

Abstract
Hemophilia B, or the “royal disease,” arises from mutations in coagulation factor IX (F9). Mutations within the F9 promoter are associated with a remarkable hemophilia B subtype, termed hemophilia B Leyden, in which symptoms ameliorate after puberty. Mutations at the −5/−6 site (nucleotides −5 and −6 relative to the transcription start site, designated +1) account for the majority of Leyden cases and have been postulated to disrupt the binding of a transcriptional activator, the identity of which has remained elusive for more than 20 years. Here, we show that ONECUT transcription factors (ONECUT1 and ONECUT2) bind to the −5/−6 site. The various hemophilia B Leyden mutations that have been reported in this site inhibit ONECUT binding to varying degrees, which correlate well with their associated clinical severities. In addition, expression of F9 is crucially dependent on ONECUT factors in vivo, and as such, mice deficient in ONECUT1, ONECUT2, or both exhibit depleted levels of F9. Taken together, our findings establish ONECUT transcription factors as the missing hemophilia B Leyden regulators that operate through the −5/−6 site.
Main Text
Hemophilia B (MIM 306900) is an X-linked recessive blood-clotting disorder that affects approximately 1 in every 30,000 males.1 It is also infamously known as the “royal disease” because it afflicted European royal families descended from Queen Victoria.2 It is caused by mutations that affect the normal expression or function of coagulation factor IX (F9 [MIM 300746]). Interestingly, mutations within the F9 proximal promoter are associated with a specific subtype, known as hemophilia B Leyden (MIM 306900), which was first reported in the Netherlands.3 Sufferers of hemophilia B Leyden express low levels of F9 up until puberty, at which point F9 levels rise and clinical symptoms improve; this phenomenon is associated with increasing androgen receptor and growth-factor activity.4–8
To date, more than 80 families affected by hemophilia B Leyden have been analyzed worldwide and 21 distinct point mutations have been detected in the F9 promoter (Figure 1).12,13 The mutations fall into three clusters: one at nucleotide −20 relative to the transcription start site (TSS, designated +1), one at around nucleotide +10, and one at nucleotides –5 and –6 (−5/−6 site). The mutations at −20 prevent binding of the transcriptional activator HNF4α (hepatocyte nuclear factor 4α, encoded by HNF4A [MIM 600281]), and the +10 mutations disrupt a C/EBPα site (CCAAT/enhancer-binding protein α, encoded by CEBPA [MIM 116897]).6,14,15 However, around half of all known Leyden mutations lie in the third cluster, and their mechanism of action has remained unresolved.12,13 We noted that the −5/−6 site resembles the recently defined consensus site for the transcription factor ONECUT1 (encoded by ONECUT1 [MIM 604164]), also known as hepatocyte nuclear factor 6 (HNF6).10 Importantly, ONECUT1 is abundant in the liver, the tissue in which F9 is produced.16 We thus sought to determine whether ONECUT1 and the related protein ONECUT2 (encoded by ONECUT2 [MIM 604894])17,18 constitute the missing Leyden regulators that operate through the −5/−6 site.
Figure 1.
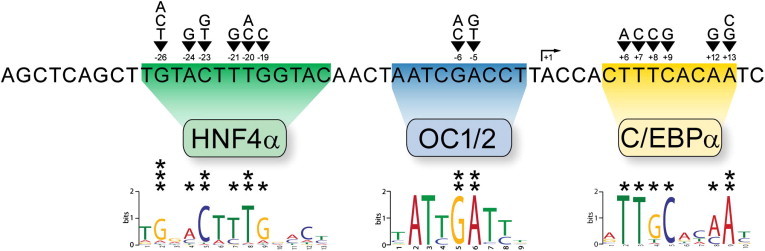
The Proximal Promoter of Human F9
The major transcription start site (TSS) is indicated by an arrow and designated +1. Point mutations associated with hemophilia B Leyden are indicated above the sequence. The transcription factors known to bind this region are shown, and the consensus sequences that they recognize are aligned below the sequence. DNA-binding consensus sequences were determined from the ChIP-Seq data reported here (ArrayExpress accession number E-MTAB-890) and previously (ArrayExpress E-TABM-722)9 with the default settings of MEME and the following parameters: -revcomp -maxw 20 -minw 6 -nmotifs 5. We selected the top 500 peaks ordered by input-corrected read depth and used the 25 base pairs centered on the identified summit of the peak as our input for the motif-discovery analysis. The position weight matrices are consistent with previously determined DNA-binding consensuses for these transcription factors.9–11 The following protein abbreviations are used: HNF4α, hepatocyte nuclear factor 4 alpha; C/EBPα, CCAAT/enhancer-binding protein alpha; and OC1/2, ONECUT1/2.
