

Abstract
Hexanucleotide repeat expansions in C9orf72 are a major cause of frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). Understanding the disease mechanisms and a method for clinical diagnostic genotyping have been hindered because of the difficulty in estimating the expansion size. We found 96 repeat-primed PCR expansions: 85/2,974 in six neurodegenerative diseases cohorts (FTLD, ALS, Alzheimer disease, sporadic Creutzfeldt-Jakob disease, Huntington disease-like syndrome, and other nonspecific neurodegenerative disease syndromes) and 11/7,579 (0.15%) in UK 1958 birth cohort (58BC) controls. With the use of a modified Southern blot method, the estimated expansion range (smear maxima) in cases was 800–4,400. Similarly, large expansions were detected in the population controls. Differences in expansion size and morphology were detected between DNA samples from tissue and cell lines. Of those in whom repeat-primed PCR detected expansions, 68/69 were confirmed by blotting, which was specific for greater than 275 repeats. We found that morphology in the expansion smear varied among different individuals and among different brain regions in the same individual. Expansion size correlated with age at clinical onset but did not differ between diagnostic groups. Evidence of instability of repeat size in control families, as well as neighboring SNP and microsatellite analyses, support multiple expansion events on the same haplotype background. Our method of estimating the size of large expansions has potential clinical utility. C9orf72-related disease might mimic several neurodegenerative disorders and, with potentially 90,000 carriers in the United Kingdom, is more common than previously realized.
Introduction
Large expansions of a noncoding GGGGCC repeat in C9orf72 have recently been identified as a leading cause of frontotemporal lobar degeneration (FTLD [MIM 600274]), amyotrophic lateral sclerosis (ALS [MIM 612069]), and the combined syndrome, FTLD-ALS.1–3 The finding is remarkable because of the high mutation prevalence in these disease syndromes and because the nature of the mutation implies a distinct mechanism of neurodegeneration. The discovery of the causal mutation and its further investigation have been hampered by the extremely large size of the expansion, which prevents amplification of the entire expansion by conventional PCR-based methods. Southern blotting has been used for approximating the true repeat size in one small series of individuals,2 so little is known about the pathogenic size range, mutation mechanisms, feasibility and accuracy of diagnostic testing, and genotype-phenotype correlations.
DNA from the expansion allele can be amplified by PCR with primers complementary to the repeat (repeat-primed PCR [rpPCR]); however, this method cannot size accurately beyond around 30 repeats.1 In a large series screened with the rpPCR-based method, mutations were found in 8% of ALS simplex cases (39% of familial ALS) and 7% of FTLD simplex cases (25% of familial FTLD).4 The phenotype associated with the expansion might extend beyond FTLD and ALS, but it is difficult to make this conclusion before the repeat size at which the mutation becomes pathogenic in different clinical syndromes is understood. Thus, aside from FTLD and ALS cases, mutations with >32 repeats have also been reported in healthy individuals,1,5 as well as in those affected by Alzheimer disease (AD)6 and corticobasal7,8 and ataxic syndromes.8 The prominence of psychiatric and amnestic symptoms in individuals with FTLD and/or ALS is a potential factor leading to diagnostic confusion.7,9
There are also important outstanding questions about mutation mechanisms. A single ancestral mutational event explaining all or the large majority of cases has been proposed because of the high prevalence of C9orf72 expansions in Finland and because mutation cases share a haplotype on the adjoining region of chromosome 9.10 An alternative hypothesis is that multiple unrelated mutational events occurred on the same SNP haplotype background as a result of a premutation allele that confers increased risk of developing a huge expansion.4,5,10,11
Here, we sought to resolve some of the outstanding genetic issues by developing a Southern blot methodology and evaluating this method by estimating expansion size in a large proportion of cases identified from a screen of the archives of a United Kingdom (UK) national referral center. Further, we capitalized on long-term cohort studies to characterize the population prevalence of large mutations and make inferences regarding mutational mechanisms and phenotypic heterogeneity.
Material and Methods
Statistics were done with the IBM SPSS Statistics 19 package. Ethical approval was obtained from the National Hospital for Neurology and Neurosurgery Research Ethics Committee. Informed consent for genetic studies was obtained from all participants. For methods of SNP and microsatellite genotyping, see the Supplemental Data, available online.
