

Abstract
Ohdo syndrome comprises a heterogeneous group of disorders characterized by intellectual disability (ID) and typical facial features, including blepharophimosis. Clinically, these blepharophimosis-ID syndromes have been classified in five distinct subgroups, including the Maat-Kievit-Brunner (MKB) type, which, in contrast to the others, is characterized by X-linked inheritance and facial coarsening at older age. We performed exome sequencing in two families, each with two affected males with Ohdo syndrome MKB type. In the two families, MED12 missense mutations (c.3443G>A [p.Arg1148His] or c.3493T>C [p.Ser1165Pro]) segregating with the phenotype were identified. Upon subsequent analysis of an additional cohort of nine simplex male individuals with Ohdo syndrome, one additional de novo missense change (c.5185C>A [p.His1729Asn]) in MED12 was detected. The occurrence of three different hemizygous missense mutations in three unrelated families affected by Ohdo syndrome MKB type shows that mutations in MED12 are the underlying cause of this X-linked form of Ohdo syndrome. Together with the recently described KAT6B mutations resulting in Ohdo syndrome Say/Barber/Biesecker/Young/Simpson type, our findings point to aberrant chromatin modification as being central to the pathogenesis of Ohdo syndrome.
Main Text
Ohdo syndrome (MIM 249620) comprises a heterogeneous group of disorders characterized by intellectual disability (ID) and typical facial features, including blepharophimosis, ptosis, a round face with a characteristic nose, and a narrow mouth.1 These blepharophimosis-ID syndromes have been classified in five distinct subgroups. The first group can be distinguished from the others because it is caused by deletions of the short arm of chromosome 3. The second group is designated as Ohdo type on the basis of the original report by Ohdo et al.1,2 These persons present with typical features of prognathism, short philtrum, and proteinuria, whereas hypotonia, abnormal growth, and limb defects are lacking. The Verloes type is a more severe condition with severe microcephaly, epilepsy, brain malformations, adducted thumbs, and abnormal genitals.1 The most clinically distinctive phenotype is the Say/Barber/Biesecker/Young/Simpson (SBBYS) type (MIM 603736), which is characterized by striking facial dysmorphisms that include a large to bulbous nasal tip; small and/or dysplastic, thick, simple, or overfolded pinnae; thick swollen cheeks; and retrognathia. In addition, hypotonia, hyperextensible joints, cryptorchidism, and a wide range of congenital malformations are present.1,3 This type was recently shown to be caused by mutations in lysine acetyltransferase 6B (KAT6B [MIM 605880]).4 Here, we report that the fifth distinct subtype of Ohdo syndrome, the Maat-Kievit-Brunner (MKB) type, which, in contrast to the other types, is characterized by X-linked inheritance and facial coarsening with thick alae nasi and a triangular face at older age,1 is caused by mutations in MED12.