To test whether ONECUT1 and ONECUT2 could recognize the −5/−6 site in the human F9 promoter, we first cloned the DNA-binding domains of ONECUT1 (residues 280–465) and ONECUT2 (residues 321–504) and fused them each individually to the C terminus of glutathione S-transferase (GST) contained in the vector pGEX-6P. Recombinant GST-fusion proteins were subsequently expressed in bacteria and were purified with glutathione beads as described previously.19 The DNA-binding activities of the resulting extracts were assessed in electrophoretic mobility shift assays (EMSAs) with a double-stranded, radiolabeled DNA probe encompassing the −5/−6 region of the human F9 promoter (5′-AACTAATCGACCTTACCA-3′). As shown in Figure 2A, strong binding was observed for both ONECUT1 and ONECUT2. We also tested probes containing the hemophilia-B-Leyden-associated mutations (−6G>C, −6G>A, −5A>T, and −5A>G). The −6G>C mutation most significantly impaired binding, −6G>A had the mildest effects, and the two −5 mutations were of intermediate severity. This broadly reflects the clinical severity of the various mutations: the −6G>C mutation produces the most severe hemophilia, −6G>A produces the mildest form, and the −5 mutations are of intermediate severity.12,13,20
Figure 2.
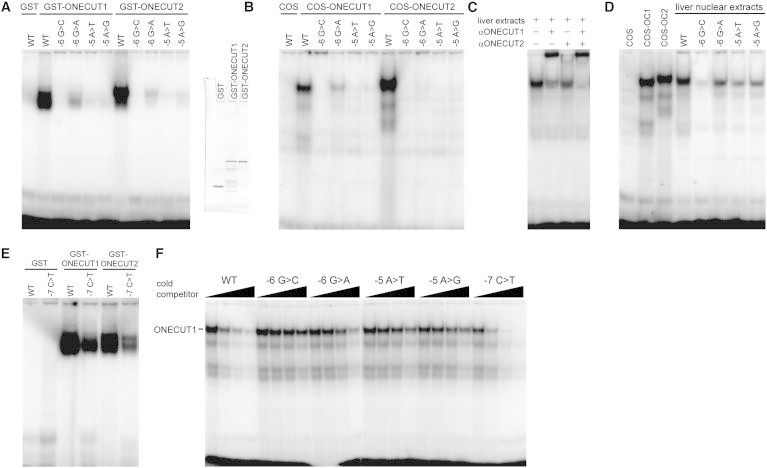
ONECUT Proteins Recognize the −5/−6 Region of the Human F9 Promoter, and Binding Is Disrupted by Leyden Mutations
(A–F) EMSAs using wild-type (WT) and mutant probes spanning the −5/−6 region.
(A) ONECUT1 and ONECUT2 GST-fusion proteins purified from bacteria. Coomassie staining is shown on the right to indicate the level and purity of the recombinant proteins used.
(B) Nuclear extracts from COS cells expressing full-length ONECUT1 and ONECUT2.
(C) Murine liver nuclear extracts with antibodies against ONECUT1 and ONECUT2.
(D) Murine liver nuclear extracts with WT and mutant probes. Nuclear extracts from COS cells transfected with ONECUT1 and ONECUT2 were included as positive controls.
(E) ONECUT1 and ONECUT2 GST-fusion proteins purified from bacteria. Equivalent levels of protein were added as in (A).
(F) A competition assay using radiolabeled WT probe (1.6 ng per lane) and cold competitor probes (0 ng, 16 ng, 160 ng, and 1.6 μg, that is, 0×, 10×, 100×, and 1,000× excesses, respectively) compares the ability of the different mutants to compete for binding by ONECUT1. Two microliters of nuclear extracts from COS cells expressing ONECUT1 was used per lane. Murine liver tissue was obtained under ethics approval number 09/128A (Animal Care and Ethics Committee, University of New South Wales). The following protein abbreviations are used: OC1, ONECUT1; and OC2, ONECUT2.