Southern Blotting
gDNA was digested overnight with AluI (20 u) and DdeI (20 u) prior to electrophoresis. DNA was transferred to positively charged nylon membrane (Roche Applied Science) by capillary blotting and was baked at 80°C for 2 hr. The hybridization probe was an oligonucleotide from Eurofins MWG Operon (Germany) and comprised five hexanucleotide repeats (GGGGCC)5 labeled 3′ and 5′ with digoxigenin (DIG). Filter hybridization was undertaken as recommended in the DIG Application Manual (Roche Applied Science) except for the supplementation of DIG Easy Hyb buffer with 100 μg/ml denatured fragmented salmon sperm DNA. After prehybridization at 48°C for 4 hr, hybridization was allowed to proceed at 48°C overnight. A total of 1 ng of labeled oligonucleotide probe was used per milliliter of hybridization solution. Membranes were washed initially in 2× standard sodium citrate (SSC) and 0.1% sodium dodecyl sulfate (SDS) while the oven was being ramped from 48°C to 65°C and then washed in fresh solution at 65°C for 15 min; further 15 min washes in 0.5× SSC, 0.1% SDS and 0.2× SSC, and 0.1% SDS at 65°C followed. Detection of the hybridized probe DNA was carried out as recommended in the DIG Application Manual with CSPD ready-to-use (Roche Applied Science) as a chemiluminescent substrate. Signals were visualized on Fluorescent Detection Film (Roche Applied Science) after 1–5 hr. All samples were electrophoresed against DIG-labeled DNA molecular-weight markers II and VII (Roche Applied Science). Hexanucleotide repeat number was estimated by visual interpolation with a plot (created in Microsoft Excel) of log10 base-pair number against migration distance and subtraction of the wild-type allele fragment size (199 bp). Maximum, minimum, and modal sizes were recorded for each individual with expanded repeats. No signal from the pathogenic range was observed with this method in 50 rpPCR samples in which no expansion > 32 repeats was detected.
Assessment of Hexanucleotide Repeat Number
Variation in the hexanucleotide repeat number was assessed in one of two ways. First, rpPCR was carried out largely as previously described.1 Expansions with a characteristic “saw-tooth” pattern were identified and put forward for Southern blotting where sufficient DNA allowed. Peaks were counted, and the first peak represented two repeats. Peak intensity was used for identifying zygosity, where homozygous samples had no obvious step in the stutter pattern, whereas a decline of about one-half was indicative of heterozygosity. Second, fluorescent-labeled PCR was followed by fragment-length analysis on an ABI 3730xl automated sequencer. The PCR used 20 ng gDNA in FastStart PCR master mix (Roche Applied Science) supplemented with 1× Q solution (Roche Applied Science), 5% dimethyl sulphoxide, 0.2 mM 7-deaza-2-deoxy guanosine triphosphate, and 1 mM MgCl2 in a 20 μl final volume. Thermal cycling included initial denaturation for 5 min and 35 subsequent cycles of 30 s denaturation at 95°C, 30 s annealing at 60°C, and 1 min elongation at 72°C.
Samples
Individual samples for mutation screening were identified from the Medical Research Council (MRC) Prion Unit research sample database, which has recorded samples referred for clinical diagnosis or research since 1990. Other referral series included samples held at the Department of Molecular Neuroscience at the University College London Institute of Neurology (Huntington disease [HD]-like syndrome), National Hospital for Neurology and Neurosurgery (ALS), North-East London and Essex Regional MND Care Centre (ALS), and Department of Clinical Neurosciences at Cambridge University (FTLD). The MRC Prion Unit receives samples from individuals with suspected prion disease, AD, FTLD, ALS, Huntington disease, and other neurodegenerative disorders that might mimic these conditions. Some of these individuals are enrolled in systematic observational cohort studies with associated high levels of diagnostic scrutiny according to recognized criteria (for example, the National Prion Monitoring Cohort study, the University College London [UCL] FTLD DNA cohort, and the UCL familial Alzheimer disease DNA cohort), whereas other samples were referred to the MRC Prion Unit by UK neurologists for research genetic studies after diagnoses were made according to local practices. Samples from individuals thought to have HD but negative for this disease upon gene testing were derived from the archives of the UCL Department of Molecular Neuroscience after referral with suspected HD from HD expansion testing; these samples largely comprise those from individuals seen at the Huntington’s Disease Multidisciplinary Clinic, National Hospital for Neurology and Neurosurgery, Queen Square. The sample cohorts investigated were therefore heterogeneous in the use of established diagnostic criteria; after the finding of a C9orf72 expansion in a sample, clinical case notes were reviewed and original clinical diagnosis were revised if necessary. Control samples were obtained from the Fondation Jean Dausset-Centre d’Etude du Polymorphisme Humain (CEPH), the European Collection of Cell Cultures (ECACC) Human Random Control DNA Panels 1–5, and the 1958 Birth Cohort (58BC) (University of Leicester). Samples with multiple brain tissues were provided by the Queen Square Brain Bank.
