

Abstract
Charcot-Marie-Tooth disease (CMT) is a heterogeneous group of inherited neuropathies. Mutations in approximately 45 genes have been identified as being associated with CMT. Nevertheless, the genetic etiologies of at least 30% of CMTs have yet to be elucidated. Using a genome-wide linkage study, we previously mapped a dominant intermediate CMT to chromosomal region 3q28–q29. Subsequent exome sequencing of two affected first cousins revealed heterozygous mutation c.158G>A (p.Gly53Asp) in GNB4, encoding guanine-nucleotide-binding protein subunit beta-4 (Gβ4), to cosegregate with the CMT phenotype in the family. Further analysis of GNB4 in an additional 88 unrelated CMT individuals uncovered another de novo mutation, c.265A>G (p.Lys89Glu), in this gene in one individual. Immunohistochemistry studies revealed that Gβ4 was abundant in the axons and Schwann cells of peripheral nerves and that expression of Gβ4 was significantly reduced in the sural nerve of the two individuals carrying the c.158G>A (p.Gly53Asp) mutation. In vitro studies demonstrated that both the p.Gly53Asp and p.Lys89Glu altered proteins impaired bradykinin-induced G-protein-coupled-receptor (GPCR) signaling, which was facilitated by the wild-type Gβ4. This study identifies GNB4 mutations as a cause of CMT and highlights the importance of Gβ4-related GPCR signaling in peripheral-nerve function in humans.
Main Text
Charcot-Marie-Tooth disease (CMT) is a group of inherited neuropathies characterized by progressive distal muscle atrophy, distal sensory loss, depressed tendon reflexes, and foot deformities; they can be categorized into either the demyelinating form or the axonal form by their electrophysiological and pathological features.1,2 Clinically, median motor nerve conduction velocity (NCV) is the most frequent parameter (with a cutoff value of 38 m/s) used for distinguishing between demyelinating and axonal CMT.3 Mutations in approximately 45 genes have been identified as being associated with CMT, but these account for fewer than 70% of all CMT cases.1,2 Within the CMTs, dominant intermediate CMT (DI-CMT) is a rare group that shows autosomal-dominant inheritance and variable NCVs, which can look like either demyelinating or axonal types of CMT within a pedigree.3–6 Previously, we characterized a three-generation Chinese family afflicted with a DI-CMT and mapped the locus of the candidate gene to chromosomal region 3q28–q29.7 In this study, 13 family members, including six affected individuals, five unaffected individuals, and two married-in spouses, were enrolled (Figure 1A). Disease status was determined by both the clinical manifestations and electrophysiological evidence.3 In brief, the age of onset ranged from 10 to 45 years, and individuals ranged in clinical severity from being asymptomatic to being wheelchair-bound. The median motor NCVs ranged from 16.5 to 45.7 m/s. Men tended to be more severely affected (Table 1). Sural nerve biopsies of two affected individuals detected loss of myelinated fibers and also demyelinating changes as evidenced by multiple onion-bulb formations. Written informed consent was obtained from all subjects, and the study protocol was approved by the institutional review board at Taipei Veterans General Hospital, Taipei, Taiwan. Genomic DNA was isolated from peripheral-blood leukocytes according to standard protocol.
Figure 1.

Pedigrees and Electropherograms of the GNB4 Mutations
(A and B) Pedigree structure of families 1 (A) and 2 (B). The probands are denoted by an arrow. The squares and circles denote males and females, respectively, and the diagonal black lines represent deceased individuals. Filled symbols represent affected members with neuropathy, and open symbols indicate unaffected individuals. The numbers in diamonds indicate the number of unaffected siblings or children. The gender and birth order have been partially hidden for the sake of confidentiality. “M” represents the GNB4 mutant allele, “W” stands for the wild-type allele, and asterisks denote members who underwent whole-exome sequencing.
(C) The electropherograms of the heterozygous GNB4 mutations, c.158G>A (p.Gly53Asp) and c.265A>G (p.Lys89Glu).
(D) RT-PCR analysis of the RNA isolated from the leukocytes of a normal control and the individual carrying GNB4 c.265A>G with the use of primers specific to GNB4 exons 5 and 6. Both samples revealed a single band at the same location (218 bp), indicating that GNB4 mRNA from the affected individual was not differentially spliced.
(E) The cDNA electropherograms of the GNB4 exon 5 and 6 junction of a normal control and the affected individual carrying GNB4 c.265A>G reveal similar splicing patterns.
Table 1.