Exome sequencing was performed in two individuals from two families affected by Ohdo syndrome MKB type (Figure 1 and Table S1, available online). This study was approved by the institutional review board Commissie Mensgebonden Onderzoek Regio Arnhem-Nijmegen. Written informed consent was obtained for all individuals participating in this study. The first family showed a clear X-linked inheritance pattern with two affected males in two generations and has been reported previously.1,10 The second family comprised two affected brothers. In both families, 250k NspI SNP array analysis (Affymetrix, Santa Clara, CA, USA) revealed no significant chromosome aberrations. Analysis of the X-chromosome-inactivation status via methylation-sensitive PCR and fragment-length analysis of the androgen-receptor CAG repeat polymorphism11 showed a nonrandom pattern of X inactivation in individual II:2 from family 1 and individual I:2 from family 2 (in >90% of the cells, the alleles submitted to the sons were inactive). In family 2, mutation analysis of FOXL2 (MIM 605597) did not show any abnormalities. For family 1, DNA was isolated from a transformed lymphoblastoid cell line of the nephew because both he and his uncle were deceased; for family 2, DNA was isolated from peripheral blood of the proband according to standard procedures. Exome enrichment was performed with the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed on the SOLiD 5500xl sequencer (Life Technologies, Carlsbad, CA, USA). Reads were aligned to the UCSC Genome Browser hg19 reference genome with the use of Life Technologies LifeScope software version 2.1. Ninety-two percent of the X chromosomal exons were covered at least three times on average in both individuals. Variants were annotated with a custom pipeline.12 Only nonsynonymous changes in the coding regions or changes affecting the canonical splice sites were analyzed. Variants present in dbSNP132 or in our in-house database containing data of 368 exomes were excluded. Assuming a hemizygous X chromosomal change, we only considered variants that were present in at least 70% of the reads on the X chromosome (Table S2). After this selection, four and five variants remained in families 1 and 2, respectively (Tables S2 and S3). Only one gene showed a private nonsynonymous variant in both families; this gene was mediator complex subunit 12 (MED12 [MIM 300188]; RefSeq accession number NM_005120.2), which encodes mediator of RNA polymerase II transcription subunit 12. Each individual had a missense mutation: c.3443G>A (p.Arg1148His) or c.3493T>C (p.Ser1165Pro). Sanger sequencing confirmed the presence of the mutation and showed segregation of the mutation with the disorder in the families (Figure 1). We subsequently performed Sanger sequencing of all 45 coding exons of MED12 on DNA from nine simplex male persons with the clinical diagnosis of Ohdo syndrome. Primer sequences and conditions are available upon request. In this cohort, we detected one additional MED12 missense mutation (c.5185C>A [p.His1729Asn]), which was shown to be de novo, in a person with the MKB type. All three missense mutations in MED12 affect evolutionary highly conserved amino acids and were predicted to be damaging to protein function (Table 1). The amino acid change p.His1729Asn is situated within the PQL domain of MED12, and the other two amino acid changes, p.Arg1148His and p.Ser1165Pro, are not situated in a known domain. The occurrence of three different hemizygous missense mutations in three unrelated families affected by Ohdo syndrome MKB type shows that mutations in MED12 are the underlying cause of this X-linked form of Ohdo syndrome. Of note, a second segregating change was identified in family 2: c.4638A>C (p.Lys1546Asn) in alpha-thalassemia/mental retardation syndrome X-linked (ATRX [MIM 300032]; RefSeq NM_000489.3), mutations in which can cause ATRX syndrome (MIM 301040) (Table S3). This change is not present in any of the available databases. It was predicted to be detrimental to protein function by PolyPhen-2 and to be neutral according to SNPs&GO.13,15 For the proband, hematological analysis including high-performance liquid chromatography and capillary electrophoresis16,17 showed no indication of alpha-thalassemia. In addition, HbH inclusions were absent in over 2,000 cells (data not shown). This does not support pathogenicity of the mutation, although it does not exclude ATRX syndrome because HbH inclusions are not seen in 12% of persons with ATRX syndrome.18 In combination with the facial dysmorphisms that differ from those of ATRX syndrome and overlap with those of the other families affected by MED12 mutations (Figure 1), this strongly suggests that the mutation in MED12 is the main cause of the phenotype in family 2. However, we cannot exclude an additional effect of the ATRX mutation on the family 2 phenotype, which is at the relatively severe end of the spectrum associated with MED12 mutations.
Figure 1.