We next assayed full-length ONECUT1 and ONECUT2 expressed in COS cells by using pCMV6-XL5 expression vectors (OriGene). COS cells were transfected and nuclear extracts were obtained as described previously.21,22 No significant retarded bands were observed in untransfected cells, but strong binding was observed when ONECUT1 or ONECUT2 was overexpressed (Figure 2B). Again, all four Leyden mutations reduced the binding observed. ONECUT1- and ONECUT2-specific antibodies (sc-13050 X and sc-51177 X, respectively; Santa Cruz Biotechnology) supershifted the retarded species, confirming the identities of these protein-DNA complexes (Figure S1, available online). The reduced binding of full-length ONECUT1 and ONECUT2 to the −5/−6 Leyden mutants was further confirmed by competition assays using radiolabeled wild-type probe and 0×, 10×, 100×, and 1,000× excesses of cold competitor probe (Figure 2F and Figures S2 and S3).
Next, we assessed nuclear extracts from murine liver (3 months of age). As shown in Figure 2C, we detected a single, distinct protein-DNA complex by using the wild-type probe. The addition of antibodies individually specific to ONECUT1 and ONECUT2 demonstrated that the retarded species contained both proteins but predominantly had ONECUT1 (Figure 2C). We then examined the binding of the complex to the Leyden mutant probes. Again, the intensity of this band was reduced when probes carrying the mutations were used (Figure 2D), and the degree of impairment correlated with the clinical severity of the mutation. Together, these data suggest that ONECUT1 and ONECUT2 (but particularly ONECUT1) constitute the majority of DNA-binding activity at the −5/−6 site in the liver and that the Leyden mutations disrupt binding of these endogenous factors.
Extensive mapping of the F9 locus has illustrated the prevalence of CpG dinucleotides as hotspots for mutation in hemophilia B cases.23 In this regard, it is interesting that although both residues −6 and −7 are within a CpG dinucleotide, no C>T transition has been reported at −7 (Figure 1). In contrast, mutations at −6 have been reported at least 34 times and account for nearly half of all Leyden individuals.12,13 Importantly, the high incidence of this mutation is not simply due to a founder effect, because different haplotypes have been shown to be associated with the −6 mutation.24 Our assertion that ONECUT proteins operate in vivo predicts that the −7C>T mutation might occur naturally but that it would not have a functional effect on F9 expression, perhaps because it would not disrupt ONECUT binding.
We used EMSAs with ONECUT1 and ONECUT2 GST-fusion proteins to test whether the anticipated mutation at −7 would impair binding of ONECUT factors (Figure 2E). We observed both ONECUT1 and ONECUT2 binding—slightly less than that observed with the wild-type sequence but significantly more than observed with the −6G>A mutation (compare with Figure 2A). Furthermore, competitor assays revealed that full-length ONECUT1 and ONECUT2 bind to probes containing the −7C>T mutation with considerably higher affinity than to any of the Leyden-associated mutations (Figure 2F and Figure S2). Thus, we conclude that −7C>T mutations would produce a minimal phenotype, and we propose that this is the reason that they have never been clinically encountered. Taken together, the investigations of the base substitutions at the −5, −6, and −7 sites concur well with the position weight matrix determined for ONECUT1 (Figure 1). The −5 and −6 sites, which are robustly conserved in the model, considerably disrupt binding when mutated, whereas the −7 site, which is more flexible and less conserved, is able to tolerate substitution.
To confirm in vivo binding at the F9 promoter, we performed ONECUT1 chromatin immunoprecipitation (ChIP) on murine liver tissue as described previously.25,26 Primers used are outlined in Table S1, and quantitative real-time PCR (qRT-PCR) runs were performed with Power SYBR Green PCR Master Mix and the 7500 Fast Real-Time PCR System (Applied Biosystems).22 Data analysis was conducted with 7500 Software v.2.0.4 (Applied Biosystems). As shown in Figure 3A, occupancy was detected immediately upstream of F9, but not at more distal locations (1.0 kb downstream and 1.5 kb upstream). Moreover, ONECUT1 was not detected at the proximal promoters of other liver-enriched genes, namely, coagulation factor VII (F7 [MIM 613878]) or factor X (F10 [MIM 613872]). We also carried out ONECUT1 ChIP sequencing (ChIP-seq) on human liver tissue. Human liver material (from two males of unknown age) was obtained from the Liver Tissue Distribution Program (National Institute of Diabetes and Digestive and Kidney Diseases contract N01-DK-9-2310) at the University of Pittsburgh and Addenbrooke’s Hospital, Cambridge under human tissue license 08/H0308/117. Immunoprecipitated material was end repaired, A tailed, ligated to single- or paired-end sequencing adapters, amplified by 18 cycles of PCR, and size selected (200–300 bp); this was followed by single-end sequencing on an Illumina Genome Analyzer II according to the manufacturer’s instructions. Data have been deposited in ArrayExpress and assigned the accession number E-MTAB-890. Analysis of the human F9 locus found a strong ONECUT1 peak over the −5/−6 site (Figures 3B and 3C). Similarly, human ChIP-seq data for HNF4α and C/EBPα, which we had previously reported (ArrayExpress E-TABM-722),9,25 revealed at the F9 promoter a high level of occupancy corresponding to the sites that are mutated in hemophilia B Leyden and to which these factors have been reported to bind in vitro (Figures 3B and 3C). Thus, ONECUT1, HNF4α, and C/EBPα all bind to the F9 promoter in vivo and together encompass all 21 reported point mutations associated with hemophilia B Leyden (Figure 1).
