

Abstract
Congenital macrothrombocytopenia (CMTP) is a heterogeneous group of rare platelet disorders characterized by a congenital reduction of platelet counts and abnormally large platelets, for which CMTP-causing mutations are only found in approximately half the cases. We herein performed whole-exome sequencing and targeted Sanger sequencing to identify mutations that cause CMTP, in which a dominant mode of transmission had been suspected but for which no known responsible mutations have been documented. In 13 Japanese CMTP-affected pedigrees, we identified six (46%) affected by ACTN1 variants cosegregating with CMTP. In the entire cohort, ACNT1 variants accounted for 5.5% of the dominant forms of CMTP cases and represented the fourth most common cause in Japanese individuals. Individuals with ACTN1 variants presented with moderate macrothrombocytopenia with anisocytosis but were either asymptomatic or had only a modest bleeding tendency. ACTN1 encodes α-actinin-1, a member of the actin-crosslinking protein superfamily that participates in the organization of the cytoskeleton. In vitro transfection experiments in Chinese hamster ovary cells demonstrated that altered α-actinin-1 disrupted the normal actin-based cytoskeletal structure. Moreover, transduction of mouse fetal liver-derived megakaryocytes with disease-associated ACTN1 variants caused a disorganized actin-based cytoskeleton in megakaryocytes, resulting in the production of abnormally large proplatelet tips, which were reduced in number. Our findings provide an insight into the pathogenesis of CMTP.
Main Text
Congenital thrombocytopenia is a highly heterogeneous group of inherited disorders showing low platelet counts. A subgroup of these disorders is distinguished morphologically by the production of abnormally large platelets and is thus referred to as congenital macrothrombocytopenia (CMTP).1–3 The clinical presentations of CMTP-affected individuals vary considerably and range from no symptoms to a severe bleeding tendency. Thus far, CMTP-causing mutations have been reported in more than 12 genes.1–3 Among these, the most common is MYH9 (MIM 160775), which is responsible for autosomal-dominant MYH9 disorders or MYH9-related disease (MIM 153650, 153640, 600208, 155100, and 605249)4,5 The second most common are GP1BA (MIM 606672), GP1BB (MIM 138720), and GP9 (MIM 173515), which are responsible for autosomal-dominant (heterozygous) and -recessive (homozygous) Bernard-Soulier syndrome (MIM 231200).6,7 Less frequent mutations have been reported in FLI1 (MIM 193067),8 FLNA (MIM 300017),9 GATA1 (MIM 305371),10 ITGA2B/ITGB3 (MIM 607759 and 173470),11,12 NBEAL2 (MIM 614169),13–15 TUBB1 (MIM 612901),16 and VWF (MIM 613160).17 However, even this array of mutations only accounts for approximately half the CMTP cases; in the remaining half, CMTP-causing mutations are unknown, which prevents a definite diagnosis of CMTP and potentially results in inappropriate treatments.1–3
To identify mutations that cause CMTP, we first performed whole-exome sequencing of genomic DNA from 11 affected and 10 unaffected individuals from six Japanese CMTP-affected families (Figure 1A) in which a dominant mode of transmission had been suspected but in which no relevant mutations in the previously reported genes had been identified (Figure S1, available online). The candidate mutations found in the exome sequencing were further examined for germline mutations in additional CMTP-affected pedigrees and/or individuals by high-throughput sequencing of PCR products from pooled DNA and subsequent validation by Sanger sequencing (Figure 1B). Written informed consent was obtained from all individuals in accordance with the Declaration of Helsinki. This study was approved by the institutional review boards of National Hospital Organization Nagoya Medical Center, The University of Tokyo, and Nagoya University.
Figure 1.