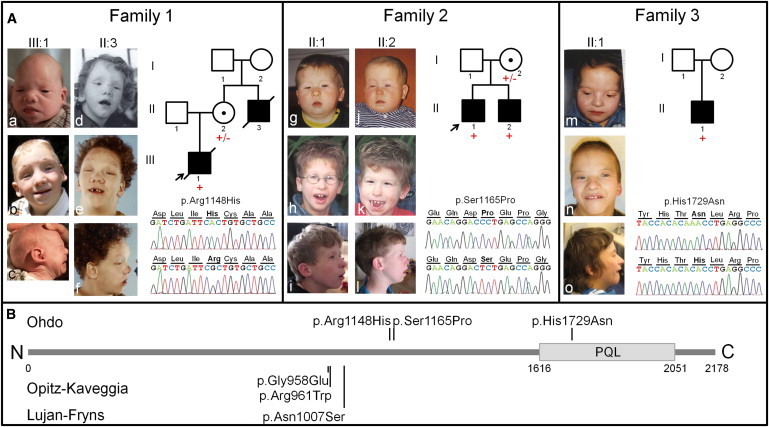
Overview of Clinical and Genetic Data of Families Affected by Ohdo Syndrome MKB Type and MED12 Mutations
(A) Photographs of individuals with MED12 mutations: family 1 proband (III-1) in infancy (Aa and Ac) and at age 4 years (Ab) and family 1 uncle (II-3) at age 4 years (Ad) and 18 years (Ae and Af); family 2 proband (II-1) at ages 1 year (Ag), 9 years (Ah), and 15 years (Ai) and family 2 brother (II-2) at ages 1 month (Aj), 7 years (Ak), and 13 years (Al); and family 3 proband (II-1) at ages 3 years (Am), 10 years (An), and 16 years (Ao). Beside the photographs, the electropherograms show the mutation in bold of an affected individual (top) and a control (bottom), and the pedigrees show results of segregation analysis. Mutant alleles are represented by a plus sign (+), and wild-type alleles are represented by a minus sign (–).
(B) Schematic overview of MED12, including the PQL domain, which is involved in β-catenin and GLI3 binding,5,6 shows previously published amino acid changes leading to Opitz-Kaveggia syndrome7,8 and Lujan-Fryns syndrome9 and the presently identified amino acid changes leading to Ohdo syndrome MKB type.
Table 1.
MED12 Mutations Causing X-linked ID
| Chromosome Positiona | cDNA change (RefSeq NM_005120.2) | Protein change (RefSeq NP_005111.2) | PhyloPb | PolyPhen-213 | SIFT14 | Reference |
|---|---|---|---|---|---|---|
| Ohdo Syndrome Maat-Kievit-Brunner (MKB) Type | ||||||
| chrX: 70,348,536 | c.3443G>A | p.Arg1148His | 5.0 | probably damaging | deleterious | family 1 |
| chrX: 70,348,981 | c.3493T>C | p.Ser1165Pro | 4.3 | probably damaging | deleterious | family 2 |
| chrX: 70,356,290 | c.5185C>A | p.His1729Asn | 5.4 | probably damaging | deleterious | family 3 |
| Opitz-Kaveggia Syndrome | ||||||
| chrX: 70,347,217 | c.2881C>T | p.Arg961Trp | 5.6 | probably damaging | deleterious | Risheg7 |
| chrX: 70,347,209 | c.2873G>A | p.Gly958Glu | 2.8 | probably damaging | deleterious | Rump8 |
| Lujan-Fryns Syndrome | ||||||
| chrX: 70,347,781 | c.3020A>G | p.Asn1007Ser | 3.1 | probably damaging | deleterious | Schwartz9 |
UCSC Genome Browser hg19.
Based on 46 vertebrate species.
Specific mutations at different positions in MED12 have previously been reported in Opitz-Kaveggia syndrome (or FG syndrome) (MIM 305450) and Lujan-Fryns syndrome (MIM 309520).7–9 Currently, ten families affected by Opitz-Kaveggia syndrome and the recurrent amino acid substitution p.Arg961Trp19 and one family affected by a p.Gly958Glu change8 have been described, whereas the p.Asn1007Ser substitution has been described in two families affected by Lujan-Fryns syndrome.9 Clinically, clear differences can be seen between Ohdo syndrome and these two syndromes (Figure S1). In contrast to the facial features associated with Opitz-Kaveggia and Lujan-Fryns syndrome, ptosis, blepharophimosis, a bulbous nasal tip, a long philtrum, and maxillar hypoplasia with full cheeks are unique to Ohdo syndrome. Moreover, dental anomalies and hearing loss are frequent, whereas anal malformations, agenesis of the corpus callosum, and macrocephaly are not present in the individuals with Ohdo syndrome. Overlapping characteristics are the following relatively nonspecific features: ID, hypotonia, behavioral problems, and some facial dysmorphisms, mainly hypertelorism, micrognathia, small low-set ears, and a high forehead, especially in family 3.