Figure 3.
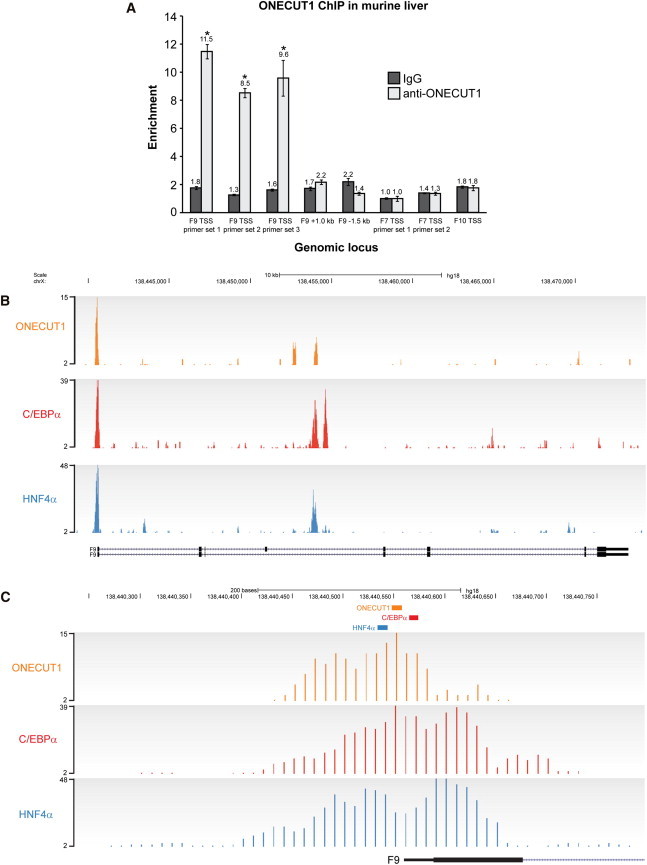
ONECUT1 Binds the F9 Promoter In Vivo along with HNF4α and C/EBPα
(A) ONECUT1 ChIP using mouse liver tissue. Primer sets are specific to the TSS of F9 and to 1.0 kb downstream and 1.5 kb upstream. Negative-control primers specific to the promoter regions of F7 and F10 were included. Enrichment was normalized to input, and the lowest values for immunoglobulin G (IgG) and anti-ONECUT1 were set to 1.0. Error bars represent the SEM, and n = 3 for each data point. ∗p < 0.05 (two-tailed t test) compared to the anti-ONECUT1 enrichment at each of the F9 distal regions and the F7 and F10 promoters.
(B–C) Human-liver-tissue ChIP-Seq tracks for ONECUT1, C/EBPα, and HNF4α across the F9 locus (B) and around the TSS (C).