A Flow Diagram of the Genomic Analysis
(A) Detection of the candidate germline variants through Genomon-exome.18
(B) Screening of ACTN1 variants with high-throughput sequencing of pooled DNA.18
For exome sequencing, genomic DNA from each member of the six pedigrees was enriched for protein-coding sequences with a SureSelect Human All Exon V3 kit (Agilent Technologies, Santa Clara, CA, USA). This was followed by massively parallel sequencing with the HiSeq 2000 platform with 100 bp paired-end reads (Illumina, San Diego, CA, USA). Candidate germline variants were detected through our in-house pipeline for exome-sequencing analysis with minor modifications for the detection of germline variants (Figure 1A).18 The obtained sequences were aligned to the UCSC Genome Browser hg19 with the Burrows-Wheeler Aligner.19 After removal of duplicate artifacts caused by PCR, the variants with an allele frequency > 0.25 were called. With a mean coverage of 117× (74×–176×), more than 92% of the 50 Mb target sequences were analyzed by greater than ten independent reads (Figure S2). In total, we identified 3,601 nucleotide variants that had not been registered in either the in-house SNP database or dbSNP131, and 360 of these cosegregated with macrothrombocytopenia in the corresponding families. The statistical analysis of the cosegregating variants indicated that only ACTN1 (MIM 102575, RefSeq accession number NM_001102.3), which encodes α-actinin-1 (ACTN1), was found to be significantly mutated in our cohort (Table S1), for which variants c.313G>A (p.Val105Ile), c.94C>A (p.Gln32Lys), and c.2255G>A (p.Arg752Gln) were confirmed in pedigrees exome-1, exome-2, and exome-3, respectively (Figures 2A and 2B and Table 1). Sanger sequencing in the other seven dominant CMTP-affected pedigrees revealed an additional three variants, c.137G>A (p.Arg46Gln), c.2212C>T (p.Arg738Trp), and c.673G>A (p.Glu225Lys), which also cosegregated with macrothrombocytopenia within the individual pedigrees (Figures 2A and 2B and Table 1). Combined, cosegregating ACTN1 variants were found in 6 (46%) out of 13 CMTP-affected pedigrees with a dominant mode of transmission (p = 1.67 × 10−6 compared to controls by a two-tailed Fisher’s exact test). No ACTN1 variants were detected among the 120 control individuals or 39 cases of sporadic CMTP, for which the mode of transmission had been unknown; the only exception was c.589C>T (p.Arg197Trp), which was found in a control individual who had a normal platelet count and size (Figure 1B and Figure S3). The multiple-sequence alignment indicated that all missense variants had highly conserved amino acid residues (Figure S4). Furthermore, different function-prediction programs suggested deleterious effects of these variants (Table S2). These results strongly suggest that these ACTN1 variants represent autosomal-dominant CMTP-causing mutations.
Figure 2.

Segregation of ACTN1 Variants, the ACTN1 Structure, α-Actinin-Actin Interaction, and the α-Actinin Domain Structure
(A) The pedigrees for the six families affected by ACTN1 variants are shown together with the Sanger-sequencing electropherograms. Filled symbols represent individuals affected by macrothrombocytopenia. The identified ACTN1 variants are shown below the symbols. The arrows indicate nucleotide changes. The following abbreviations are used: WT, wild-type; and NA, not available.
(B) Genomic organization of ACTN1, variants in which are identified in individuals with CMTP.
(C) α-actinin cross-links actin filaments into actin-filament bundles.
(D) α-actinin consists of an N-terminal actin-binding domain (ABD), composed of two calponin homology domains (CHD), four spectrin repeats (R1–R4), and a C-terminal calmodulin-like domain (CaM). Two molecules form an antiparallel dimer. The arrows indicate the positions of the identified ACTN1 variants.
Table 1.