Clinical Features of the Individuals with GNB4 Mutations
|
Family 1 (c.158G>A [p.Gly53Asp]) |
Family 2 (c.265A>G [p.Lys89Glu]) |
||||||
|---|---|---|---|---|---|---|---|
| III-2 | III-3 | IV-1 | IV-2 | IV-3 | IV-4 | II-1 | |
| Age of onset (years) | 13 | 45 | 10 | 13 | 17 | 22 | 5 |
| Age at exam (years) | 49 | 48 | 23 | 18 | 17 | 22 | 9 |
| Gender | male | female | male | male | female | female | female |
| Upper-limb weakness (MRC) | distal: 4 | normal | normal | normal | normal | normal | normal |
| Lower-limb weakness (MRC) | distal: 2 | distal: 4- to 4 | distal: 4- to 4 | distal: 4 | distal: 4+ | distal: 4+ | distal: 4 |
| Upper-limb muscle atrophy | distal: moderate | normal | distal: mild | normal | normal | normal | normal |
| Lower-limb muscle atrophy | distal: severe | distal: mild | distal: severe | distal: mild | distal: mild | distal: mild | distal: moderate |
| Knee and ankle DTRs | absent | absent | absent | absent | absent | reduced or absent | absent |
| Gait | bound in wheelchair | steppage | steppage | steppage | normal | normal | steppage |
| Pinprick sensation | reduced | mildly reduced | mildly reduced | mildly reduced | normal | normal | mildly reduced |
| Vibration | reduced | mildly reduced | mildly reduced | mildly reduced | normal | normal | mildly reduced |
| Median nerve MNCV (m/s) | 25.2 | 35.4 | 16.5 | 28.6 | 44.2 | 45.7 | 20 |
| Median nerve cMAP (mV) | 7.7 | 8.3 | 3.9 | 3.2 | 9.3 | 5.9 | 2.6 |
| Sural nerve SNAP | NR | NR | NR | NR | NR | 7 | NR |
| Sural nerve biopsy | demyelination | demyelination | ND | ND | ND | ND | ND |
The following abbreviations are used: MRC, Medical Research Council scale; DTR, deep tendon reflex; MNCV, motor nerve conduction velocity; cMAP, compound motor action potential; SNAP, sensory nerve action potential; NR, not recordable; and ND, not done.
About 165,000 exonic regions from the genomic DNA of the two affected first cousins of family 1 (IV-1 and IV-4 in Figure 1A) were captured and enriched with the Agilent SureSelect Human All Exon v.2 kit. The DNA-sequencing libraries were prepared with an Illumina Paired-End DNA Sample Preparation kit. A total of 100 bp paired-end reads were obtained by sequencing on an Illumina Genome Analyzer GAIIx. All sequence reads were mapped to the human reference genome (GRCh37 patch 2) with the Burrows-Wheeler Aligner.8 Removal of PCR duplicates and variant calling were performed with SAMtools/BCFTools.9 Only the variants with Phred-like scores of at least 30.0 were collected for the investigation of candidate variants of high confidence. On average, 6 Gb of sequence was generated from each individual, resulting in a coverage depth of 83× per base per targeted region. After analysis (Table S1, available online), 64 heterozygous coding variants were found in both individuals; these variants were not present in dbSNP, the 1000 Genomes Project,10 or the in-house exome data from ten non-CMT ethnic controls. Targeted sequencing of eleven other members of family 1 revealed that only the c.158G>A (p.Gly53Asp) variant in GNB4 (MIM 610863; RefSeq accession number NM_021629.3), encoding guanine-nucleotide-binding protein (G protein) subunit beta-4 (Gβ4), perfectly segregated with the CMT phenotype (Figures 1A and 1C). This variant was also located within the chromosomal region 3q28-q29, which was the previously mapped locus for this type of CMT.
To support that mutations in GNB4 cause CMT and to investigate the frequency of GNB4 mutations in our CMT population, we further sequenced all nine coding exons of GNB4 in another 88 unrelated individuals with molecularly unassigned CMT (Table S2); these individuals included 66 with the demyelinating variant and 22 with the axonal variant. Among them, one additional de novo mutation, c.265A>G (p.Lys89Glu) (Figures 1B and 1C) in the heterozygous form, was identified. Neither the GNB4 c.158G>A mutation nor the c.265A>G mutation was present in the Exome Variant Server of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project or in 1,920 ethnically matched control chromosomes. Gly53 and Lys89 are highly conserved residues in Gβ4 (Figure S1). SNPs&GO,11 Panther,12 and Polyphen213 predicted both mutations to be deleterious to Gβ4 functioning14 (Table S3). Because the c.265A>G mutation occurs at the nucleotide adjacent to the exon-intron boundary (Figure 1C), we analyzed the mRNA from leukocytes of the individual carrying this mutation by RT-PCR and nucleotide sequencing. No splicing alteration was found (Figures 1D and 1E). We also utilized a minigene assay to demonstrate that the GNB4 c.265A>G mutation did not change the mRNA splicing pattern (Figure S2). Because the CMT individuals in this study were selected from 251 pedigrees affected by CMT after the cases with clear genetic diagnosis were excluded,15 the frequency of GNB4 mutations in our CMT population was approximately 0.8% (2/251).