(C) The HNF4α, ONECUT1, and C/EBPα binding sites at nucleotides −20, −5/−6, and +10, respectively, are denoted by colored boxes. Murine liver tissue was obtained under ethics approval number 09/128A (Animal Care and Ethics Committee, University of New South Wales). ChIP sequencing reads and input samples were aligned with MAQ with default parameters to NCBI Genome browser build 36. All sequence, genome-annotation, and comparative-genomics data were taken from Ensembl release 52. After alignment, binding events were discovered with a dynamic programming algorithm (SWEMBL) with the parameter “-R 0.005 -i –S” as previously described.27
We next tested the functional effect of ONECUT1 and ONECUT2 on the F9 promoter by means of reporter assays in HepG2 cells as described previously.22 We used a fragment that encompassed a minimal region (−189 to +21) of the human F9 promoter and that had previously been shown to recapitulate key aspects of the Leyden phenotype in transfection studies and transgenic mice.15,28 This region was cloned into the firefly luciferase vector pGL4.10[luc2] (Promega). Cells in 6-well plates were transfected (FuGENE6, Roche Diagnostics) with 1 μg triplicate DNA preparations of pGL4.10[luc2] (wild-type or Leyden mutants of the –189 to +21 region) together with 100 ng of the Renilla luciferase vector pGL4.74[hRluc/TK] (Promega) as a transfection control. Expression of ONECUT1 and ONECUT2 was achieved with 1 μg pCMV6-XL5 (ONECUT1 or ONECUT2) (OriGene). We found that both ONECUT1 and ONECUT2 could potently transactivate the wild-type promoter but had less activity on the Leyden mutant promoters (Figure 4A and Figure S4). We also found that compared to the wild-type promoter, the −7C>T promoter did not appreciably reduce transactivation (Figure 4A), consistent with the view that such a mutation does not functionally impair ONECUT binding. Lastly, it should also be noted that C/EBPα and HNF4α are expressed in HepG2 cells.29,30 This raises the possibility that in these cells, the ONECUT proteins might synergistically activate the F9 promoter constructs in combination with either or both of these endogenous factors.
Figure 4.
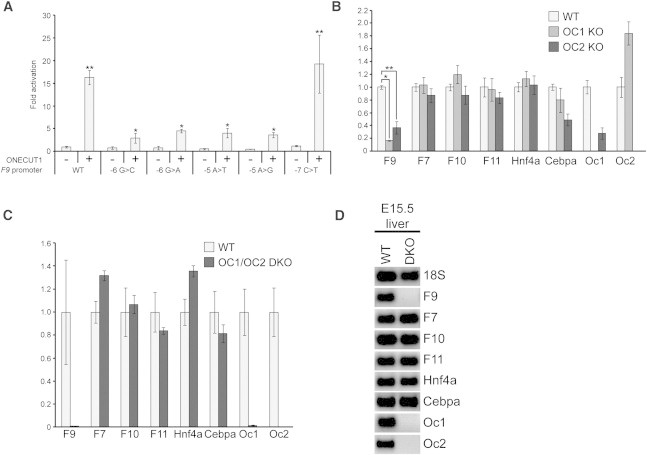
ONECUT Proteins Are Functionally Required for F9 Expression
(A) A Luciferase reporter assay in HepG2 cells. Wild-type (WT) and mutant F9 promoter fragments were tested in the presence or absence of ONECUT1. Triplicate, independent DNA preparations for the promoter constructs were used. Firefly levels were normalized with the use of Renilla, and the value for the WT promoter in the absence of ONECUT1 was set to 1.0. ∗p < 0.02 for −5/−6 mutant promoters relative to the WT promoter, and ∗∗p < 0.05 for the WT and −7C>T promoters in the presence compared to the absence of ONECUT1 (two-tailed t tests). n = 3 for each data point or genotype, and error bars represent the SEM.
(B and C) qRT-PCR analysis of the expression of F9 and other liver genes in embryonic day (E) 17.5 livers from WT and Onecut1−/− and Onecut2−/− single-knockout (KO) mice (B) and qRT-PCR analysis of E15.5 livers from WT and Onecut1−/−/Onecut2−/− double-knockout (DKO) mice (C). Expression was normalized to 18S rRNA for all genes, and WT levels were set to 1.0. Murine liver tissue was obtained under ethics approval number 09/128A (Animal Care and Ethics Committee, University of New South Wales). ∗p < 0.002, ∗∗p < 0.02 (two-tailed t tests). n = 3 for each data point or genotype, and error bars represent the SEM. The following protein abbreviations are used: OC1, ONECUT1; and OC2, ONECUT2.
(D) Agarose gel showing ethidium-bromide-stained amplicons from (C).