ACTN1 Mutations and Platelet Characteristics
| Family | DNA Mutation | Protein Alteration | Platelet Count ( × 109/l) | Platelet Size (μm)a | Bleeding Tendency | Duke Bleeding Time (min) | Platelet Aggregation ADP (%) | Initial Diagnosis | |
|---|---|---|---|---|---|---|---|---|---|
| Exome-1 | mother | c.313G>A | p.Val105Ile | 80 | 3.7 ± 1.1 | none | - | - | - |
| proband | c.313G>A | p.Val105Ile | 91 | 3.8 ± 1.4 | none | - | - | immune thrombocytopenia | |
| Exome-2 | father | c.94C>A | p.Gln32Lys | 113 | 2.7 ± 0.9 | none | - | - | congenital thrombocytopenia |
| proband | c.94C>A | p.Gln32Lys | 54 | 3.1 ± 1.1 | none | 4.5 | 34 (2 μM) | congenital thrombocytopenia | |
| brother | c.94C>A | p.Gln32Lys | 124 | 2.9 ± 0.9 | epistaxis | - | - | congenital thrombocytopenia | |
| Exome-3 | mother | c.2255G>A | p.Arg752Gln | 100 | ND | none | - | - | immune thrombocytopenia |
| proband | c.2255G>A | p.Arg752Gln | 77 | 2.9 ± 0.6 | none | - | 49 (2 μM) | CMTP | |
| Sanger-1 | father | c.137G>A | p.Arg46Gln | 131 | 4.2 ± 1.3 | none | 1 | 76 (2 μM) | - |
| proband | c.137G>A | p.Arg46Gln | 120 | 3.9 ± 1.6 | none | - | 66 (2 μM) | CMTP | |
| sister | c.137G>A | p.Arg46Gln | 97 | 3.6 ± 1.4 | none | - | 74 (2 μM) | CMTP | |
| Sanger-2 | proband | c.2212C>T | p.Arg738Trp | 77 | 3.7 ± 1.5 | none | 2 | - | CMTP |
| Sanger-3 | proband | c.673G>A | p.Glu225Lys | 80 | 3.0 ± 0.9 | epistaxis | - | 59 (2.5 μM) | CMTP |
| mother | c.673G>A | p.Glu225Lys | 132 | 2.8 ± 0.8 | none | - | - | - | |
| Mean ± SD | - | - | - | 98.1 ± 23.2 | 3.4 ± 0.5 | - | - | - | - |
The following abbreviations are used: ND, not done; and CMTP, congenital macrothrombocytopenia.
Determined by a microscopic observation of 200 platelets on a stained peripheral-blood smear. Control size = 2.5 ± 0.3 μm (n = 31).
Individuals with mutant ACTN1 alleles had approximately half the normal platelet counts and a 30% increase in platelet size (Table 1 and Figure 3A). The platelet size in ACTN1-mutated individuals was smaller than that in individuals with a MYH9 disorder or homozygous Bernard-Soulier syndrome but was similar to that in individuals with heterozygous Bernard-Soulier syndrome or GPIIb/IIIa-associated macrothrombocytopenia (data not shown). Electron microscopy showed no other abnormalities (Figure 3B). The macrothrombocytopenia was accompanied by anisocytosis. Bleeding diathesis was absent or mild if present: only two individuals (15%) experienced occasional epistaxis, and the bleeding time was within the normal limit (Table 1). In addition, no apparent in vitro defects in platelet functions, including normal platelet aggregation (Table 1), clot retraction, and platelet spreading on glass surfaces, were noted in ACTN1-mutated individuals (Figure S5). Flow cytometry showed increased expression of platelet GPIb/IX and GPIIb/IIIa, which were thought to reflect the increased platelet size (data not shown).
Figure 3.

The Platelet Morphology
(A) Peripheral-blood smears were stained with May-Grünwald Giemsa for a normal control, the exome-1 proband, and the Sanger-3 proband. The affected individuals showed macrothrombocytopenia accompanied by anisocytosis. The number in each panel shows the mean platelet size (n = 200). Images were obtained with a BX50 microscope with a 100×/1.35 numeric aperture oil objective (Olympus, Tokyo, Japan). Images of the slides were acquired with a DP70 digital camera and DP manager software (Olympus). The original magnification is ×1,000.
(B) The ultrastructure of platelets from a normal control, the exome-3 proband, and the Sanger-3 proband. Platelet-rich plasma was prepared from acid-citrate-dextrose-citrated whole blood that was fixed in 2% glutaraldehyde and postfixed in 1% osmium tetroxide. The samples were then dehydrated in a graded ethanol series and n-butyl glycidyl ether and embedded in an epoxy resin (Epon 812; TAAB, Berkshire, UK). Ultrathin sections (0.1 μm) were doubly stained with 2% uranyl acetate and 1% lead citrate and were then observed with a transmission electron microscope (H-7650, Hitachi, Tokyo, Japan) at an accelerating voltage of 80 kV. The original magnification is ×1,500. The scale bar represents 2 μm.