MED12 is a component of the multisubunit RNA polymerase II transcriptional Mediator.20 This complex is involved in transcriptional regulation by conveying information from gene-specific regulatory elements to the RNA polymerase II transcription machinery.21 Mediator consists of four distinct modules.22,23 The head, middle, and tail modules together form the core Mediator complex that interacts directly with RNA polymerase II and both general and gene-specific transcription factors.24 MED12, along with MED13, CCNC, and CDK8, composes a fourth “kinase” module that reversibly associates with the core complex.25 This module was primarily thought to be involved in transcriptional repression through blocking the binding of the core Mediator complex to RNA polymerase II because Mediator containing this module is less active than Mediator lacking this module.23,26 However, the kinase module was recently also found to be involved in transcriptional activation.27 Through MED12, the kinase module modulates, for example, Wnt/β-catenin signaling5 and Gli3-dependent sonic hedgehog signaling.6,28 In addition, MED12 is involved in epigenetic silencing of neuronal gene expression imposed by the RE1-silencing transcription factor REST.29 In this regard, we previously showed that the MED12 interface in Mediator links REST with euchromatic histone-lysine N-methyltransferase 2 (EHMT2) in epigenetic repression of neuronal genes in nonneuronal cells.29 Importantly, we also found that the MED12 mutations identified in Opitz-Kaveggia and Lujan-Fryns syndrome disrupted REST-imposed extraneuronal gene silencing by impairing REST-directed recruitment of Mediator to RE1-silencing elements.29
To explore the pathogenicity of the MED12 missense mutations identified in Ohdo syndrome, we monitored the impact of two of the three identified mutations on repression of REST target genes. To this end, we comparatively examined siRNA-resistant wild-type (WT) FLAG-tagged MED12 (FLAG-MED12) and its corresponding p.Arg1148His and p.Ser1165Pro mutant derivatives for their respective abilities to suppress enhanced REST-target-gene expression triggered by RNAi-mediated depletion of endogenous MED12 in HEK293 cells (Figure 2). As expected, MED12 knockdown triggered derepression of REST target genes, including cholinergic receptor, muscarinic 4 (CHRM4 [MIM 118495]), synaptosomal-associated protein, 25 kDa (SNAP25 [MIM 600322]), and synapsin I (SYN1 [MIM 313440]). Introduction of WT FLAG-MED12 in these cells reversed this effect; in contrast, the p.Arg1148His and p.Ser1165Pro mutants were significantly compromised in this ability (Figure 2A). This indicates that the MED12 mutations in Ohdo syndrome impair the repressive function of MED12. Neither amino acid change deleteriously affected the incorporation of MED12 into Mediator (Figure 2C) or its direct interaction with G9a (Figure 2D), indicating that the Ohdo syndrome MED12 mutations do not disrupt the function of MED12 as a stable G9a interface in Mediator. It is thus possible that the Ohdo syndrome mutations in MED12 impair recruitment of Mediator to RE1-bound REST, as was shown previously for the Opitz-Kaveggia and Lujan-Fryns mutations in MED12.29 Impaired Mediator recruitment to RE1 elements could explain how these amino acid substitutions disrupt REST-imposed epigenetic restrictions on neuronal gene expression, given that MED12 and/or Mediator is essential to link RE1-bound REST with EHMT2.
Figure 2.