The results demonstrate that ONECUT proteins can bind to and activate the F9 promoter in experimental systems. To determine whether F9 expression is dependent on ONECUT proteins in vivo, we analyzed tissue from Onecut−/− mice.31,32 Because ONECUT1-deficient mice typically die shortly after birth, we assessed F9 expression in fetal liver. Total RNA was extracted from fetal livers and used as a template for cDNA synthesis by standard methods. qRT-PCR was performed as described above and with primer sequences shown in Table S2. We observed clear reductions in F9 expression in both Onecut1−/− and Onecut2−/− single knockouts (6-fold and 3-fold, respectively) at day 17.5 of gestation (Figure 4B). Given that both ONECUT1 and ONECUT2 can bind the −5/−6 site and could thus potentially compensate in each other’s absence, we next examined tissues from Onecut1−/− Onecut2−/− double-knockout animals. These animals die in utero, so we analyzed the liver at embryonic day 15.5, when F9 was already expressed and the embryos were still viable. We observed a dramatic reduction (>100-fold) in F9 levels in Onecut1−/− Onecut2−/− double-knockout samples (Figures 4C and 4D). Importantly, we found near-normal expression of a panel of other liver-specific genes, including F7, F10, and coagulation factor XI (F11 [MIM 264900]), and of the other transcription factors (HNF4α6,14 and C/EBPα12) known to drive F9, indicating that the deficit in F9 is specific and not due to a general failure in liver development at this embryonic stage (Figures 4C and 4D).
In conclusion, we have established that the missing regulators in hemophilia B Leyden are the ONECUT transcription factors ONECUT1 and ONECUT2. These proteins bind to the proximal promoter and drive expression of F9. Their binding, and hence functional activity, is disrupted by −5/−6 base substitutions, which are found in more than half of all individuals with hemophilia B Leyden. We have shown here that the different Leyden mutations at −5/−6 disrupt ONECUT binding to varying degrees in a manner that correlates well with their clinical severities. It should be noted, however, that other factors might also influence the severity of hemophilia in individual cases, and we thus caution against solely using the mutations to predict clinical outcome.
The comprehensive compilation of hemophilia B mutations worldwide provides what might be the most complete mapping of a human gene promoter.12,13 Detailed consensus sequences have now been determined for HNF4α, ONECUT1, and C/EBPα9–11 (data presented here), and the mutations associated with hemophilia B Leyden fit remarkably well with predictions from these consensus sequences (Figure 1). In the position weight matrices for these transcription factors, the most robustly conserved bases are, by and large, those that are mutated in cases of hemophilia.
These proteins are also relevant in terms of understanding the liver-restricted expression of F9. HNF4α, ONECUT1, and C/EBPα are well known as major regulators of hepatocyte gene expression,33 and this regulatory module satisfyingly explains the liver-restricted expression pattern of F9. Their cognate DNA elements are very tightly clustered within the F9 promoter, and the fact that disruption of any single site severely disrupts promoter activity suggests that these transcription factors must cooperate to ensure proper F9 expression.
Taken together, these findings contribute to a thorough characterization of the human F9 promoter and its transacting factors and provide an illustrative example of how disrupted local promoter architecture can result in human disease.
Acknowledgments
This research was supported by the Australian Research Council and National Health and Medical Research Council (M.C., A.P.W.F., K.S.M., J.B., and R.C.M.P.), the European Research Council and EMBO Young Investigator Program (D.T.O.), Cancer Research UK and University of Cambridge (M.D.W. and D.T.O.), the Wellcome Trust (D.T.O., B.B., and P.F.), the D.G. Higher Education and Scientific Research of the French Community of Belgium (F.P.L.), the Fund for Scientific Medical Research Belgium (F.P.L.), and the European Molecular Biology Laboratory (P.F.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Archive Ensemble release 52, http://Dec2008.archive.ensembl.org/