The affected protein, α-actinin, is a member of the actin-crosslinking protein superfamily that participates in the organization of the cytoskeleton.20,21 Among the four known isoforms of α-actinin, ACTN2 (MIM 102573) and ACTN3 (MIM 102574) are relatively specific to muscle cells, whereas ACTN1 and ACTN4 (MIM 604638) are widely expressed in nonmuscle cells. Platelets and megakaryocytes mainly express ACTN1 and, to a lesser extent, ACTN4.22 The α-actinins exist as antiparallel dimers with an actin-binding domain (ABD) at the N terminus and cross-link actin filaments in bundles (Figure 2C). All of the identified ACTN1 mutations reside within the functional domain (the ABD and the C-terminal calmodulin-like domain), but not within the spacer spectrin repeats (Figure 2D). Therefore, ACTN1 variants that have differences in their structure and/or function might exert a dominant-negative effect, causing disorganization of actin filaments, although the localization and expression of ACTN1 in the individuals’ platelets was similar to that in controls (Figure S6). However, ACTN4 missense mutations, which are implicated in familial focal segmental glomerulosclerosis (MIM 603278), promote the aggregation of α-actinin-4 molecules and abrogate the integrity of actin filaments in transfected cells.23,24
We therefore first evaluated the effects of mutant ACTN1 on the organization of actin filaments by expressing each of the seven identified ACTN1 variants in Chinese hamster ovary (CHO) cells. A full-length ACTN1 sequence was amplified from normal platelet cDNA and constructed into mammalian expression vector pcDNA3.1 (Invitrogen, San Diego, CA, USA) with a 5′ Myc tag sequence. Six altered ACTN1 constructs (p.Gln32Lys, p.Arg46Gln, p.Val105Ile, p.Glu225Lys, p.Arg738Trp, and p.Arg752Gln) identified exclusively in the affected individuals and one construct (p.Arg197Trp) also identified in a control individual were prepared. Plasmid DNA was transfected into CHO cells with the PolyFect transfection reagent (QIAGEN, Hilden, Germany). Twenty-four hours after transfection, the ACTN1-transduced cells were replated on fibronectin (10 μg/ml)-coated coverslips for an immunofluorescence analysis.16 As shown in Figure 4, cells transduced with wild-type ACTN1 showed well-organized, fine actin-filament networks, where ACTN1 colocalized in a large part onto the actin filaments, whereas unbound ACTN1 was finely distributed within the cytoplasm. In contrast, except for the p.Arg197Trp variant found in a control individual, all ACTN1 variants caused varying degrees of disorganization of the actin filaments in variant-transduced cells, in which ACTN1 colocalized with less fine, shortened actin filaments and in which unbound ACTN1 was coarsely distributed within the cytoplasm (some cells showed punctuated or condensed staining).
Figure 4.

The Subcellular Localization of Altered ACTN1 in CHO Cells
CHO cells were transiently transfected with Myc-tagged wild-type or mutant ACTN1 cDNAs. After the cells were replated on fibronectin-coated coverslips, they were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. They were then stained with Myc-tagged antibody (Invitrogen), followed by Alexa-488-labeled goat anti-mouse IgG, Alexa-555-conjugated phalloidin (Invitrogen), and DAPI. Images were obtained with a BX50 fluorescence microscope with a 40×/1.00 numerical aperture oil objective lens. The cells shown are representative of eight independent experiments.
We further investigated whether altered ACTN1 also causes disorganization of the actin cytoskeleton in megakaryocytes by expressing wild-type and altered ACTN1 (p.Gln32Lys and p.Val105Ile) in primary mouse fetal liver-derived megakaryocytes by retrovirus-mediated gene transfer.11 We subcloned wild-type and mutant ACTN1 cDNAs into retroviral vector pGCDNsamIRES/EGFP with a 5′ Myc tag sequence and transfected them into 293 gpg packaging cells to obtain viral stocks.25 We used the vesicular stomatitis virus G protein to pseudotype the envelope of the virus. Mouse fetal liver cells harvested from embryonic day 13.5 embryos were transduced with either vehicle vector or pGCDNsamIRES/EGFP ACTN1 and were cultured with 40 ng/ml recombinant mouse thrombopoietin. The Experimental Animal Committee of Nagoya Medical Center approved the animal studies.