The GNB4 c.265A>G (p.Lys89Glu) mutation was found in a 9-year-old girl who had presented with slowly progressive weakness of her distal lower limbs since the age of 5 years. She had started to walk at a normal age. Physical examination at age 9 years revealed high-arched feet, hammer toes, atrophy and weakness of the intrinsic muscles of the feet (score 4−/5 on the Medical Research Council Scale), impaired dorsiflexion of the feet (score 4/5), generalized areflexia, and mildly diminished sensation for all modalities in regions distal to the ankles despite a lack of sensory complaints. Nerve conduction studies demonstrated the presence of demyelinating sensorimotor polyneuropathy with axonal loss. The median and tibial motor NCVs were 20 and 15 m/s, respectively. Neither parent had the CMT phenotype or the GNB4 c.265A>G mutation (Figure 1B).
G proteins are heterotrimeric and are composed of α, β, and γ subunits; they are important to cellular signal transduction. They transmit extracellular signals into cells by acting in concert with different G-protein-coupled receptors (GPCRs); these GPCRs can be activated by many different signal factors, such as neurotransmitters, hormones, and cytokines.16 In humans, there are 16 Gα subunits, 5 Gβ subunits, 12 Gγ subunits, and a number of splice variants, which thus contribute to a large diversity of G protein heterotrimers.17 In the inactive state, the guanosine-diphosphate (GDP)-bound Gα is associated with the Gβγ dimer. When a GPCR is activated by a ligand, it catalyzes the G protein to exchange the GDP for guanosine triphosphate (GTP) on Gα, which results in the dissociation of the Gα from the Gβγ.16 The Gα-GTP and released Gβγ dimer each then transduce signals to a wide range of effectors, including adenylyl cyclases, phospholipases, ion channels, phosphodiesterases, and many more proteins,17,18 thereby regulating various intracellular signaling pathways and cellular functions. As a result of this wide range of G protein signaling activities, GNB4, which encodes Gβ4, is likely to be important to a number of cellular signal-transduction systems.
To understand the location of Gβ4 in peripheral nerves, we analyzed the sural nerves of one neurologically normal control and one individual (III:3 in Figure 1A) with the c.158G>A (p.Gly53Asp) GNB4 mutation by double immunofluorescence staining. Gβ4 was found to be colocalized with neurofilament heavy chain and S100 (Figure 2 and Table S4), indicating that Gβ4 is expressed in both axons and Schwann cells. Intriguingly, some myelinated fibers of the normal control had a target-like appearance because of the presence of dense Gβ4 staining in the axons and the outer rim of the myelin, but not in the compacted myelin sheath (Figure 2). Onion-bulb formations in the sural nerve of individual III:3 showed a “rosette” pattern with Gβ4 staining in the axons and the cytoplasm of surrounding Schwann cells (Figure 2).
Figure 2.

Gβ4 Expression in Both Axons and Schwann Cells of Peripheral Nerves
Double immunostaining of Gβ4 with neurofilament heavy chain (NF-H) (A) or S100 (B) with sural nerve samples from a neurologically normal control and family 1 individual III:3, who harbors the GNB4 c.158G>A (p.Gly53Asp) mutation. The colocalization of Gβ4 with NF-H and S100 indicates that Gβ4 is expressed in both axons and Schwann cells. Some myelinated fibers of the normal control have a target-like appearance as a result of dense Gβ4 staining in the axons and the outer rim of the myelin, but not in the compacted myelin sheath (arrows). Onion-bulb formations in the sural nerve of individual III:3 have a “rosette” pattern with Gβ4 staining in the axons and the cytoplasm of surrounding Schwann cells (arrowheads). The scale bars represent 10 um.
We next compared Gβ4 expression in the sural nerve samples from two individuals (III:2 and III:3) with the GNB4 c.158G>A (p.Gly53Asp) mutation, one individual with CMT1A, and one neurologically normal control by immunohistochemistry (IHC). In addition to staining Gβ4, we also doubly stained the cell nuclei with TDP-43 antibody as a reference (Table S4). Immunostaining revealed that Gβ4 was abundantly expressed in the axons and Schwann cells of the sural nerve of the normal control (Figure 3A). Some myelinated fibers had a target-like appearance (Figure 3A). Onion-bulb formations in the sural nerve of the CMT1A control were also densely stained with Gβ4 antibody (Figure 3B). In contrast, Gβ4 staining was significantly weaker in the sural nerves of the two individuals with the GNB4 mutation than in those of the normal control and the individual with CMT1A (Figures 3C and 3D).
Figure 3.