ArrayExpress, http://www.ebi.ac.uk/arrayexpress/
Factor IX Mutation Database, http://www.factorix.org
MEME Suite, http://meme.nbcr.net/meme
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Accession Numbers
The ArrayExpress accession number for the ONECUT1 ChIP-Seq data reported in this paper is E-MTAB-890. ChIP-Seq experiments for C/EBPα and HNF4α have previously been reported under ArrayExpress accession number E-TABM-722.9
References
- 1.Mannucci P.M., Tuddenham E.G. The hemophilias—From royal genes to gene therapy. N. Engl. J. Med. 2001;344:1773–1779. doi: 10.1056/NEJM200106073442307. [DOI] [PubMed] [Google Scholar]
- 2.Rogaev E.I., Grigorenko A.P., Faskhutdinova G., Kittler E.L., Moliaka Y.K. Genotype analysis identifies the cause of the “royal disease”. Science. 2009;326:817. doi: 10.1126/science.1180660. [DOI] [PubMed] [Google Scholar]
- 3.Veltkamp J.J., Meilof J., Remmelts H.G., van der Vlerk D., Loeliger E.A. Another genetic variant of haemophilia B: Haemophilia B Leyden. Scand. J. Haematol. 1970;7:82–90. doi: 10.1111/j.1600-0609.1970.tb01873.x. [DOI] [PubMed] [Google Scholar]
- 4.Briët E., Bertina R.M., van Tilburg N.H., Veltkamp J.J. Hemophilia B Leyden: A sex-linked hereditary disorder that improves after puberty. N. Engl. J. Med. 1982;306:788–790. doi: 10.1056/NEJM198204013061306. [DOI] [PubMed] [Google Scholar]
- 5.Kurachi S., Huo J.S., Ameri A., Zhang K., Yoshizawa A.C., Kurachi K. An age-related homeostasis mechanism is essential for spontaneous amelioration of hemophilia B Leyden. Proc. Natl. Acad. Sci. USA. 2009;106:7921–7926. doi: 10.1073/pnas.0902191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crossley M., Ludwig M., Stowell K.M., De Vos P., Olek K., Brownlee G.G. Recovery from hemophilia B Leyden: An androgen-responsive element in the factor IX promoter. Science. 1992;257:377–379. doi: 10.1126/science.1631558. [DOI] [PubMed] [Google Scholar]
- 7.Rimmer E.K., Seftel M.D., Israels S.J., Houston D.S. Unintended benefit of anabolic steroid use in hemophilia B leiden. Am. J. Hematol. 2012;87:122–123. doi: 10.1002/ajh.22190. [DOI] [PubMed] [Google Scholar]
- 8.Brady J.N., Notley C., Cameron C., Lillicrap D. Androgen effects on factor IX expression: In-vitro and in-vivo studies in mice. Br. J. Haematol. 1998;101:273–279. doi: 10.1046/j.1365-2141.1998.00694.x. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laudadio I., Manfroid I., Achouri Y., Schmidt D., Wilson M.D., Cordi S., Thorrez L., Knoops L., Jacquemin P., Schuit F. A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation. Gastroenterology. 2012;142:119–129. doi: 10.1053/j.gastro.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Bolotin E., Liao H., Ta T.C., Yang C., Hwang-Verslues W., Evans J.R., Jiang T., Sladek F.M. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology. 2010;51:642–653. doi: 10.1002/hep.23357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giannelli F., Green P.M., Sommer S.S., Poon M., Ludwig M., Schwaab R., Reitsma P.H., Goossens M., Yoshioka A., Figueiredo M.S., Brownlee G.G. Haemophilia B: Database of point mutations and short additions and deletions—Eighth edition. Nucleic Acids Res. 1998;26:265–268. doi: 10.1093/nar/26.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rallapalli, P.M., Kemball-Cook, G., Tuddenham, E.G., Gomez, K., and Perkins, S.J. (2013). Factor IX Mutation Database, http://www.factorix.org. [DOI] [PubMed]
- 14.Reijnen M.J., Sladek F.M., Bertina R.M., Reitsma P.H. Disruption of a binding site for hepatocyte nuclear factor 4 results in hemophilia B Leyden. Proc. Natl. Acad. Sci. USA. 1992;89:6300–6303. doi: 10.1073/pnas.89.14.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crossley M., Brownlee G.G. Disruption of a C/EBP binding site in the factor IX promoter is associated with haemophilia B. Nature. 1990;345:444–446. doi: 10.1038/345444a0. [DOI] [PubMed] [Google Scholar]
- 16.Rausa F., Samadani U., Ye H., Lim L., Fletcher C.F., Jenkins N.A., Copeland N.G., Costa R.H. The cut-homeodomain transcriptional activator HNF-6 is coexpressed with its target gene HNF-3 beta in the developing murine liver and pancreas. Dev. Biol. 1997;192:228–246. doi: 10.1006/dbio.1997.8744. [DOI] [PubMed] [Google Scholar]