Results
A total of 2,974 individuals composed six disease cohorts (FTLD, AD, ALS, sporadic Creutzfeldt-Jakob disease [sCJD] [simplex cases with CJD], HD-like syndrome, or other neurodegenerative diseases). The purpose of the extended screen of individuals was to characterize the phenotypic range and provide varied case samples for subsequent genotype-phenotype correlation. The number of rpPCR individual samples estimated to have >32 repeats was 28/375 (7.5%) for FTLD, 11/904 (1.2%) for AD, 29/360 (8.1%) for ALS, 1/470 (0.2%) for sCJD, 9/444 (2.0%) for other neurodegenerative diseases, and 7/421 (1.7%) for HD-like syndrome for a total of 85 C9orf72 expansion samples (two samples were identified retrospectively to be in the cohorts for both HD-like syndrome and FLTD and were removed from the former category). Eighteen FTLD cases from the UCL FTLD DNA cohort have been described in detail elsewhere but are included here for comparison purposes.9 Mean age of onset was 54.6 years and did not differ between cohorts; an autosomal-dominant inheritance pattern of early-onset neurodegenerative disease (at least one other relative) was documented in 29%. Notable atypical clinical presentations and/or features included psychiatric symptoms (treatment with major tranquilizers in at least three) and movement disorders (Parkinsonism in two, chorea in several in the HD-like-syndrome cohort, and myoclonus prompting consideration of CJD in one). Upon review of the case notes from the AD series, in which more details were available, C9orf72 cases had clinical features that overlapped those of FLTD, suggesting that diagnostic overlap between AD and FTLD might explain the finding of expansions in the AD cohort; there were no autopsy findings available.
To determine the frequency of C9orf72 expansion in the healthy control population, we also performed rpPCR in the 58BC (Table 1). Because individuals in this cohort are now 54 years old, we performed a retrospective case review to exclude individuals with a clinical diagnosis. After excluding one individual with ALS, we determined that the prevalence of the C9orf72 expansion in the control population was 11/7,598 (1 in 691 or 0.15% [95% confidence interval 0.07%–0.26%]). In case-control comparison, as expected, an expansion > 32 repeats was associated with FTLD and ALS (p < 10−28, Fisher’s exact test), AD (p = 4.3 × 10−6), HD-like syndrome (p = 2 × 10−5), and other neurodegenerative diseases (p = 4.3 × 10−7), but not CJD.
Table 1.
Frequency of Large C9orf72 Expansions in Cases and Controls
|
Cases |
Controls |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FTLD | ALS | Other Neurodegenerative Disease | sCJD | AD | HD-like Syndrome | All Cases | ECACC | 1958 Birth Cohort | All Controls | |
| Total screened | 375 | 360 | 444 | 470 | 904 | 421 | 2,974 | 474 | 7,105 | 7,579 |
| Expansion detected by rpPCR | 28 | 29 | 9 | 1 | 11 | 7 | 85 | 2 | 9 | 11 |
| Frequency | 0.075 | 0.081 | 0.02 | 0.002 | 0.012 | 0.017 | 0.029 | 0.004 | 0.001 | 0.0015 |
| Expansion confirmed by Southern blotting | 17/17 | 19/19 | 6/7a | 1/1 | 8/8 | 6/6 | 57/58 | 2/2 | 9/9 | 11/11 |
Details of the study cohort show the total number of cases and controls screened by rpPCR and the number of C9orf72 expansions detected in each cohort and their frequency. The subset of those in whom rpPCR detected expansions > 32 repeats and who were then screened by Southern blotting are also shown. The following abbreviations are used: FTLD, frontotemporal lobar degeneration; ALS, amyotrophic lateral sclerosis; sCJD, sporadic Creutzfeldt-Jakob disease; AD, Alzheimer disease; HD, Huntington disease; ECACC, European Collection of Cell Cultures; and rpPCR, repeat-primed PCR.
One case with an apparent expansion of 80+ repeats by rpPCR did not show large expansion by Southern blotting, although there was not enough material for repeat analysis.
We then looked at the stability of the C9orf72 hexanucleotide repeat region in the CEPH family series (Tables S1 and S2). No large expansions (>32 repeats) were identified via rpPCR. However, in 1,046 transmissions, three changes in repeat size between generations were identified. In the CEPH families, the largest repeat (22 repeats) changed size twice in the same family: from 21 in the paternal grandparent to 22 in the father and from 22 in the father to 20 in the son (Table S2). There were no unstable maternal transmissions. The overall intergenerational repeat change rate was 0.29%. Interestingly, all intergenerational changes occurred from a starting repeat length > 10. These changes were verified by repeat rpPCR and fluorescent-labeled PCR size fractionation (although we cannot exclude alteration of flanking sequences).
To determine whether there was a specific haplotype on which the intergenerational repeat changes in the CEPH family occurred, we genotyped neighboring SNPs and microsatellites and empirically determined haplotypes by using family data. All intergenerational changes occurred on the same haplotype, which we named the rs3849942A haplotype. The shared haplotype and the larger starting repeat length (but within the normal range), support the inference that either one or both of these features might confer instability of repeat length.