Gβ4 Immunohistochemistry and Toluidine-Blue Staining of Sural Nerves
Double immunostaining of Gβ4 (red) and TDP-43 (dark blue) in sural nerve samples from a neurologically normal control (A), a disease control with CMT type 1A (CMT1A) (B), and family 1 individuals III:2 (C) and III:3 (D), who harbor the GNB4 c.158G>A (p.Gly53Asp) mutation. Some myelinated fibers in the normal control feature a target-like appearance (A, arrows). The Gβ4 staining of onion-bulb formations appears weaker in the two subjects with the GNB4 mutation (C and D, arrowheads) than in the individual with CMT1A (B, arrowheads). The sural nerves of the two individuals with the GNB4 mutation have obviously weaker Gβ4 staining than do those in the normal control and the individual with CMT1A. The cell nuclei were stained with TDP-43 antibody as a reference. The toluidine-blue staining of sural nerve samples of individuals III:2 and III:3 from family 1 reveals a loss of myelinated fibers and a presence of multiple onion-bulb formations (C and D), associated with a demyelinating change. The scale bars represent 10 um.
To test the hypothesis that GNB4 is important for peripheral nerve functioning, we investigated Gβ4 expression in the sciatic nerves of the rats 3 days after sciatic nerve transection, nerve conditioning with a 20 s transient crush injury, or a sham operation. The nerve tissue 10 mm in length and distal to the injured site was analyzed for Gβ4 expression by immunoblotting and IHC. Compared with Gβ4 expression in the sham-operated group, the expression of Gβ4 appeared to increase in the nerve conditioning group and decrease in the nerve transection group (Figure S3). These findings suggest that GNB4 plays a role in peripheral nerve regeneration and support its importance in peripheral nerve functioning.
To investigate the effect of the GNB4 mutations on protein stability and G-protein-coupled signaling by Gβ4, we constructed Gβ4 expression plasmids to allow further in vitro analysis. The coding region of GNB4 from a human GNB4 cDNA clone (Invitrogen; GenBank accession number AF300648.1) was subcloned into pcDNA3.1+ (Invitrogen). Mutations c.158G>A (p.Gly53Asp) and c.265A>G (p.Lys89Glu) were introduced by site-directed mutagenesis according to the Quick-Change method (Stratagene) (Table S5). pECFP-C1 was purchased from Clontech. A human PLCβ2 expression clone, pCMV5-PLCβ2, was a kind gift from E.M. Ross (Southwestern Medical Center, University of Texas, TX, USA). All constructs were verified by DNA sequencing.
In order to carry out a protein-stability assay, we incubated human embryonic kidney (HEK) 293T cells maintained in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone) containing 10% fetal bovine serum (FBS; Invitrogen) at 37°C and in 5% CO2 and transfected them with plasmids expressing wild-type, p.Gly53Asp, or p.Lys89Glu forms of Gβ4 by using calcium-phosphate precipitation.19 At 48 hr after transfection, the cells were treated with 100 μg/ml cyclohexamide for 0, 2, 4, 8, or 24 hr. Total-protein cell extracts were analyzed by immunoblotting (40 μg of protein was loaded from each cell lysate). The immunoblot signals were used for comparing the relative degradation profiles between the Gβ4 variants and actin. Representative graphs and quantification obtained from six independent experiments are shown in Figure S4. These results show that the degradation rates of the two altered forms of Gβ4 were not significantly different from that of their wild-type counterpart, suggesting that these alterations did not affect protein stability.