On posttransduction day 3, large megakaryocytes were enriched with a 1.5/3% BSA gradient and were then incubated on fibrinogen-coated coverslips for 45 min in the presence of 100 nM phorbol myristate acetate.26 Cells were fixed, permeabilized, and stained with phalloidin, and then green-fluorescent-protein (GFP)-positive megakaryocytes were observed under a fluorescence microscope. Megakaryocytes adhere to fibrinogen-coated surfaces through the integrin αIIbβ3 receptor and undergo cytoskeletal reorganization. Cells transduced with wild-type ACTN1 reorganized the actin cytoskeleton with the development of organized actin filaments, which were circumferential and parallel to the cell periphery. However, the circumferential actin-filament network was less organized in the megakaryocytes transduced with altered ACTN1 than in control cells (Figure 5A). These findings indicate that CMTP-associated ACTN1 mutations dominantly affect the actin-filament assembly and might result in abnormal cytoskeletal organization.
Figure 5.
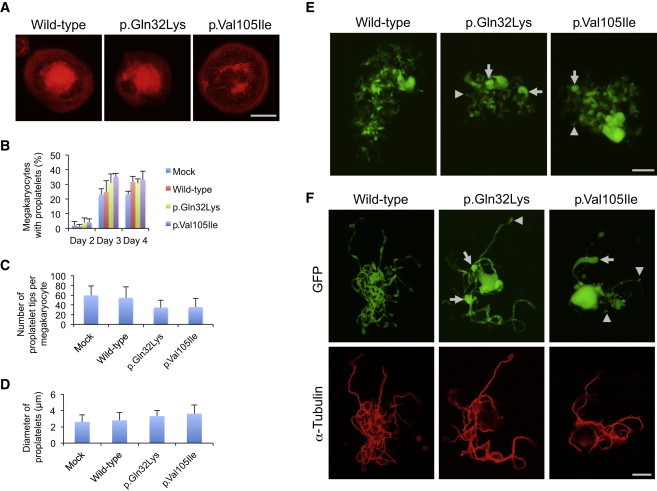
Abnormal Proplatelet Formation in Megakaryocytes Transduced with Altered ACTN1
(A) The organization of the actin filaments in megakaryocytes adhered to fibrinogen. The scale bar represents 50 μm.
(B) The percentage of megakaryocytes extending proplatelets was evaluated under an IX71 fluorescence microscope with a 20×/0.40 objective lens (Olympus) 2–4 days after infection. For each specimen, at least 100 megakaryocytes were evaluated.
(C) The number of proplatelet tips per megakaryocyte was decreased in megakaryocytes transduced with mutant ACTN1.
(D) The size of proplatelet tips was increased in megakaryocytes transduced with altered ACTN1.
(E) Representative megakaryocytes extending proplatelets 3 days after infection. Note that the size of the proplatelet tips was variable in the megakaryocytes transduced with altered ACTN1 (arrows indicate large tips, and arrowheads indicate small tips). The scale bar represents 50 μm.
(F) The proplatelet morphology and α-tubulin localization of EGFP-positive megakaryocytes. Infection day 3 cultures were cytospun onto glass slides. Cells were then fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and stained with GFP antibody (D153-3; Medical & Biological Laboratories, Nagoya, Japan) and α-tubulin antibody (RB-9281; Lab Vision, Fremont, CA), followed by Alexa-488-labeled goat anti-rat IgG and Alexa-555-labeled goat anti-rabbit IgG (Invitrogen). Images were obtained with a BX50 microscope with a 100×/1.35 numeric aperture oil objective lens (Olympus). The size of the proplatelet tips was variable in megakaryocytes transduced with altered ACTN1 (arrows indicate large tips, and arrowheads indicate small tips). The scale bar represents 20 μm. Representative images from four independent experiments are shown.