As reported by others, we found strong linkage disequilibrium (LD) between repeat length and a neighboring SNP, rs3849942 (see Figure 1).4,5,10 A total of 5,200 Wellcome Trust Case Control Consortium 2 control individuals were assessed with fastPHASE, generating haplotypes across chromosome 9 region 27,471,905–27,562,634 (∼91 kb). Of 10,400 haplotypes, 2,597 (25%) were rs3849942A. Of the 2,597 rs3849942A haplotypes we detected, 2,435 (94%) were identical to each other and the disease-related haplotype described by Mok et al.10 The disease-associated SNP haplotype is therefore common in the UK population.
Figure 1.
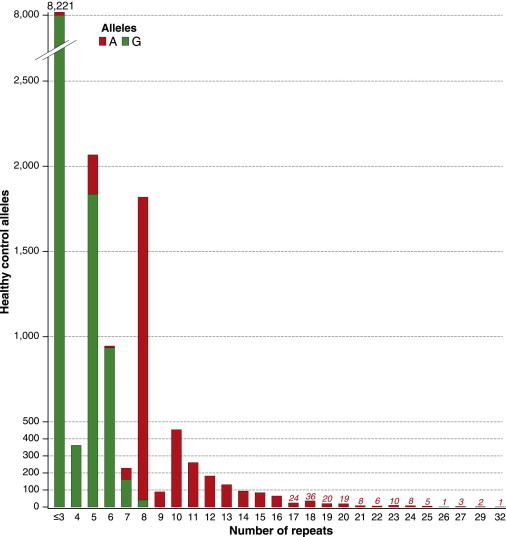
Histogram of C9orf72 Repeat Size in the Healthy Population
Histogram showing frequency of C9orf72 repeat sizes from 1–32 in 58BC UK controls and the entire CEPH collection. rs3849942G-associated repeats are shown in green, and those associated with rs3849942A (“risk” haplotype marker) are shown in red. The phase of genotypes with repeat size was calculated for the CEPH individuals, and frequencies were then applied to the 58BC data.
The outstanding question is whether all cases share an ancient common ancestor or whether the rs3849942A haplotype confers increased risk of mutation. We sought to distinguish these possibilities by testing for evidence of recent shared ancestry between cases diagnosed in one region by looking at ten microsatellites over 13.1 Mb surrounding C9orf72 (two microsatellites were within 300 kb of C9orf72). The moment an expansion mutation occurs, it is linked with all microsatellite variation on the same chromosome; however, over subsequent generations, this mutation-associated haplotype breaks down as a result of both recombination occurring between C9orf72 and the microsatellite and alteration of the microsatellite repeat length by mutation. We found eight different microsatellite alleles linked to C9orf72 expansions at two microsatellites within 300 kb; they had an estimated recombination rate with C9orf72 of less than once in 100 generations.12 We empirically estimated the total number of possible microsatellite haplotypes in a subset of 48 expansion cases for whom complete haplotype data existed. We found at least 60 different haplotypes on the basis of the incompatibility of genotypes. Using the same empirical methods, we made similar estimates in 48 CEPH parents and predicted at least 76 haplotypes (not statistically significantly different from those of the cases). Haplotyping using genotypes from children of the same CEPH parents revealed that all 96 haplotypes were unique, implying that all or a very high proportion of haplotypes in the case series were also unique. The microsatellite allele frequencies associated with C9orf72 expansions as a group were indistinguishable from those of controls, including those linked with rs3849942A (Table S1). These data provide strong evidence against recent shared ancestry of a large proportion of C9orf72 expansion individuals in the UK.
We modified the Southern blotting method of DeJesus-Hernandez et al.2 with the aim of enhancing the expansion signal (see Material and Methods and Figures 2–4) by using a more complete restriction-endonuclease digestion of genomic DNA and a (GGGGCC)5 DIG probe rather than one specific to adjacent DNA sequence. A more sensitive blotting methodology would allow direct estimates of document size ranges (smears) and expansion size in a large and more representative sample series and would allow genotype-phenotype correlations. We found no large expansions in 50 of those in whom rpPCR did not detect mutations > 32 repeats (Figure S2), and we confirmed large expansions in 68/69 of those in whom rpPCR did detect expansions, demonstrating the high specificity of the modified protocol (Figure S3). We were unable to retest the single sample in which the Southern blot did not confirm a large expansion because of a lack of material. Out of the 68 of those in whom rpPCR detected expansions > 32 repeats, 67 gave patterns of varying forms of long smears interrupted by one or more modal points (see Figures 2–4 for individual estimates of repeat size). The blot of the remaining individual did not manifest as a smear but as two distinct bands (Figure S4). For statistical analysis, we compared multiple estimates of repeat size on the basis of smear maxima (range 790–4,400) and minima (400–1,500), smear midpoint (700–3,000), and mode (630–3,800) or modal points (630–2,200; 20 samples had more than one mode). DNA from lymphocyte cell lines (LCLs) was associated with smaller repeats sizes and a distinct multimodal banding pattern (Figures 3 and 4), which we assumed relates to the pauciclonal origins of DNA in cell lines. Surprisingly, all of the control participants in whom rpPCR detected expansions > 32 repeats had large expansions (>400 repeat smear minima) and overlapped the range seen in cases. Three control samples were available from blood, and all were typical of cases (Figure 4).
