

Abstract
It is more convenient and practical to collect rectal swabs than stool specimens to study carriage of colon pathogens. In this study, we examined the ability to use rectal swabs rather than stool specimens to quantify Klebsiella pneumoniae carbapenemase (KPC)-producing carbapenem-resistant Enterobacteriaceae (CRE). We used a quantitative real-time PCR (qPCR) assay to determine the concentration of the blaKPC gene relative to the concentration of 16S rRNA genes and a quantitative culture-based method to quantify CRE relative to total aerobic bacteria. Our results demonstrated that rectal swabs are suitable for quantifying the concentration of KPC-producing CRE and that qPCR showed higher correlation between rectal swabs and stool specimens than the culture-based method.
INTRODUCTION
Klebsiella pneumoniae carbapenemase (KPC)-type enzymes are β-lactamases, capable of hydrolyzing all known β-lactam antibiotics (1). The spread of the blaKPC genes has led to the emergence of carbapenem-resistant Enterobacteriaceae (CRE), mostly K. pneumoniae, as important nosocomial pathogens causing outbreaks in various parts of the world (2). Infections by these pathogens are severe, with an estimated case fatality rate of 35%, and thus considered a clinical and public health threat (3).
Active surveillance of high-risk patients has been advocated in areas where CRE are endemic or where there are ongoing outbreaks (4). Although stool samples are considered the “gold standard” specimen for studying gut bacteria in general (5, 6) and for detecting CRE in particular (7), hospital epidemiologists, clinical microbiologists, and researchers frequently use rectal swabs due to practical considerations, such as ease of collection, handling, and processing (8–11). Several studies compared the qualitative sensitivity of rectal swabs versus stool specimens for the recovery and detection of various pathogens, such as Campylobacter fetus subsp. jejuni, vancomycin-resistant Enterococcus faecium (VRE), and Lawsonia intracellularis, by culturing and/or molecular quantification methods (12–14). These studies had mixed results, with some studies reporting comparable test sensitivity with either sample type (12–14) and others reporting significantly lower sensitivity with rectal swabs (15, 16). For quantitative testing, rectal swabs are believed to be inadequate due to the high variation in the quantity of fecal material on each swab and the difficulties in determining this quantity (17–20).
As the human colon flora is a diverse ecosystem, which is populated by both anaerobic and aerobic microorganisms (21), development of quantitative methods, such as culture- and quantitative real-time PCR (qPCR)-based techniques is important for studying the bacterial composition of this complex ecosystem. The use of these methods can lead to a thorough knowledge about gastrointestinal community composition, the effects of antibiotics, and their roles in health and disease.
Here, we suggest an alternate methodology for quantifying pathogens that would overcome the problem of the unknown quantity of fecal material on the rectal swab by assessing the relative concentration of the target bacteria compared to total bacteria. We used two quantification methods: a culture-based method comparing the number of CRE to total aerobic culturable bacterial growth and a quantitative real-time PCR method for detection and quantification of blaKPC genes and 16S rRNA genes as a total bacterial reference gene. We evaluated whether, by using these methods, rectal swabs are adequate substitute for stool samples to quantify KPC-producing CRE (KPC-CRE).
MATERIALS AND METHODS
Sample collection.
Specimens were collected from 37 hospitalized patients known to be KPC-producing CRE carriers; they were identified as carriers by blaKPC PCR-based testing (22). Stool samples and rectal swabs (perirectal swabs were not allowed) were simultaneously collected from each CRE carrier and immediately transported to the lab. Rectal samples were collected using eSwab (Copan, Brescia, Italy) by inserting the swab 1 cm into the rectum while rotating the swab. Following sampling, the swab was placed in the transport tube (tube supplied by the manufacturer) containing 1 ml sterile Amies transport medium and was vortexed for 1 min at maximum speed upon arrival to the lab. In parallel, the weight of the stool sample (wet weight) was determined, diluted 1:10 (wt/vol) with 0.9% saline, and vortexed for 5 min at maximum speed to generate a stool suspension. Thirty-seven stool and rectal swab specimens were subjected to analysis; 22 pairs were analyzed in parallel using both culture-based method and qPCR-based analysis. The remaining 15 pairs were analyzed only by one method due to insufficient material submitted (in most cases, the stool sample, not the rectal swab, was the specimen type with insufficient quantity). In addition, a known CRE carrier patient was sampled 5 times over a 1-month period, and the specimens were analyzed for temporal changes.
Culture-based quantification of KPC-CRE.
After preliminary testing to ascertain the optimal conditions, the stool suspension (500 μl) and the eSwab Amies transport medium containing bacteria (100 μl) were used for performing viable bacterial counts by serial 10-fold dilutions in 0.9% saline. Bacteria were plated on tryptic soy agar plates supplemented with 5% sheep blood (HyLabs, Rehovot, Israel) to determine the total culturable aerobic bacteria (TAB), and bacteria were plated on CHROMagar KPC plates (HyLabs, Rehovot, Israel) to determine the CRE bacteria. Viable bacterial counts were determined after 18 h of growth at 37°C, and the ratio of CFU/ml of CRE to TAB was determined and expressed as CRE/TAB or log CRE/TAB.
Molecular biology-based quantification of KPC-CRE. (i) DNA extraction from pure cultures, rectal swabs, and stool samples.
The reference bacterial strains used for molecular quantification are listed in Table S1 in the supplemental material. These strains were used to evaluate the performance (sensitivity and specificity) of the PCR primer-probe sets.