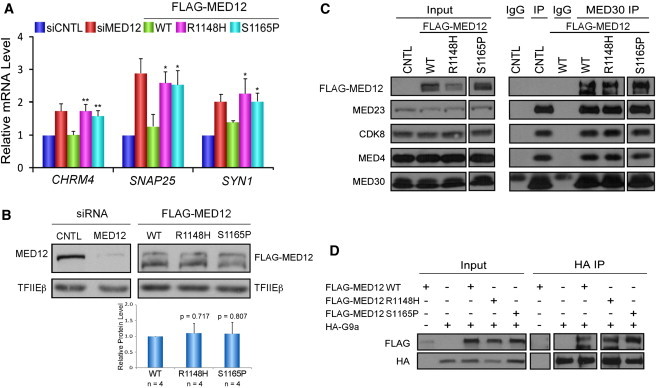
Ohdo Syndrome Mutations in MED12 Disrupt Its Gene-Repression Function
(A) HEK293 cells were transfected with control (siCNTL) or MED12-specific (siMED12) siRNAs. Where indicated (FLAG-MED12), MED12-knockdown cells were transfected with FLAG-tagged siRNA-resistant WT, p.Arg1148His (R1148H), or p.Ser1165Pro (S1165P) MED12 expression plasmids. RNA-expression levels of CHRM4, SNAP25, or SYN1 were determined by quantitative RT-PCR. mRNA levels are expressed relative to mRNA levels in control siRNA-transfected cells. Data represent the mean ± the SEM of at least three independent experiments performed in duplicate. Asterisks denote statistically significant differences compared to WT FLAG-MED12 (Student’s t test, ∗p < 0.05, ∗∗p < 0.01). FLAG-tagged MED12 mutants p.Arg1148His and p.Ser1165Pro were unable to repress CHRM4, SNAP25, and SYN1 expression.
(B) Nuclear extracts5 from a representative transient expression assay were resolved by SDS-PAGE and processed by immunoblot analysis with antibodies specific to MED12, the FLAG epitope on FLAG-MED12 derivatives, or the TFIIEβ that was used as an internal loading control. Representative immunoblots show that siMED12 significantly diminished expression of MED12. Cotransfection with FLAG-MED12 restored MED12 expression for the WT and both mutants. For the doublet bands, the band with the higher molecular mass was previously shown to represent full-length MED12.28 The relative levels of ectopically expressed FLAG-MED12 WT and mutant proteins averaged over four independent experiments are given below the immunoblots and were calculated first by normalization of FLAG-MED12 immunoblot signals to internal-control TFIIEβ signals (within the linear range of detection; Figure S2) and subsequent division of the normalized expression levels of FLAG-MED12 mutants by those of FLAG-MED12 WT. Immunoblot signals were quantified with ImagQuant TL software. No statistically significant differences in expression levels were observed (Student’s t test; p values are given above the bars).
(C) Nuclear extracts from untransfected (CNTL) or transfected HEK293 cells transiently expressing FLAG epitope-tagged WT, p.Arg1148His, or p.Ser1165Pro MED12 derivatives were subjected to immunoprecipitation (IP) with IgG or antibodies specific to MED30, as indicated. Mediator immunoprecipitates were resolved by SDS-PAGE and processed by immunoblot analysis with antibodies specific to the FLAG epitope or the indicated Mediator subunits. Input corresponds to 10% of the nuclear extracts subjected to IP. FLAG-tagged MED12 mutants p.Arg1148His and p.Ser1165Pro were incorporated into Mediator comparably to WT MED12.
(D) HA-tagged G9a was expressed without or with FLAG-tagged MED12 WT, p.Arg1148His, or p.Ser1165Pro derivatives in HEK293 cells prior to the processing of nuclear extracts by IP with antibodies specific to the HA epitope. Immunoprecipitates were resolved by SDS-PAGE and processed by immunoblot analysis with FLAG- or HA-specific antibodies, as indicated. Input corresponds to 10% of the nuclear extracts subjected to IP. FLAG-tagged MED12 mutants p.Arg1148His and p.Ser1165Pro bound to G9a comparably to WT MED12.