Figure 2.
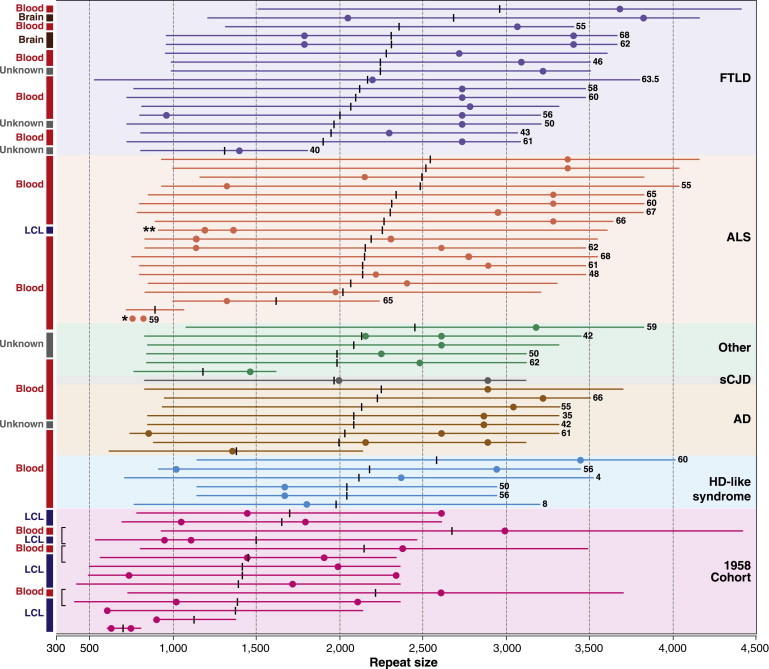
Schematic of Southern Blotting Data for Large C9orf72 Repeat Expansions
A schematic of Southern blot data for 57 cases and 11 controls shows C9orf72 repeats sizes across seven cohorts. Individual blot data are represented by colored bars, modes are indicated by similarly colored dots, and the midpoint of size is represented by a vertical black bar. Ages of onset, when available, are given in years at the right-hand end of individual bars. DNA was extracted from tissues, as shown on the left. In three controls, data are shown for DNA extracted from a lymphocyte cell line (LCL) and peripheral blood (blood). Brackets indicate that two adjacent bars are from the same individual. A single asterisk indicates an unusual ALS case with a doublet of bands of relatively low size, and double asterisks indicate a single 58BC individual with a large repeat size from LCL DNA and a diagnosis of ALS.
Figure 3.

Example Southern Blots of Various Large C9orf72 Repeat Expansions
A Southern blot showing C9orf72 repeat expansions in eight cases and one ECACC control and two 58BC controls demonstrates typical banding patterns and lower size in LCL DNA than in peripheral-blood DNA. Control DNA without an expansion is also shown. Cases 1 and 2 show Southern blotting of DNA from three different brain regions. The asterisk indicates GGGGCC containing a short-tandem-repeat genome motif unrelated to C9orf72. The missing bands in case 1 (brain stem) is most likely due to a reduced amount of gDNA.
Figure 4.
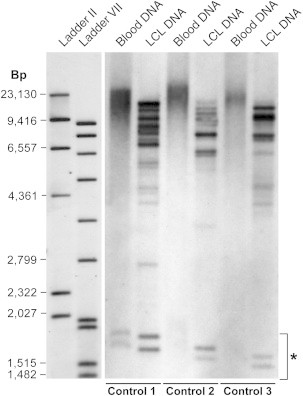
Marked Difference in Observed Expansion Size for LCL and Peripheral-Blood DNA
Southern blot showing data from three 58BC controls with C9orf72 expansions for both peripheral-blood DNA and LCL DNA. Typical LCL banding patterns can be seen and might represent pauciclonality of LCL DNA. Repeats associated with LCL DNA are smaller in size than repeats seen in peripheral-blood DNA; the latter repeats are similar in size to those in case DNA. The asterisk indicates GGGGCC containing a short-tandem-repeat genome motif unrelated to C9orf72.
Minima, maxima, midpoint, and modal estimates of repeat size were all statistically significantly correlated with some aspects of clinical phenotype; however, importantly, there were no differences between any two disease cohorts by any measure of repeat size (p > 0.1 for all pairwise comparisons, Tukey’s post hoc test, ANOVA). Repeat sizes of LCL DNA (largely controls) were smaller than those of blood-extracted DNA by all measures (p < 0.01, Tukey’s post hoc test, ANOVA). The modal point of repeat size correlated with age at clinical onset (increasing age, increasing repeat size, Pearson correlation 0.38, p = 0.02); however, other repeat-size metrics did not significantly correlate. In two cases, we blotted DNA extracted from the frontal cortex, brain stem, and cerebellum and observed marked differences among the different brain regions (Figure 3), although more samples will need to be analyzed for consistency and a statistical analysis.
