research Centre (F.M.), the Paul D. Wellstone Muscular Dystrophy Cooperative Research Centre Grant (1U54NS053672; K.P.C., T.W., and F.M.), and the Medical Research Council (MRC) Neuromuscular Centre (F.M.). E.S. is a PhD student supported by the MRC, Great Ormond Street Children’s Charity, and the Child Health Research Appeal Trust (CHRAT) (F.M.). K.J.C. is a PhD student supported by the Wellcome Trust. A.R.F. is a Clinical Research Fellow supported by the Muscular Dystrophy Campaign (F.M.). M.C.M. was supported by a Development Grant from the Muscular Dystrophy Association and by the William Randolph Hearst Fund and is currently the recipient of a Junior Faculty Career Development Award from the Manton Centre for Orphan Disease Research and a K99/R00 Transition to Independence award from the NIH (NICHD, K99HD067379). Sequencing at Boston Children’s Hospital was supported by the Intellectual and Developmental Disabilities Research Centres (CHB DDRC, P30HD19655). Sequencing at the Broad Institute was supported by a grant from NIH and the American Recovery & Reinvestment Act (NIMH RC2MH089952). K.P.C. and C.A.W. are Investigators of the Howard Hughes Medical Institute. We are very grateful to A. Eddaoudi and his team for their excellent support of the flow cytometry core facility at Great Ormond Street Hospital.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
HomoloGene (NCBI), http://www.ncbi.nlm.nih.gov/homologene
Human Genome Variation Society, http://www.hgvs.org/mutnomen/
ImageJ, http://rsbweb.nih.gov/ij/
Mutalyzer, https://mutalyzer.nl/index
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UK10K Consortium, http://www.uk10k.org/
ZFIN, http://zfin.org
References
- 1.Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., Slaughter C.A., Sernett S.W., Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 4.Talts J.F., Andac Z., Göhring W., Brancaccio A., Timpl R. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 1999;18:863–870. doi: 10.1093/emboj/18.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Durbeej M., Larsson E., Ibraghimov-Beskrovnaya O., Roberds S.L., Campbell K.P., Ekblom P. Non-muscle alpha-dystroglycan is involved in epithelial development. J. Cell Biol. 1995;130:79–91. doi: 10.1083/jcb.130.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durbeej M., Henry M.D., Ferletta M., Campbell K.P., Ekblom P. Distribution of dystroglycan in normal adult mouse tissues. J. Histochem. Cytochem. 1998;46:449–457. doi: 10.1177/002215549804600404. [DOI] [PubMed] [Google Scholar]
- 7.Durbeej M., Henry M.D., Campbell K.P. Dystroglycan in development and disease. Curr. Opin. Cell Biol. 1998;10:594–601. doi: 10.1016/s0955-0674(98)80034-3. [DOI] [PubMed] [Google Scholar]
- 8.Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A., Celli J., van Beusekom E., van der Zwaag B., Kayserili H., Merlini L., Chitayat D., Dobyns W.B. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Reeuwijk J., Janssen M., van den Elzen C., Beltran-Valero de Bernabé D., Sabatelli P., Merlini L., Boon M., Scheffer H., Brockington M., Muntoni F. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida A., Kobayashi K., Manya H., Taniguchi K., Kano H., Mizuno M., Inazu T., Mitsuhashi H., Takahashi S., Takeuchi M. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 12.Brockington M., Blake D.J., Prandini P., Brown S.C., Torelli S., Benson M.A., Ponting C.P., Estournet B., Romero N.B., Mercuri E. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Longman C., Brockington M., Torelli S., Jimenez-Mallebrera C., Kennedy C., Khalil N., Feng L., Saran R.K., Voit T., Merlini L. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum. Mol. Genet. 2003;12:2853–2861. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- 14.Barone R., Aiello C., Race V., Morava E., Foulquier F., Riemersma M., Passarelli C., Concolino D., Carella M., Santorelli F. DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann. Neurol. 2012;72:550–558. doi: 10.1002/ana.23632. [DOI] [PubMed] [Google Scholar]