It is of particular interest that MED12 is implicated in chromatin modification through H3K9 methylation, given that Ohdo syndrome SBBYS type has recently been shown to be caused by mutations in KAT6B, a component of the MOZ/MORF complex that has histone H3 acetyltransferase activity.30 Apart from their role in Ohdo syndrome, both MED12 and KAT6B have also been implicated in tumor development. In acute myeloid leukemia, translocations leading to a fusion gene of KAT6B and opsin 1 (cone pigments) long-wave-sensitive have been identified,31 and MED12 mutations have recently been identified in prostate cancer.32 With that in mind, it is noteworthy that one of our family members died of prostate cancer at the age of 25 years. Moreover, both genes are implicated in uterine leiomyomata. KAT6B was previously mapped to the breakpoint of recurrent 10q22 aberrations,33 which are present in 2% of uterine leiomyomata,34 and exon 2 of MED12 is mutated in 70% of uterine leiomyomata.35 For uterine leiomyomata, involvement of other genes has been suggested, but only a few have been identified.36 Therefore, the observation that both MED12 and KAT6B are chromatin-modifying enzymes implicated in Ohdo syndrome and the development of uterine leiomyomata and other tumors strongly suggests that both genes are functionally related.
Ohdo syndrome, especially SBBYS type, is generally considered a de novo dominant disorder with a low recurrence risk. Our study shows that Ohdo syndrome can also be an X-linked disorder. Moreover, by screening only nine simplex male individuals with Ohdo syndrome, we identified one additional pathogenic MED12 mutation. Although Ohdo syndrome MKB type can be distinguished from Ohdo syndrome SBBYS type at older age through the triangular face and the increasingly bulbous nose with thick alae nasi, its facial phenotype can be very similar to that of Ohdo syndrome SBBYS type at a young age. Therefore, we recommend sequencing of MED12 in all young male individuals with Ohdo syndrome because this can have great implications for the recurrence risk in the respective families.
In conclusion, we show that mutations in MED12 cause X-linked Ohdo syndrome. The identification of this gene in Ohdo syndrome, together with KAT6B, points to aberrant chromatin modification as central to the pathogenesis of Ohdo syndrome.
Acknowledgments
We are grateful to the families for their participation. We thank the Genomic Disorders Group Nijmegen for the technical support in performing the exome-sequencing experiments. This work was supported by the European Commission (AnEUploidy Project grant 037627 under FP6 to A.T.V.v.S., B.B.A.d.V., H.G.B., and A.H. and GENCODYS grant 241995 under FP7 to A.T.V.v.S. and B.B.A.d.V.), the Netherlands Organisation for Health Research and Development (ZON-MW grant 917-86-319 to B.B.A.d.V. and ZON-MW grant 917-66-363 to A.H.), the National Institutes of Health (National Institute of Mental Health grant R01MH085320 to T.G.B.), and Hersenstichting Nederland (2010(1)-30 to A.d.B.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Verloes A., Bremond-Gignac D., Isidor B., David A., Baumann C., Leroy M.A., Stevens R., Gillerot Y., Héron D., Héron B. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am. J. Med. Genet. A. 2006;140:1285–1296. doi: 10.1002/ajmg.a.31270. [DOI] [PubMed] [Google Scholar]