Discussion
We have screened a large case and control series and developed a Southern blotting methodology to understand the prevalence of the C9orf72 expansion and its pathogenicity and extend genotype-phenotype correlations. Whereas earlier studies have suggested a healthy control upper limit of 30 repeats, we found that large expansions (>400 repeats) in C9orf72 are not infrequent in the UK population (around 1 in 600 individuals). This is considerably more prevalent than would be expected from epidemiological studies. Surprisingly, control individual expansions were indistinguishable from those of case samples. Despite a wide range of sizes and smear morphologies, expansion metrics did not differ in diagnostic categories. Finally, we provide evidence in support of multiple mutational events in human history.
In order to approximate the size of pathogenic expansions, we developed a Southern blot methodology that utilized a DIG-labeled oligonucleotide probe comprising five hexanucleotide repeats (GGGGCC)5. Our concept was that this probe with multiple hybridization sites within the repeat expansion would give a stronger signal than would a single-copy probe hybridizing to the restriction fragment containing the repeats. The choice of two frequently cutting restriction endonucleases with restriction sites that closely flank the repeat region produced highly fragmented gDNA (∼200–300 bp modal size). This allowed the oligonucleotide repeat probe to have hybridization specificity for the C9orf72 expansion. Further, our suggestion is that blot findings should only be interpreted in conjunction with an abnormal expansion detected by the rpPCR method. No hybridization signal was detected for restriction fragments above 1,700 bp in 50 controls, allowing for unambiguous and sensitive detection of C9orf72 expansions greater than ∼275 repeats.
The refined methodology allows for sizing of as little as 1–3 μg gDNA. It also allows for a more accurate definition of the range observed in gDNA samples extracted from tissue. In LCL DNA from controls carrying large expansions, the method detects multiple bands of variable intensity, highlighting the pauciclonality that exists in such lines.13,14 There might be a disparity in expansion range between our data and those utilizing other blot methods. This is most likely due to a combination of factors. First, it is now apparent that DNA extracted from cell lines is not reliable for the sizing of the expansion. Second, the sensitivity of our assay increases the range of expansion observed on the blot, whereas hybridizations using single-copy probes are likely to reflect a modal size. Third, it has been reported that some DNA fragments containing repeats have unusual structures and thus result in abnormal migration in agarose when compared with more typical gDNA fragments.15 The amount of flanking sequence in the fragment containing the expansion might also influence structure.16 Therefore, it remains a possibility that overall repeat number could appear different with the use of a different Southern methodology. As a precaution, we emphasize the relative size of expansions rather than the exact number of repeats. Although control and case expansions were indistinguishable from one another, sequence differences within the expansions remain a possibility. The single individual with a discrete banding pattern (Figure S4) might indicate the presence of such heterogeneity in some C9orf72 expansions.
The prevalence of large expansions in the UK population is intriguing. Lifetime risk of ALS has been estimated at ∼1 in 430.17,18 Lifetime risk of FTLD is less well understood, but the incidence measured in two studies was 3.5 and 4.1 per 100,000 in the 45–64 year age cohort;19,20 this is comparable to that of ALS,21 implying a similar lifetime risk. With the use of C9orf72-mutation frequencies based on a recent large study and estimates of the proportion of ALS and FTLD cases showing evidence of familial disease,4,22,23 the lifetime risk of C9orf72-associated FTLD or ALS is approximately 1 in 2,000. Although the uncertainties in the true lifetime risk of FTLD prevent a formal statistical comparison with the frequency of C9orf72 expansions, the estimate differs considerably from the approximately 1 in 700 estimate of our population study. There are several potential explanations for this discrepancy: first, the lifetime risk of FTLD might in fact be much greater than that of ALS; second, many clinical syndromes caused by C9orf72 expansions are not diagnosed as FTLD or ALS; and third, the penetrance of the expansion is much lower than that predicted by family studies of currently ascertained cases, perhaps because of additional genetic risk factors. Our case screen supports the second suggestion given that C9orf72 expansions were found in all neurodegenerative-disease categories we tested and a third of our case series had diagnoses other than FTLD and/or ALS. Several of these syndromes (notably Alzheimer-type dementia) are highly prevalent conditions in old-age populations, which might therefore harbor large numbers of C9orf72 cases. These data emphasize the potential importance of the C9orf72 expansion in neurodegeneration in that our estimates suggest that there might be approximately 90,000 mutation carriers in the UK.