- 15.Lefeber D.J., Schönberger J., Morava E., Guillard M., Huyben K.M., Verrijp K., Grafakou O., Evangeliou A., Preijers F.W., Manta P. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am. J. Hum. Genet. 2009;85:76–86. doi: 10.1016/j.ajhg.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefeber D.J., de Brouwer A.P., Morava E., Riemersma M., Schuurs-Hoeijmakers J.H., Absmanner B., Verrijp K., van den Akker W.M., Huijben K., Steenbergen G. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet. 2011;7:e1002427. doi: 10.1371/journal.pgen.1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willer T., Lee H., Lommel M., Yoshida-Moriguchi T., de Bernabe D.B., Venzke D., Cirak S., Schachter H., Vajsar J., Voit T. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 2012;44:575–580. doi: 10.1038/ng.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cirak S., Foley A.R., Herrmann R., Willer T., Yau S., Stevens E., Torelli S., Brodd L., Kamynina A., Vondracek P., UK10K Consortium ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain. 2013;136:269–281. doi: 10.1093/brain/aws312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roscioli T., Kamsteeg E.J., Buysse K., Maystadt I., van Reeuwijk J., van den Elzen C., van Beusekom E., Riemersma M., Pfundt R., Vissers L.E. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 2012;44:581–585. doi: 10.1038/ng.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manzini M.C., Tambunan D.E., Hill R.S., Yu T.W., Maynard T.M., Heinzen E.L., Shianna K.V., Stevens C.R., Partlow J.N., Barry B.J. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am. J. Hum. Genet. 2012;91:541–547. doi: 10.1016/j.ajhg.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vuillaumier-Barrot S., Bouchet-Séraphin C., Chelbi M., Devisme L., Quentin S., Gazal S., Laquerrière A., Fallet-Bianco C., Loget P., Odent S. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am. J. Hum. Genet. 2012;91:1135–1143. doi: 10.1016/j.ajhg.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez-Mallebrera C., Torelli S., Feng L., Kim J., Godfrey C., Clement E., Mein R., Abbs S., Brown S.C., Campbell K.P. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol. 2009;19:596–611. doi: 10.1111/j.1750-3639.2008.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Reeuwijk J., Brunner H.G., van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin. Genet. 2005;67:281–289. doi: 10.1111/j.1399-0004.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- 24.Brockington M., Yuva Y., Prandini P., Brown S.C., Torelli S., Benson M.A., Herrmann R., Anderson L.V., Bashir R., Burgunder J.M. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- 25.Godfrey C., Clement E., Mein R., Brockington M., Smith J., Talim B., Straub V., Robb S., Quinlivan R., Feng L. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 26.Bouchet C., Gonzales M., Vuillaumier-Barrot S., Devisme L., Lebizec C., Alanio E., Bazin A., Bessières-Grattagliano B., Bigi N., Blanchet P. Molecular heterogeneity in fetal forms of type II lissencephaly. Hum. Mutat. 2007;28:1020–1027. doi: 10.1002/humu.20561. [DOI] [PubMed] [Google Scholar]
- 27.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubowitz V., Sewry C.A., Oldfors A. Fourth Edition. SAUNDERS Elsevier; Oxford, UK: 2012. Muscle Biopsy: A Practical Approach. [Google Scholar]
- 29.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 30.Michele D.E., Barresi R., Kanagawa M., Saito F., Cohn R.D., Satz J.S., Dollar J., Nishino I., Kelley R.I., Somer H. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 31.Rojek J.M., Campbell K.P., Oldstone M.B., Kunz S. Old World arenavirus infection interferes with the expression of functional alpha-dystroglycan in the host cell. Mol. Biol. Cell. 2007;18:4493–4507. doi: 10.1091/mbc.E07-04-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brockington M., Torelli S., Sharp P.S., Liu K., Cirak S., Brown S.C., Wells D.J., Muntoni F. Transgenic overexpression of LARGE induces α-dystroglycan hyperglycosylation in skeletal and cardiac muscle. PLoS ONE. 2010;5:e14434. doi: 10.1371/journal.pone.0014434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper S.T., Lo H.P., North K.N. Single section Western blot: improving the molecular diagnosis of the muscular dystrophies. Neurology. 2003;61:93–97. doi: 10.1212/01.wnl.0000069460.53438.38. [DOI] [PubMed] [Google Scholar]