We next investigated the functional impact of the Gβ4 alterations on GPCR signaling. In this functional study, we used bradykinin to activate the B2-receptor-coupled Gqα signaling pathway in a PLCβ2-overexpressing COS-7 cell model.20,21 Activation of PLCβ2 by Gqα signaling leads to hydrolysis of phosphatidylinositol 4,5-bisphosphate and the formation of the second messengers inositol trisphosphate (IP3) and diacylglycerol, which is followed by IP3-induced intracellular Ca2+ release. Thus, IP3 production and cytosolic calcium changes were used to reflect GPCR signaling activity. In brief, COS-7 cells were maintained in DMEM containing 10% FBS at 37°C and in 5% CO2; LipofectAmine-2000 (Invitrogen) was used for transfecting these cells at a 1:1:1 ratio with plasmids expressing ECFP, expressing PLCβ2, and expressing wild-type, p.Gly53Asp, or p.Lys89Glu forms of Gβ4 or empty vectors. Forty-eight hours after transfection, the cells were harvested for subsequent second-messenger analysis. To measure IP3 production, we subcultured the cells into a 96-well plate at a density of 2 × 104 cells per well with 20 μl PBS. The cells were then stimulated with 100 nM bradykinin for 1 min and lysed with 0.2 N perchloric acid. Intracellular IP3 production was then measured with a HitHunter IP3 Fluorescence Polarization Assay Kit (DiscoveRx) as outlined in the manufacturer’s manual. To measure cytosolic calcium changes, we incubated cells on coverslips with 5 μM of the calcium indicator dye fura-2 AM (Molecular Probes) in loading buffer (LB) for 45 min at 37°C and then washed them with LB. Cells coexpressing ECFP with a similar expression level (mean pixel intensity = 526.6 ± 32.8 AU; n = 148) were selected for the subsequent measurements. These cells were stimulated with a pulse of 100 nM bradykinin for 10 s while the fluorescence of fura-2 was simultaneously monitored under an inverted microscope (IX-71, Olympus Optical) coupled with a monochromator (Polychrome IV, TILL Photonics, Gräfelfing, Germany) with a 40× oil-immersion objective (UApo/340 40×/1.35 numeric aperture oil objective, Olympus Optical).22 Use of the 340/380 nm excitation ratio with fura-2 allowed measurements of the intracellular calcium concentration ([Ca2+]i).23,24
The study revealed that the function of both the p.Gly53Asp and p.Lys89Glu altered forms of Gβ4 was impaired, that is, they showed reduced bradykinin-induced PLCβ2 activation, namely an inhibition of IP3 production and a lower increase in cytosolic calcium. Compared to control cells, PLCβ2-overexpressing cells showed a bradykinin-induced increase in IP3 production by 25% (25.7% ± 6.2%, n = 3). When cells coexpressed wild-type Gβ4 and PLCβ2, their IP3 generation was found to be about 3.5-fold higher than that of the PLCβ2-overexpressing cells (87.9 ± 11.1% versus 25.7% ± 6.2%). However, compared to the control, the p.Gly53Asp and p.Lys89Glu altered forms of Gβ4 reduced bradykinin-induced IP3 production by 42.1% ± 2.8% and 45.7% ± 6.0%, respectively (Figure 4A). Similar results were observed when the bradykinin-induced [Ca2+]i increases were monitored. Bradykinin induced a similar increase in [Ca2+]i when the controls, the PLCβ2-overexpressing cells, and the cells coexpressing PLCβ2 and wild-type Gβ4 were compared. However, when the cells coexpressing either PLCβ2 and p.Gly53Asp Gβ4 or PLCβ2 and p.Lys89Glu Gβ4 were examined, there were no significant changes in [Ca2+]i upon bradykinin stimulation (Figures 4B and 4C). These findings indicate that the mutations in GNB4 cause a defect in the GPCR signaling cascade and impair the activation of PLCβ2.
Figure 4.
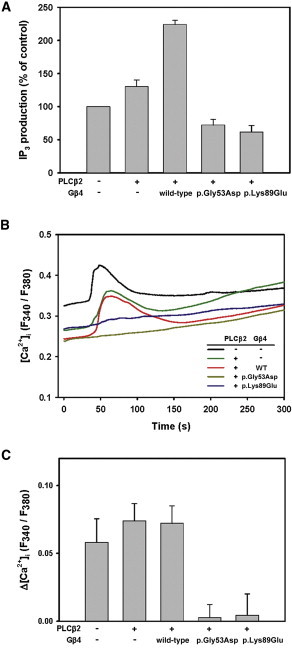
Inhibition of the GPCR Signaling Results from GNB4 Mutations
(A) The p.Gly53Asp and p.Lys89Glu altered forms of Gβ4 had an inhibition of inositol trisphosphate (IP3) production upon bradykinin stimulation. COS-7 cells were transfected with pcDNA3.1 (control, first bar on the left), pcDNA3.1-PLCβ2 alone, or pcDNA3.1-PLCβ2 and pcDNA3.1-Gβ4 or altered forms of Gβ4. The IP3 generated upon stimulation with 100 nM bradykinin for 1 min was then measured. The IP3 production level was normalized against the control as 100%. The data shown are the mean ± SEM from three independent transfections.
(B) Absence of changes in intracellular calcium ([Ca2+]i) upon bradykinin stimulation in the cells expressing altered forms of Gβ4. COS-7 cells were transfected with pcDNA3.1 (control), pcDNA3.1-PLCβ2 alone, or pcDNA3.1-PLCβ2 and pcDNA3.1-Gβ4 or altered forms of Gβ4 in conjunction with pECFP-C1. Changes in the F340/F380 ratio of fura-2 upon 100 nM bradykinin stimulation were monitored by a fluorescence microscope. Bradykinin stimulation started at 30 s and lasted for 10 s. The [Ca2+]i changes are shown as the F340/F380 ratio of fura-2 fluorescence.