According to the current model of platelet production, differentiated megakaryocytes extend long, branching multiple microtubule-based protrusions, called proplatelets, into the bone-marrow sinusoids, which are composed of platelet-sized swellings, or tips, in tandem arrays that are mutually connected by thin cytoplasmic bridges.27 Proplatelets are then released into the blood stream, where terminal production of mature platelets takes place through several intermediate forms.28 The proplatelet formation and their release from megakaryocytes, as well as the subsequent production of mature platelets in the periphery, are spatially and temporally regulated by dynamic reorganization of the cytoskeletons and signal transduction.29 Several lines of evidence suggest that macrothrombocytopenias are caused by defects in the regulation of terminal platelet production.30 In fact, most of the previously reported defects responsible for CMTP affect genes involved in the signaling pathways and/or components of the cytoskeleton;30 these defects include mutations in integrin αIIbβ3 (ITGA2B/ITGB3),11,12 GPIb/IX (GP1BA, GP1BB, and GP9),5,6 nonmuscle myosin heavy chain IIA (MYH9),4,5 actin-binding protein filamin A (FLNA),9 and β1-tubulin (TUBB1).16
To further explore the effects of mutant ACTN1 on proplatelet formation, after transducing ACTN1 variants into primary mouse fetal liver-derived megakaryocytes, we monitored proplatelet formation in enhanced-GFP (EGFP)-positive megakaryocytes in suspension by inverted fluorescence microscopy. The cultures were then spread onto glass slides and subsequently analyzed by immunofluorescence staining. During the 4 day culture period, the proportion of proplatelet-formation-positive megakaryocytes was not different among wild-type-, p.Gln32Lys-, and p.Val105Ile-transduced megakaryocytes, suggesting that ACTN1 variants do not accelerate or inhibit the rate of proplatelet formation and/or platelet production (Figure 5B). However, the number of proplatelet tips tended to be decreased, and the size of the tips increased in the megakaryocytes transduced with both altered forms of ACTN1 (Figures 5C and 5D).
During proplatelet formation, microtubules function to propel proplatelet elongation, and the actin cytoskeleton plays a critical role in bending and bifurcating the proplatelet shaft to increase the number of proplatelet tips. Cytochalasin-mediated inhibition of actin polymerization has been shown to interfere with proplatelet branching.27 Moreover, mouse models for null alleles of actin-modulating genes, such as cofilin-1 (Cfl1), and a hypomorphic allele of WD repeat domain 1 (Wdr1) exhibit macrothrombocytopenia, indicating that dynamic changes in the actin cytoskeleton are required for normal platelet production.31,32 Because ACTN1 plays a pivotal role in crosslinking actin filaments,20,21 defective ACTN1 variants could cause deregulated actin-filament organization, as shown in CHO cells and cultured megakaryocytes (Figures 4 and 5A). Furthermore, we found that the platelet tips in wild-type-ACTN1-transduced megakaryocytes were uniform in size, whereas they were heterogeneous in the variant-transduced cells (Figures 5E and 5F). These results are consistent with the thrombocytopenia and increased platelet size (macrothrombocytopenia) accompanied by anisocytosis in the affected individuals, demonstrating that abnormal ACTN1 affects proplatelet formation.
In addition to playing a role in actin bundling, α-actinins have been shown to interact with a number of other cytoskeletal and receptor proteins.20,21 In platelets, ACTN1 binds to β integrins and can mediate their signaling.33 Thus, similar to the activating mutations in ITGA2B/ITGB3,11,12 ACTN1 mutations could also affect proplatelet formation through abnormal integrin signaling.
In summary, we identified ACTN1 mutations that cause autosomal-dominant CMTP. In our cohort, ACNT1 variants accounted for 5.5% of the dominant forms of CMTP cases, representing the fourth most common cause of Japanese CMTP. Our finding clearly demonstrates the effectiveness of whole-exome sequencing of CMTP-affected pedigrees for clarifying their genetic basis, although the limitation of the available pedigree samples for exome sequencing prevented further identification of the causative genes for the remaining families. Such studies also facilitate the differential diagnosis of congenital macrothrombocytopenia and can provide important information about the mechanisms of platelet production.
Acknowledgments
This work was supported by Research on Measures for Intractable Diseases Project (H23-012), a matching fund subsidy from the Ministry of Health Labour and Welfare, the Japan Society for the Promotion of Science (Grant-in-Aid for Scientific Research 20591161 and 23591429), and the National Hospital Organization Research Fund (Network Research Grant for Congenital Thrombocytopenia). The supercomputing resources were provided by the Human Genome Center, the Institute of Medical Science, The University of Tokyo. We thank R.C. Mulligan for the 293 gp and 293 gpg cells, M. Onodera for pGCDNsamIRES/EGFP, K. Oguri and Y. Kusano for discussions on the expression analysis, and Y. Kito for technical assistance.