Although the presence and size of C9orf72 expansions did not differ between diagnostic groups, we did identify a correlation between the age of onset and expansion size. From three brain regions in two cases, we also found evidence of marked and consistent differences within an individual, indicating considerable scope for heterogeneity in specific cell types; large smears and variable patterns were seen in blood-extracted DNA. These findings are likely to be due to somatic instability. Variation in expansion size between brain regions and with age is consistent with the age-dependent expansions displayed in transgenic mouse models of the human FXN locus;24 these models also implicate somatic mutation. This might be an explanation for the phenotypic heterogeneity and incomplete penetrance of C9orf72-expansion diseases.
The considerable instability of the expansion suggested by somatic mutation and in the CEPH families raises questions about the hypothesis regarding a single ancestral mutational event.4,10 We used genotyping of the surrogate marker rs3849942 for the haplotype associated with expansions to make inferences about the stability and origin of the expansion in UK population history. In keeping with previous reports, we found a distinct difference between the size distribution of repeats in controls linked with rs3849942A and those in controls linked with rs3849942G; longer repeats were linked to rs3849942A.2 Additionally, all 11 control samples with expansions greater than 400 repeats were either heterozygous or homozygous for rs3849942A. In the CEPH pedigrees, we found three mutations of repeat size between generations, and the fact that all occurred on the rs3849942A haplotype further indicates that the repeat region on this haplotype is less stable. Further, using microsatellite analysis, we found no evidence of recent shared ancestry in the UK. Two of the microsatellites genotyped were within 300 kb of C9orf72 and would be expected to show residual LD if a single mutational event in Finland resulted in a large proportion of UK cases. Taken together, our data are compatible with the hypothesis that larger but normal-range repeats, which are more unstable, have generated very large expansion mutations in unrelated individuals many times in human history. This hypothesis is compatible with the considerable prevalence of mutations in countries distant from Finland.25
The absence of a strong correlation between expansion size and key aspects of the clinical phenotype was surprising. In the gain-of-function model of disease etiology, perhaps related to the sequestering of RNA binding proteins in RNA foci, it might be expected that larger expansions would result in a more severe phenotype. There are several possible explanations for the lack of such effects in our data: (1) a false-negative association might be due to small sample size or the inability (in many cases) to analyze tissue directly from the principle site of pathology, (2) the clinical phenotype might be modified by other genetic, environmental, or stochastic factors, and (3) if the loss-of-function hypothesis is correct, there might be complete loss of expression of C9orf72 beyond a certain expansion size.3
In summary, we have developed a reliable method of approximating the C9orf72 expansion size, and it might have clinical diagnostic utility. Our data emphasize the importance of this mutation in neurodegeneration and common neurodegenerative-disease syndromes outside of the FTLD and ALS spectrum and provide direct evidence of repeat instability, somatic mutation, and multiple mutational events in unrelated individuals.
Acknowledgments
This work was funded by the Medical Research Council (MRC, UK). We are grateful for the support of cohort individuals and their families and to numerous physicians for sending DNA samples to the MRC Prion Unit for research genetic studies. Some of the work was conducted at University College London Hospitals National Health Service Trust Biomedical Research Centre (BRC), supported by BRC funding of the National Prion Monitoring Cohort study and researchers at the National Institute for Health Research (NIHR) Queen Square Dementia BRU. J.D.W. is supported by a Wellcome Trust Senior Clinical Fellowship (091673/Z/10/Z). J.B.R. is supported by a Wellcome Trust Senior Research Fellowship (088324), and J.B.R. and J.M.B. are supported by the NIHR Cambridge Biomedical Research Centre. Ray Young assisted with figure design. Some of this work was supported by the Motor Neuron Disease Association. Nicole Gurunlian provided technical assistance related to the amyotrophic lateral sclerosis samples. We acknowledge the use of genotype data from the British 1958 Birth Cohort DNA collection, funded by MRC grant G0000934 and Wellcome Trust grant 068545/Z/02. We thank the MRC London Neurodegenerative Diseases Brain Bank, Institute of Psychiatry, King’s College London and the Queen Square Brain Bank for Neurological Disorders, University College London Institute of Neurology, London for providing tissue.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., ITALSGEN Consortium A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gijselinck I., Van Langenhove T., van der Zee J., Sleegers K., Philtjens S., Kleinberger G., Janssens J., Bettens K., Van Cauwenberghe C., Pereson S. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 4.Majounie E., Renton A.E., Mok K., Dopper E.G.P., Waite A., Rollinson S., Chiò A., Restagno G., Nicolaou N., Simon-Sanchez J., Chromosome 9-ALS/FTD Consortium; French research network on FTLD/FTLD/ALS; ITALSGEN Consortium Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith B., Newhouse S., Shatunov A., Vance C., Topp S., Miller J., Weale M., Al Chalabi A., Shaw C. The C9ORF72 expansion mutation has a single European founder that arose from a background haplotype associated with repeat instability. Dement. Geriatr. Cogn. Disord. 2012;33:87–88. [Google Scholar]