(C) Maximal [Ca2+]i changes upon bradykinin stimulation. The maximal [Ca2+]i changes were obtained by subtraction of the averaged fluorescence ratio in the 10 s before stimulation from the maximal fluorescence ratio. The data shown are the mean ± SEM from 26–34 cells for each group (148 cells in total). The cells were obtained from at least three independent transfections.
Utilizing exome sequencing, bioinformatic analysis, and mutational analysis, we have identified two mutations that affect Gβ4-encoding GNB4 and are present in subjects with CMT. Both GNB4 mutations alter the evolutionarily conserved amino acid residues, are absent from the NHLBI Exome Variant Server, and do not occur in 1,920 ethnic control chromosomes. Immunohistochemical studies revealed that Gβ4 is abundantly expressed in peripheral nerves, including both axons and Schwann cells. In vitro studies demonstrated that both GNB4 mutations impair bradykinin-induced GPCR signaling. Our findings provide strong genetic and functional evidence supporting that mutations in GNB4 are the cause of this type of CMT.
According to the tissue expression databases of UniGene25 and GeneNote (now retired),26 Gβ4 is widely expressed in many human tissues, including the spinal cord and peripheral nerves. Gβ4 has also been found to be expressed in peripheral nerve myelin by proteome analyses.27 We have been able to consistently demonstrate that Gβ4 is readily detected in the axons and Schwann cells of sural nerves, which indicates that the Gβ4 signal pathway is essential for peripheral nerve functioning. Decreased Gβ4 immunostaining in the sural nerves of the two CMT individuals with GNB4 mutations further highlights the important role of Gβ4 dysfunction in the pathogenesis of this hereditary neuropathy.
Our functional studies have shown that both the p.Gly53Asp and p.Lys89Glu Gβ4 alterations impair GPCR signaling pathways via a dominant-negative effect. When compared to cells expressing PLCβ2 alone, cells coexpressing PLCβ2 and p.Gly53Asp or p.Lys89Glu altered Gβ4 showed a significantly reduced IP3 production and an absence of cytosolic calcium change when stimulated with bradykinin. This phenomenon suggests that both altered forms of Gβ4 have a negative effect on the endogenous wild-type Gβ4 molecules already present in the cells and that this affects GPCR signaling. Gly53 and Lys89 are highly conserved across the Gβ protein family (including Gβ1–Gβ5) across the various kingdoms (including mammals, fungi, nematode, and amoeba). This suggests that they both most likely play critical roles in GPCR signaling.28
The functional defect caused by the two mutations appears to be related to their roles in the specific binding of Gα to the Gβγ dimer. On the basis of the three-dimensional (3D) structure of the native heterotrimer, Giα1β1γ2, the first two WD40 repeats of Gβ1 interact with the N-terminal helix of Giα1, and it has been suggested that this interaction confers a certain degree of specificity to Gα-Gβγ complex formation.29 Gβ4 shares 96% amino acid sequence homology with Gβ1.30 Gly53 and Lys89 are located within the first and second WD40 repeats, respectively, of the β propeller, and according to high-resolution structure analysis, Gly53 contacts one residue and Lys89 contacts multiple residues within the N-terminal helix of Giα1 (Figure S5).29 Therefore, it is likely that the interaction between Gβ4 and Gα, but not the interaction between Gβ4 and Gγ, is affected in both the p.Gly53Asp and p.Lys89Glu Gβ4 altered proteins. Although the dimerization of Gβ and Gγ is obligatory in nature,31 the interaction between Gγ and the altered forms of Gβ4 might render a decrease in the functional wild-type Gβ4-Gγ dimer and thus exert a dominant-negative effect that abolishes relevant GPCR signaling.
All of the individuals carrying the GNB4 c.158G>A (p.Gly53Asp) or c.265A>G (p.Lys89Glu) mutation feature a typical CMT phenotype that involves distal motor and sensory function; however, the subject carrying the c.265A>G (p.Lys89Glu) mutation had an earlier age of onset, indicating that she was more severely affected. The c.265A>G (p.Lys89Glu) mutation seemed to affect Gβ4 functioning more than the c.158G>A (p.Gly53Asp) mutation in our small subject group. This is supported by the 3D structure of the heterotrimeric Giα1β1γ2. Whereas Gly53 of Gβ1 only interacts with Leu23 of Giα via van der Waals interactions, Lys89 interacts with multiple residues, namely Giα via hydrogen bond with Ser16, via a salt bridge with Asp20, and via van der Waals interactions with Ile19 and Leu23 (Figure S5).29 Thus, Lys89 might be more important than Gly53 in terms of the association between subunits Gα and Gβ, and this might therefore cause the c.265A>G (p.Lys89Glu) mutation to produce a more severe disease phenotype.