- 34.Malicki J., Jo H., Wei X., Hsiung M., Pujic Z. Analysis of gene function in the zebrafish retina. Methods. 2002;28:427–438. doi: 10.1016/s1046-2023(02)00262-1. [DOI] [PubMed] [Google Scholar]
- 35.Parsons M.J., Campos I., Hirst E.M., Stemple D.L. Removal of dystroglycan causes severe muscular dystrophy in zebrafish embryos. Development. 2002;129:3505–3512. doi: 10.1242/dev.129.14.3505. [DOI] [PubMed] [Google Scholar]
- 36.Robu M.E., Larson J.D., Nasevicius A., Beiraghi S., Brenner C., Farber S.A., Ekker S.C. p53 activation by knockdown technologies. PLoS Genet. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hiruma T., Togayachi A., Okamura K., Sato T., Kikuchi N., Kwon Y.D., Nakamura A., Fujimura K., Gotoh M., Tachibana K. A novel human β1,3-N-acetylgalactosaminyltransferase that synthesizes a unique carbohydrate structure, GalNAcbeta1-3GlcNAc. J. Biol. Chem. 2004;279:14087–14095. doi: 10.1074/jbc.M310614200. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida-Moriguchi T., Yu L., Stalnaker S.H., Davis S., Kunz S., Madson M., Oldstone M.B., Schachter H., Wells L., Campbell K.P. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison R., Hitchen P.G., Panico M., Morris H.R., Mekhaiel D., Pleass R.J., Dell A., Hewitt J.E., Haslam S.M. Glycoproteomic characterization of recombinant mouse α-dystroglycan. Glycobiology. 2012;22:662–675. doi: 10.1093/glycob/cws002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobyns W.B., Pagon R.A., Armstrong D., Curry C.J., Greenberg F., Grix A., Holmes L.B., Laxova R., Michels V.V., Robinow M. Diagnostic criteria for Walker-Warburg syndrome. Am. J. Med. Genet. 1989;32:195–210. doi: 10.1002/ajmg.1320320213. [DOI] [PubMed] [Google Scholar]
- 41.Cormand B., Pihko H., Bayés M., Valanne L., Santavuori P., Talim B., Gershoni-Baruch R., Ahmad A., van Bokhoven H., Brunner H.G. Clinical and genetic distinction between Walker-Warburg syndrome and muscle-eye-brain disease. Neurology. 2001;56:1059–1069. doi: 10.1212/wnl.56.8.1059. [DOI] [PubMed] [Google Scholar]
- 42.Gerety S.S., Wilkinson D.G. Morpholino artifacts provide pitfalls and reveal a novel role for pro-apoptotic genes in hindbrain boundary development. Dev. Biol. 2011;350:279–289. doi: 10.1016/j.ydbio.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Y.Y., White R.J., Torelli S., Cirak S., Muntoni F., Stemple D.L. Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum. Mol. Genet. 2011;20:1763–1775. doi: 10.1093/hmg/ddr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bassett D.I., Bryson-Richardson R.J., Daggett D.F., Gautier P., Keenan D.G., Currie P.D. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development. 2003;130:5851–5860. doi: 10.1242/dev.00799. [DOI] [PubMed] [Google Scholar]
- 45.Godfrey C., Foley A.R., Clement E., Muntoni F. Dystroglycanopathies: coming into focus. Curr. Opin. Genet. Dev. 2011;21:278–285. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Herzog C., Has C., Franzke C.W., Echtermeyer F.G., Schlötzer-Schrehardt U., Kröger S., Gustafsson E., Fässler R., Bruckner-Tuderman L. Dystroglycan in skin and cutaneous cells: beta-subunit is shed from the cell surface. J. Invest. Dermatol. 2004;122:1372–1380. doi: 10.1111/j.0022-202X.2004.22605.x. [DOI] [PubMed] [Google Scholar]
- 47.Gill D.J., Chia J., Senewiratne J., Bard F. Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J. Cell Biol. 2010;189:843–858. doi: 10.1083/jcb.201003055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.