- 6.Majounie E., Abramzon Y., Renton A.E., Perry R., Bassett S.S., Pletnikova O., Troncoso J.C., Hardy J., Singleton A.B., Traynor B.J. Repeat expansion in C9ORF72 in Alzheimer’s disease. N. Engl. J. Med. 2012;366:283–284. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snowden J.S., Rollinson S., Thompson J.C., Harris J.M., Stopford C.L., Richardson A.M.T., Jones M., Gerhard A., Davidson Y.S., Robinson A. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135:693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindquist S., Duno M., Batbayli M., Puschmann A., Braendgaard H., Mardosiene S., Svenstrup K., Pinborg L., Vestergaard K., Hjermind L. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin. Genet. 2012 doi: 10.1111/j.1399-0004.2012.01903.x. Published online May 31, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Mahoney C.J., Beck J., Rohrer J.D., Lashley T., Mok K., Shakespeare T., Yeatman T., Warrington E.K., Schott J.M., Fox N.C. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135:736–750. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mok K., Traynor B.J., Schymick J., Tienari P.J., Laaksovirta H., Peuralinna T., Myllykangas L., Chiò A., Shatunov A., Boeve B.F. Chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol. Aging. 2012;33:209.e3–209.e8. doi: 10.1016/j.neurobiolaging.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. 2012;11:297–298. doi: 10.1016/S1474-4422(12)70046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong A., Gudbjartsson D.F., Sainz J., Jonsdottir G.M., Gudjonsson S.A., Richardsson B., Sigurdardottir S., Barnard J., Hallbeck B., Masson G. A high-resolution recombination map of the human genome. Nat. Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 13.Plagnol V., Uz E., Wallace C., Stevens H., Clayton D., Ozcelik T., Todd J.A. Extreme clonality in lymphoblastoid cell lines with implications for allele specific expression analyses. PLoS ONE. 2008;3:e2966. doi: 10.1371/journal.pone.0002966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryan J.L., Kaufmann W.K., Raab-Traub N., Oglesbee S.E., Carey L.A., Gulley M.L. Clonal evolution of lymphoblastoid cell lines. Lab. Invest. 2006;86:1193–1200. doi: 10.1038/labinvest.3700472. [DOI] [PubMed] [Google Scholar]
- 15.Mirkin S.M. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 16.Sakamoto N., Chastain P.D., Parniewski P., Ohshima K., Pandolfo M., Griffith J.D., Wells R.D. Sticky DNA: Self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol. Cell. 1999;3:465–475. doi: 10.1016/s1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- 17.Armon C. Sports and trauma in amyotrophic lateral sclerosis revisited. J. Neurol. Sci. 2007;262:45–53. doi: 10.1016/j.jns.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 18.Alonso A., Logroscino G., Jick S.S., Hernán M.A. Incidence and lifetime risk of motor neuron disease in the United Kingdom: A population-based study. Eur. J. Neurol. 2009;16:745–751. doi: 10.1111/j.1468-1331.2009.02586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mercy L., Hodges J.R., Dawson K., Barker R.A., Brayne C. Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology. 2008;71:1496–1499. doi: 10.1212/01.wnl.0000334277.16896.fa. [DOI] [PubMed] [Google Scholar]
- 20.Knopman D.S., Petersen R.C., Edland S.D., Cha R.H., Rocca W.A. The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology. 2004;62:506–508. doi: 10.1212/01.wnl.0000106827.39764.7e. [DOI] [PubMed] [Google Scholar]
- 21.The Scottish Motor Neuron Disease Register: A prospective study of adult onset motor neuron disease in Scotland. Methodology, demography and clinical features of incident cases in 1989. J. Neurol. Neurosurg. Psychiatry. 1992;55:536–541. doi: 10.1136/jnnp.55.7.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanby M.F., Scott K.M., Scotton W., Wijesekera L., Mole T., Ellis C.E., Leigh P.N., Shaw C.E., Al-Chalabi A. The risk to relatives of patients with sporadic amyotrophic lateral sclerosis. Brain. 2011;134:3454–3457. doi: 10.1093/brain/awr248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohrer J.D., Guerreiro R., Vandrovcova J., Uphill J., Reiman D., Beck J., Isaacs A.M., Authier A., Ferrari R., Fox N.C. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451–1456. doi: 10.1212/WNL.0b013e3181bf997a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark R.M., De Biase I., Malykhina A.P., Al-Mahdawi S., Pook M., Bidichandani S.I. The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model. Hum. Genet. 2007;120:633–640. doi: 10.1007/s00439-006-0249-3. [DOI] [PubMed] [Google Scholar]
- 25.Ishiura H., Takahashi Y., Mitsui J., Yoshida S., Kihira T., Kokubo Y., Kuzuhara S., Ranum L.P., Tamaoki T., Ichikawa Y. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch. Neurol. 2012;69:1154–1158. doi: 10.1001/archneurol.2012.1219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.