The diagnosis of DI-CMT in family 1 was based on the NCVs and the sural nerve biopsies of subjects III:2 and III:3, who had slow NCVs consistent mainly with demyelinating neuropathy. Because NCV cannot faithfully reflect the underlying pathological changes, the full pathological spectrum of Gβ4-related neuropathy is yet to be delineated. Given that Gβ4 is highly expressed in both axons and Schwann cells, mutations in GNB4 might result in myelinopathy and/or axonopathy; this might partially explain the phenotypic heterogeneity of the family 1 individuals, in whom variable NCVs that blurred the division between demyelinating and axonal CMT were documented.
In the family carrying the GNB4 c.158G>A (p.Gly53Asp) mutation, men tended to be more severely affected than women. If this proves to be true in a larger cohort, it will imply that the Gβ4 signal-transduction pathway might be influenced by gender-preferential genes in the peripheral nerve tissues. One recent study uncovered 159 genes that show male or female preference when expressed in the brain.32 Our study suggests the interesting possibility of identifying genes with sex-preferential expression in peripheral nerves.
The fact that no significant difference in protein stability exists between wild-type and altered forms of Gβ4 suggests that the pathomechanism of the GNB4 mutations is not due to a haploinsufficiency of the Gβ4 molecules. This finding is consistent with the possible dominant-negative mechanism. Because the stability of Gβ4 is not affected by the GNB4 mutations, the reduced Gβ4 expression in the sural nerves of the individuals with GNB4 mutation suggests that the altered Gβ4 might be toxic. If the Gβ4-related GPCR signaling function is vital to some cells, the dominant-negative effect of the altered Gβ4 could be lethal or toxic to them. Alternatively, the altered Gβ4 might have gained a toxic function that is unrelated to the dominant-negative effect. Such a toxic function could result in a situation where cells expressing the most altered form of Gβ4 are likely to die first. In such a circumstance, the remaining cells in the sural nerves of these individuals would be those with relatively lower Gβ4 expression. This speculation is supported by the lower cell and nerve-fiber densities in the sural nerve samples of the two individuals with a GNB4 mutation than in those in the control samples (Figure 3).
We have also screened for mutations in the genes encoding the Gγ subunits that are known to be expressed in the peripheral nerves; these genes included GNG3 (MIM 608941), GNG4 (MIM 604388), GNG7 (MIM 604430), GNG8, and GNG10 (MIM 604389) across 87 molecularly unassigned and unrelated CMT individuals. So far, we have not identified any relevant mutation in them (data not shown). The possibility that other proteins that interact with Gβ4 might also be pivotal to the etiology of some of these hereditary neuropathies cannot be ruled out.
In conclusion, our findings demonstrate that mutations in GNB4 cause CMT, and this emphasizes the importance of Gβ4-related GPCR signaling to peripheral nerve functioning in humans.
Acknowledgments
The authors thank the individuals who participated in this study, as well as E.M. Ross of the University of Texas for kindly providing a human PLCβ2 expression clone, pCMV5-PLCβ2. This work was supported by research grants from the National Science Council, Taiwan, ROC (NSC97-2314-B-075-064-MY3, NSC98-2320-B-010-029-MY3, NSC99-2314-B-010-013-MY3, and NSC100-2314-B-075-020-MY2), the Taiwan Ministry of Education Aim for the Top University Program (V100-E6-006, V101E7-005, and V102E9-006), the Brain Research Center, National Yang-Ming University, and the High-throughput Genome Analysis Core Facility and Bioinformatics Consortium of Taiwan of the National Core Facility Program for Biotechnology, Taiwan (NSC100-2319-B-010-001 and NSC100-2319-B-010-002).
Contributor Information
Lung-Sen Kao, Email: lskao@ym.edu.tw.