- 2.Ohdo S., Madokoro H., Sonoda T., Hayakawa K. Mental retardation associated with congenital heart disease, blepharophimosis, blepharoptosis, and hypoplastic teeth. J. Med. Genet. 1986;23:242–244. doi: 10.1136/jmg.23.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Day R., Beckett B., Donnai D., Fryer A., Heidenblad M., Howard P., Kerr B., Mansour S., Maye U., McKee S. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin. Genet. 2008;74:434–444. doi: 10.1111/j.1399-0004.2008.01087.x. [DOI] [PubMed] [Google Scholar]
- 4.Clayton-Smith J., O’Sullivan J., Daly S., Bhaskar S., Day R., Anderson B., Voss A.K., Thomas T., Biesecker L.G., Smith P. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim S., Xu X., Hecht A., Boyer T.G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006;281:14066–14075. doi: 10.1074/jbc.M602696200. [DOI] [PubMed] [Google Scholar]
- 6.Zhou H., Kim S., Ishii S., Boyer T.G. Mediator modulates Gli3-dependent Sonic hedgehog signaling. Mol. Cell. Biol. 2006;26:8667–8682. doi: 10.1128/MCB.00443-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Risheg H., Graham J.M., Jr., Clark R.D., Rogers R.C., Opitz J.M., Moeschler J.B., Peiffer A.P., May M., Joseph S.M., Jones J.R. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat. Genet. 2007;39:451–453. doi: 10.1038/ng1992. [DOI] [PubMed] [Google Scholar]
- 8.Rump P., Niessen R.C., Verbruggen K.T., Brouwer O.F., de Raad M., Hordijk R. A novel mutation in MED12 causes FG syndrome (Opitz-Kaveggia syndrome) Clin. Genet. 2011;79:183–188. doi: 10.1111/j.1399-0004.2010.01449.x. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz C.E., Tarpey P.S., Lubs H.A., Verloes A., May M.M., Risheg H., Friez M.J., Futreal P.A., Edkins S., Teague J. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J. Med. Genet. 2007;44:472–477. doi: 10.1136/jmg.2006.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maat-Kievit A., Brunner H.G., Maaswinkel-Mooij P. Two additional cases of the Ohdo blepharophimosis syndrome. Am. J. Med. Genet. 1993;47:901–906. doi: 10.1002/ajmg.1320470618. [DOI] [PubMed] [Google Scholar]
- 11.Spath M.A., Nillesen W.N., Smits A.P., Feuth T.B., Braat D.D., van Kessel A.G., Yntema H.G. X chromosome inactivation does not define the development of premature ovarian failure in fragile X premutation carriers. Am. J. Med. Genet. A. 2010;152A:387–393. doi: 10.1002/ajmg.a.33243. [DOI] [PubMed] [Google Scholar]
- 12.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 13.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calabrese R., Capriotti E., Fariselli P., Martelli P.L., Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 16.Dacie J.V., Lewis S.M. Churchill Livingstone; Edinburgh: 1991. Practical Haematology. [Google Scholar]
- 17.Van Delft P., Lenters E., Bakker-Verweij M., de Korte M., Baylan U., Harteveld C.L., Giordano P.C. Evaluating five dedicated automatic devices for haemoglobinopathy diagnostics in multi-ethnic populations. Int. J. Lab. Hematol. 2009;31:484–495. doi: 10.1111/j.1751-553X.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- 18.Cassidy S.B., Allanson J.E. John Wiley & Sons, Inc.; New Jersey: 2010. Management of genetic syndromes. [Google Scholar]
- 19.Clark R.D., Graham J.M., Jr., Friez M.J., Hoo J.J., Jones K.L., McKeown C., Moeschler J.B., Raymond F.L., Rogers R.C., Schwartz C.E. FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing. Genet. Med. 2009;11:769–775. doi: 10.1097/GIM.0b013e3181bd3d90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boube M., Joulia L., Cribbs D.L., Bourbon H.M. Evidence for a mediator of RNA polymerase II transcriptional regulation conserved from yeast to man. Cell. 2002;110:143–151. doi: 10.1016/s0092-8674(02)00830-9. [DOI] [PubMed] [Google Scholar]