- 17.Jacquemin P., Lannoy V.J., Rousseau G.G., Lemaigre F.P. OC-2, a novel mammalian member of the ONECUT class of homeodomain transcription factors whose function in liver partially overlaps with that of hepatocyte nuclear factor-6. J. Biol. Chem. 1999;274:2665–2671. doi: 10.1074/jbc.274.5.2665. [DOI] [PubMed] [Google Scholar]
- 18.Dusing M.R., Maier E.A., Aronow B.J., Wiginton D.A. Onecut-2 knockout mice fail to thrive during early postnatal period and have altered patterns of gene expression in small intestine. Physiol. Genomics. 2010;42:115–125. doi: 10.1152/physiolgenomics.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith D.B., Johnson K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 20.Vidaud D., Tartary M., Costa J.M., Bahnak B.R., Gispert-Sanchez S., Fressinaud E., Gazengel C., Meyer D., Goossens M., Lavergne J.M. Nucleotide substitutions at the -6 position in the promoter region of the factor IX gene result in different severity of hemophilia B Leyden: Consequences for genetic counseling. Hum. Genet. 1993;91:241–244. doi: 10.1007/BF00218264. [DOI] [PubMed] [Google Scholar]
- 21.Crossley M., Whitelaw E., Perkins A., Williams G., Fujiwara Y., Orkin S.H. Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol. Cell. Biol. 1996;16:1695–1705. doi: 10.1128/mcb.16.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Funnell A.P., Maloney C.A., Thompson L.J., Keys J., Tallack M., Perkins A.C., Crossley M. Erythroid Krüppel-like factor directly activates the basic Krüppel-like factor gene in erythroid cells. Mol. Cell. Biol. 2007;27:2777–2790. doi: 10.1128/MCB.01658-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sommer S.S. Assessing the underlying pattern of human germline mutations: Lessons from the factor IX gene. FASEB J. 1992;6:2767–2774. doi: 10.1096/fasebj.6.10.1634040. [DOI] [PubMed] [Google Scholar]
- 24.Morgan G.E., Figueiredo M.S., Winship P.R., Baker R., Bolton-Maggs P.H., Brownlee G.G. The high frequency of the -6G—>A factor IX promoter mutation is the result both of a founder effect and recurrent mutation at a CpG dinucleotide. Br. J. Haematol. 1995;89:672–674. doi: 10.1111/j.1365-2141.1995.tb08388.x. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt D., Wilson M.D., Spyrou C., Brown G.D., Hadfield J., Odom D.T. ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009;48:240–248. doi: 10.1016/j.ymeth.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odom D.T., Dowell R.D., Jacobsen E.S., Gordon W., Danford T.W., MacIsaac K.D., Rolfe P.A., Conboy C.M., Gifford D.K., Fraenkel E. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt D., Schwalie P.C., Wilson M.D., Ballester B., Gonçalves A., Kutter C., Brown G.D., Marshall A., Flicek P., Odom D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012;148:335–348. doi: 10.1016/j.cell.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boland E.J., Liu Y.C., Walter C.A., Herbert D.C., Weaker F.J., Odom M.W., Jagadeeswaran P. Age-specific regulation of clotting factor IX gene expression in normal and transgenic mice. Blood. 1995;86:2198–2205. [PubMed] [Google Scholar]
- 29.Friedman A.D., Landschulz W.H., McKnight S.L. CCAAT/enhancer binding protein activates the promoter of the serum albumin gene in cultured hepatoma cells. Genes Dev. 1989;3:1314–1322. doi: 10.1101/gad.3.9.1314. [DOI] [PubMed] [Google Scholar]
- 30.Greenberg D., Miao C.H., Ho W.T., Chung D.W., Davie E.W. Liver-specific expression of the human factor VII gene. Proc. Natl. Acad. Sci. USA. 1995;92:12347–12351. doi: 10.1073/pnas.92.26.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clotman F., Jacquemin P., Plumb-Rudewiez N., Pierreux C.E., Van der Smissen P., Dietz H.C., Courtoy P.J., Rousseau G.G., Lemaigre F.P. Control of liver cell fate decision by a gradient of TGF beta signaling modulated by Onecut transcription factors. Genes Dev. 2005;19:1849–1854. doi: 10.1101/gad.340305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacquemin P., Durviaux S.M., Jensen J., Godfraind C., Gradwohl G., Guillemot F., Madsen O.D., Carmeliet P., Dewerchin M., Collen D. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol. Cell. Biol. 2000;20:4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemaigre F.P. Mechanisms of liver development: concepts for understanding liver disorders and design of novel therapies. Gastroenterology. 2009;137:62–79. doi: 10.1053/j.gastro.2009.03.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.