Yi-Chung Lee, Email: ycli@vghtpe.gov.tw.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
GRCh37 patch 2, ftp://ftp.ensembl.org/pub/release-61/
NHLBI Exome Variant Server Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Panther, http://www.pantherdb.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SAMtools, http://samtools.sourceforge.net/
UniGene, http://www.ncbi.nlm.nih.gov/unigene/
References
- 1.Murphy S.M., Laura M., Fawcett K., Pandraud A., Liu Y.T., Davidson G.L., Rossor A.M., Polke J.M., Castleman V., Manji H. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry. 2012;83:706–710. doi: 10.1136/jnnp-2012-302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saporta A.S., Sottile S.L., Miller L.J., Feely S.M., Siskind C.E., Shy M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011;69:22–33. doi: 10.1002/ana.22166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harding A.E., Thomas P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103:259–280. doi: 10.1093/brain/103.2.259. [DOI] [PubMed] [Google Scholar]
- 4.Davis C.J., Bradley W.G., Madrid R. The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies. I. Clinical, genetic and electrophysiological findings and classification. J. Genet. Hum. 1978;26:311–349. [PubMed] [Google Scholar]
- 5.Verhoeven K., Villanova M., Rossi A., Malandrini A., De Jonghe P., Timmerman V. Localization of the gene for the intermediate form of Charcot-Marie-Tooth to chromosome 10q24.1-q25.1. Am. J. Hum. Genet. 2001;69:889–894. doi: 10.1086/323742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Züchner S., Noureddine M., Kennerson M., Verhoeven K., Claeys K., De Jonghe P., Merory J., Oliveira S.A., Speer M.C., Stenger J.E. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat. Genet. 2005;37:289–294. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]
- 7.Lee Y.C., Lee T.C., Lin K.P., Lin M.W., Chang M.H., Soong B.W. Clinical characterization and genetic analysis of a possible novel type of dominant Charcot-Marie-Tooth disease. Neuromuscul. Disord. 2010;20:534–539. doi: 10.1016/j.nmd.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calabrese R., Capriotti E., Fariselli P., Martelli P.L., Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 12.Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thusberg J., Olatubosun A., Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011;32:358–368. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- 15.Lin K.P., Soong B.W., Yang C.C., Huang L.W., Chang M.H., Lee I.H., Antonellis A., Lee Y.C. The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS ONE. 2011;6:e29393. doi: 10.1371/journal.pone.0029393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilman A.G. G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 17.Oldham W.M., Hamm H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 18.Dupré D.J., Robitaille M., Rebois R.V., Hébert T.E. The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu. Rev. Pharmacol. Toxicol. 2009;49:31–56. doi: 10.1146/annurev-pharmtox-061008-103038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi G., Kleinklaus A.K., Marrion N.V., Trimmer J.S. Properties of Kv2.1 K+ channels expressed in transfected mammalian cells. J. Biol. Chem. 1994;269:23204–23211. [PubMed] [Google Scholar]
- 20.Offermanns S., Simon M.I. G alpha 15 and G alpha 16 couple a wide variety of receptors to phospholipase C. J. Biol. Chem. 1995;270:15175–15180. doi: 10.1074/jbc.270.25.15175. [DOI] [PubMed] [Google Scholar]
- 21.Rosskopf D., Nikula C., Manthey I., Joisten M., Frey U., Kohnen S., Siffert W. The human G protein beta4 subunit: gene structure, expression, Ggamma and effector interaction. FEBS Lett. 2003;544:27–32. doi: 10.1016/s0014-5793(03)00441-1. [DOI] [PubMed] [Google Scholar]
- 22.Grynkiewicz G., Poenie M., Tsien R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 23.Pan C.Y., Tsai L.L., Jiang J.H., Chen L.W., Kao L.S. The co-presence of Na+/Ca2+-K+ exchanger and Na+/Ca2+ exchanger in bovine adrenal chromaffin cells. J. Neurochem. 2008;107:658–667. doi: 10.1111/j.1471-4159.2008.05637.x. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y.C., Fann M.J., Chang W.H., Tai L.H., Jiang J.H., Kao L.S. Regulation of sodium-calcium exchanger activity by creatine kinase under energy-compromised conditions. J. Biol. Chem. 2010;285:28275–28285. doi: 10.1074/jbc.M110.141424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheeler D.L., Church D.M., Federhen S., Lash A.E., Madden T.L., Pontius J.U., Schuler G.D., Schriml L.M., Sequeira E., Tatusova T.A., Wagner L. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yanai I., Benjamin H., Shmoish M., Chalifa-Caspi V., Shklar M., Ophir R., Bar-Even A., Horn-Saban S., Safran M., Domany E. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- 27.Patzig J., Jahn O., Tenzer S., Wichert S.P., de Monasterio-Schrader P., Rosfa S., Kuharev J., Yan K., Bormuth I., Bremer J. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J. Neurosci. 2011;31:16369–16386. doi: 10.1523/JNEUROSCI.4016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaudet R., Bohm A., Sigler P.B. Crystal structure at 2.4 angstroms resolution of the complex of transducin betagamma and its regulator, phosducin. Cell. 1996;87:577–588. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]
- 29.Wall M.A., Posner B.A., Sprang S.R. Structural basis of activity and subunit recognition in G protein heterotrimers. Structure. 1998;6:1169–1183. doi: 10.1016/s0969-2126(98)00117-8. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz-Velasco V., Ikeda S.R., Puhl H.L. Cloning, tissue distribution, and functional expression of the human G protein beta 4-subunit. Physiol. Genomics. 2002;8:41–50. doi: 10.1152/physiolgenomics.00085.2001. [DOI] [PubMed] [Google Scholar]
- 31.Higgins J.B., Casey P.J. In vitro processing of recombinant G protein gamma subunits. Requirements for assembly of an active beta gamma complex. J. Biol. Chem. 1994;269:9067–9073. [PubMed] [Google Scholar]
- 32.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.