Supplemental Data
Web Resources
The URLs for the data presented herein are as follows:
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/index.shtml
ClustalW2 - Multiple Sequence Alignment, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Genomon-exome, http://genomon.hgc.jp/exome/en/index.html
Mutation Taster, http://www.mutationtaster.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2
SAMtools, http://samtools.sourceforge.net/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Balduini C.L., Savoia A. Genetics of familial forms of thrombocytopenia. Hum. Genet. 2012;131:1821–1832. doi: 10.1007/s00439-012-1215-x. [DOI] [PubMed] [Google Scholar]
- 2.Kunishima S., Saito H. Congenital macrothrombocytopenias. Blood Rev. 2006;20:111–121. doi: 10.1016/j.blre.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Nurden A.T., Freson K., Seligsohn U. Inherited platelet disorders. Haemophilia. 2012;18(Suppl 4):154–160. doi: 10.1111/j.1365-2516.2012.02856.x. [DOI] [PubMed] [Google Scholar]
- 4.Kunishima S., Saito H. Advances in the understanding of MYH9 disorders. Curr. Opin. Hematol. 2010;17:405–410. doi: 10.1097/MOH.0b013e32833c069c. [DOI] [PubMed] [Google Scholar]
- 5.Balduini C.L., Pecci A., Savoia A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br. J. Haematol. 2011;154:161–174. doi: 10.1111/j.1365-2141.2011.08716.x. [DOI] [PubMed] [Google Scholar]
- 6.Kunishima S., Kamiya T., Saito H. Genetic abnormalities of Bernard-Soulier syndrome. Int. J. Hematol. 2002;76:319–327. doi: 10.1007/BF02982690. [DOI] [PubMed] [Google Scholar]
- 7.Berndt M.C., Andrews R.K. Bernard-Soulier syndrome. Haematologica. 2011;96:355–359. doi: 10.3324/haematol.2010.039883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hart A., Melet F., Grossfeld P., Chien K., Jones C., Tunnacliffe A., Favier R., Bernstein A. Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity. 2000;13:167–177. doi: 10.1016/s1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- 9.Nurden P., Debili N., Coupry I., Bryckaert M., Youlyouz-Marfak I., Solé G., Pons A.C., Berrou E., Adam F., Kauskot A. Thrombocytopenia resulting from mutations in filamin A can be expressed as an isolated syndrome. Blood. 2011;118:5928–5937. doi: 10.1182/blood-2011-07-365601. [DOI] [PubMed] [Google Scholar]
- 10.Nichols K.E., Crispino J.D., Poncz M., White J.G., Orkin S.H., Maris J.M., Weiss M.J. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet. 2000;24:266–270. doi: 10.1038/73480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunishima S., Kashiwagi H., Otsu M., Takayama N., Eto K., Onodera M., Miyajima Y., Takamatsu Y., Suzumiya J., Matsubara K. Heterozygous ITGA2B R995W mutation inducing constitutive activation of the αIIbβ3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood. 2011;117:5479–5484. doi: 10.1182/blood-2010-12-323691. [DOI] [PubMed] [Google Scholar]
- 12.Nurden A.T., Fiore M., Nurden P., Pillois X. Glanzmann thrombasthenia: A review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118:5996–6005. doi: 10.1182/blood-2011-07-365635. [DOI] [PubMed] [Google Scholar]
- 13.Kahr W.H., Hinckley J., Li L., Schwertz H., Christensen H., Rowley J.W., Pluthero F.G., Urban D., Fabbro S., Nixon B. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat. Genet. 2011;43:738–740. doi: 10.1038/ng.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunay-Aygun M., Falik-Zaccai T.C., Vilboux T., Zivony-Elboum Y., Gumruk F., Cetin M., Khayat M., Boerkoel C.F., Kfir N., Huang Y. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat. Genet. 2011;43:732–734. doi: 10.1038/ng.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albers C.A., Cvejic A., Favier R., Bouwmans E.E., Alessi M.C., Bertone P., Jordan G., Kettleborough R.N., Kiddle G., Kostadima M. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat. Genet. 2011;43:735–737. doi: 10.1038/ng.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunishima S., Kobayashi R., Itoh T.J., Hamaguchi M., Saito H. Mutation of the β1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood. 2009;113:458–461. doi: 10.1182/blood-2008-06-162610. [DOI] [PubMed] [Google Scholar]