- 21.Malik S., Roeder R.G. Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem. Sci. 2005;30:256–263. doi: 10.1016/j.tibs.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Dotson M.R., Yuan C.X., Roeder R.G., Myers L.C., Gustafsson C.M., Jiang Y.W., Li Y., Kornberg R.D., Asturias F.J. Structural organization of yeast and mammalian mediator complexes. Proc. Natl. Acad. Sci. USA. 2000;97:14307–14310. doi: 10.1073/pnas.260489497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taatjes D.J., Näär A.M., Andel F., 3rd, Nogales E., Tjian R. Structure, function, and activator-induced conformations of the CRSP coactivator. Science. 2002;295:1058–1062. doi: 10.1126/science.1065249. [DOI] [PubMed] [Google Scholar]
- 24.Conaway R.C., Conaway J.W. Origins and activity of the Mediator complex. Semin. Cell Dev. Biol. 2011;22:729–734. doi: 10.1016/j.semcdb.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borggrefe T., Davis R., Erdjument-Bromage H., Tempst P., Kornberg R.D. A complex of the Srb8, -9, -10, and -11 transcriptional regulatory proteins from yeast. J. Biol. Chem. 2002;277:44202–44207. doi: 10.1074/jbc.M207195200. [DOI] [PubMed] [Google Scholar]
- 26.Knuesel M.T., Meyer K.D., Bernecky C., Taatjes D.J. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009;23:439–451. doi: 10.1101/gad.1767009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conaway R.C., Conaway J.W. Function and regulation of the Mediator complex. Curr. Opin. Genet. Dev. 2011;21:225–230. doi: 10.1016/j.gde.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H., Spaeth J.M., Kim N.H., Xu X., Friez M.J., Schwartz C.E., Boyer T.G. MED12 mutations link intellectual disability syndromes with dysregulated GLI3-dependent Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA. 2012;109:19763–19768. doi: 10.1073/pnas.1121120109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding N., Zhou H., Esteve P.O., Chin H.G., Kim S., Xu X., Joseph S.M., Friez M.J., Schwartz C.E., Pradhan S., Boyer T.G. Mediator links epigenetic silencing of neuronal gene expression with x-linked mental retardation. Mol. Cell. 2008;31:347–359. doi: 10.1016/j.molcel.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ullah M., Pelletier N., Xiao L., Zhao S.P., Wang K., Degerny C., Tahmasebi S., Cayrou C., Doyon Y., Goh S.L. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell. Biol. 2008;28:6828–6843. doi: 10.1128/MCB.01297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X.J., Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene. 2007;26:5408–5419. doi: 10.1038/sj.onc.1210609. [DOI] [PubMed] [Google Scholar]
- 32.Barbieri C.E., Baca S.C., Lawrence M.S., Demichelis F., Blattner M., Theurillat J.P., White T.A., Stojanov P., Van Allen E., Stransky N. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore S.D., Herrick S.R., Ince T.A., Kleinman M.S., Dal Cin P., Morton C.C., Quade B.J. Uterine leiomyomata with t(10;17) disrupt the histone acetyltransferase MORF. Cancer Res. 2004;64:5570–5577. doi: 10.1158/0008-5472.CAN-04-0050. [DOI] [PubMed] [Google Scholar]
- 34.Ozisik Y.Y., Meloni A.M., Surti U., Sandberg A.A. Involvement of 10q22 in leiomyoma. Cancer Genet. Cytogenet. 1993;69:132–135. doi: 10.1016/0165-4608(93)90089-5. [DOI] [PubMed] [Google Scholar]
- 35.Mäkinen N., Mehine M., Tolvanen J., Kaasinen E., Li Y., Lehtonen H.J., Gentile M., Yan J., Enge M., Taipale M. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011;334:252–255. doi: 10.1126/science.1208930. [DOI] [PubMed] [Google Scholar]
- 36.Okolo S. Incidence, aetiology and epidemiology of uterine fibroids. Best Pract. Res. Clin. Obstet. Gynaecol. 2008;22:571–588. doi: 10.1016/j.bpobgyn.2008.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.