- 17.Jackson S.C., Sinclair G.D., Cloutier S., Duan Z., Rand M.L., Poon M.C. The Montreal platelet syndrome kindred has type 2B von Willebrand disease with the VWF V1316M mutation. Blood. 2009;113:3348–3351. doi: 10.1182/blood-2008-06-165233. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 19.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otey C.A., Carpen O. α-actinin revisited: A fresh look at an old player. Cell Motil. Cytoskeleton. 2004;58:104–111. doi: 10.1002/cm.20007. [DOI] [PubMed] [Google Scholar]
- 21.Sjöblom B., Salmazo A., Djinović-Carugo K. α-actinin structure and regulation. Cell. Mol. Life Sci. 2008;65:2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haudek V.J., Slany A., Gundacker N.C., Wimmer H., Drach J., Gerner C. Proteome maps of the main human peripheral blood constituents. J. Proteome Res. 2009;8:3834–3843. doi: 10.1021/pr801085g. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan J.M., Kim S.H., North K.N., Rennke H., Correia L.A., Tong H.Q., Mathis B.J., Rodríguez-Pérez J.C., Allen P.G., Beggs A.H., Pollak M.R. Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 24.Yao J., Le T.C., Kos C.H., Henderson J.M., Allen P.G., Denker B.M., Pollak M.R. α-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol. 2004;2:e167. doi: 10.1371/journal.pbio.0020167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nabekura T., Otsu M., Nagasawa T., Nakauchi H., Onodera M. Potent vaccine therapy with dendritic cells genetically modified by the gene-silencing-resistant retroviral vector GCDNsap. Mol. Ther. 2006;13:301–309. doi: 10.1016/j.ymthe.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 26.Shiraga M., Ritchie A., Aidoudi S., Baron V., Wilcox D., White G., Ybarrondo B., Murphy G., Leavitt A., Shattil S. Primary megakaryocytes reveal a role for transcription factor NF-E2 in integrin α IIb β 3 signaling. J. Cell Biol. 1999;147:1419–1430. doi: 10.1083/jcb.147.7.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Italiano J.E., Jr., Lecine P., Shivdasani R.A., Hartwig J.H. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J. Cell Biol. 1999;147:1299–1312. doi: 10.1083/jcb.147.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thon J.N., Montalvo A., Patel-Hett S., Devine M.T., Richardson J.L., Ehrlicher A., Larson M.K., Hoffmeister K., Hartwig J.H., Italiano J.E., Jr. Cytoskeletal mechanics of proplatelet maturation and platelet release. J. Cell Biol. 2010;191:861–874. doi: 10.1083/jcb.201006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartwig J.H., Italiano J.E., Jr. Cytoskeletal mechanisms for platelet production. Blood Cells Mol. Dis. 2006;36:99–103. doi: 10.1016/j.bcmd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Thon J.N., Italiano J.E., Jr. Does size matter in platelet production? Blood. 2012;120:1552–1561. doi: 10.1182/blood-2012-04-408724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bender M., Eckly A., Hartwig J.H., Elvers M., Pleines I., Gupta S., Krohne G., Jeanclos E., Gohla A., Gurniak C. ADF/n-cofilin-dependent actin turnover determines platelet formation and sizing. Blood. 2010;116:1767–1775. doi: 10.1182/blood-2010-03-274340. [DOI] [PubMed] [Google Scholar]
- 32.Kile B.T., Panopoulos A.D., Stirzaker R.A., Hacking D.F., Tahtamouni L.H., Willson T.A., Mielke L.A., Henley K.J., Zhang J.G., Wicks I.P. Mutations in the cofilin partner Aip1/Wdr1 cause autoinflammatory disease and macrothrombocytopenia. Blood. 2007;110:2371–2380. doi: 10.1182/blood-2006-10-055087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tadokoro S., Nakazawa T., Kamae T., Kiyomizu K., Kashiwagi H., Honda S., Kanakura Y., Tomiyama Y. A potential role for α-actinin in inside-out αIIbβ3 signaling. Blood. 2011;117:250–258. doi: 10.1182/blood-2009-10-246751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.