A bacterial colony from each of these strains was suspended in 100 μl molecular biology-grade water (Bio-Lab, Jerusalem, Israel) and was used for lysate preparation (20 min at 100°C followed by 10 min at −20°C). Specifically, 3.6 ml of the stool suspension and 300 μl of the eSwab Amies transport medium containing bacteria were subjected to bacterial DNA extraction using the UltraClean fecal DNA isolation kit (MO BIO, Carlsbad, CA) and BiOstic bacteremia DNA isolation kit (MO BIO, Carlsbad, CA), respectively.
(ii) Design of primers and probes.
The primers and probe used in this study are listed in Table 1. An alternative set of primers and probe for blaKPC has been reported (25); however, since the primer and probe set listed in Table 1 had already been confirmed in our laboratory (see below), we elected to continue using them. The blaKPC primers and probe set were designed for conserved regions based on alignment of blaKPC sequences deposited at the NCBI site (http://www.ncbi.nlm.nih.gov/), and their specificities were confirmed in silico using the BLAST network service (26; http://blast.ncbi.nlm.nih.gov/).
Table 1.
qPCR primers and probe used in this study
| Primer or probe | Group specificity | Sequence (5′→3′) | Reference |
|---|---|---|---|
| Primersa | |||
| 515F | Eubacteria | GTGCCAGCAGCCGCGGTAA | 23 |
| 685R | Eubacteria | TCTACGCATTTCACCGCTAC | 24 |
| blaKPC15F | Gut KPC producers | CCGTCTAGTTCTGCTGTCTTGT | This study |
| blaKPC177R | Gut KPC producers | GTAACTTACAGTTGCGCCTGAG | This study |
| blaKPC probe | Gut KPC producers | CTGACCAACCTCGTCG | This study |
The direction of the primer is indicated at the end of the primer name as follows: F for forward and R for reverse.
(iii) Conventional PCR.
For specificity testing, conventional PCR was used on bacterial lysates and fecal DNA as mentioned above. The PCR mixtures (25 μl) contained 0.125 μl of HotStarTaq DNA polymerase (Qiagen, Hilden, Germany), 2.5 μl of 10× buffer (Qiagen, Hilden, Germany), 2.25 mM MgCl2 (Qiagen, Hilden, Germany), 0.8 μM of each primer, 0.2 mM of each deoxynucleoside triphosphate (dNTP) (Qiagen, Hilden, Germany), and 1 μl of template DNA. Amplifications were performed with an initial denaturation step of 15 min at 95°C, followed by 35 cycles, with each cycle consisting of denaturation for 30 s at 94°C, annealing for 20 s at 60°C, and elongation for 20 s at 72°C. A final extension step was performed at 72°C for 5 min. The PCR product was purified, cloned into pDrive cloning vector (Qiagen, Hilden, Germany) and sequenced (Macrogen Inc., Seoul, South Korea). The identities of the cloned fragments were determined using the BLAST network service.
(iv) qPCR.
Quantitative real-time PCR was performed in two singleplex assays, one for assessing the total bacteria using the 16S rRNA gene primers set 515F and 685R (SYBR green reaction; Table 1) and the other for assessing colonic KPC producers with the newly designed KPC primers and TaqMan MGB probe (Table 1). For standards for each singleplex reaction, we used the pDrive cloning vector containing the 16S rRNA gene or the blaKPC-3 gene. The standards were diluted to generate samples ranging from approximately 1 to 1 × 106 copies per PCR. Each singleplex reaction tube contained 4 μl of DNA, 0.25 μM each blaKPC primer or 0.2 μM each 16S rRNA gene primer (Table 1), 10 μl of 2 × ABsolute blue QPCR ROX mix (Thermo Fisher Scientific, Epsom, United Kingdom) (for the TaqMan reaction) or 2 × ABsolute blue SYBR green ROX mix (Thermo Fisher Scientific, Epsom, United Kingdom) (for the SYBR green reaction), 0.2 μM TaqMan probe (Table 1), and molecular biology-grade water up to 20 μl. Cycling conditions consisted of one holding period at 95°C for 15 min, followed by 50 cycles, with each cycle consisting of 10 s at 95°C, 15 s (for the SYBR green reaction) or 60 s (for the TaqMan reaction) at 60°C, and 15 s at 72°C for the SYBR green reaction only. All reactions were carried out in a Rotor-Gene 6000 (Corbett Life Science, Mortlake, Australia) in duplicate samples. The results were expressed as a ratio of the number of copies of the blaKPC gene/number of copies of 16S rRNA genes (blaKPC/16S rRNA genes) or log blaKPC/16S rRNA genes.
(v) Specificity, sensitivity, and inhibition of the qPCR assays.
Suspensions (in molecular biology-grade water [Bio-Lab, Jerusalem, Israel]) of overnight colonies of Klebsiella pneumoniae carrying the blaKPC-3 gene (KPC-Kpn), Klebsiella oxytoca, Salmonella sp., Proteus sp., Citrobacter koseri, Enterobacter cloacae, and Enterococcus gallinarum were normalized to an optical density at 600 nm (OD600) of 1 using a spectrophotometer (UVmini-10240; Shimadzu, MD), approximately 1 × 108 CFU/ml, and bacterial lysates were prepared as mentioned above. The specificity of the real-time TaqMan assay was determined on KPC-Kpn bacterial lysate as the target and on a bacterial mixture containing the target and nontarget microorganisms. The specificity of the 16S rRNA gene qPCR SYBR reaction was determined using the bacterial lysates of K. oxytoca, Proteus sp., Enterobacter cloacae, and Enterococcus gallinarum separately and in combination. To determine the effect of fecal material on the sensitivities of our assays, DNA extracted from rectal swabs of KPC carriers was diluted 10-fold from 100 to 10−2, spiked with lysate of the target bacteria KPC-Kpn (TaqMan assay) or with K. oxytoca (16S rRNA gene qPCR SYBR reaction) and with the mixtures mentioned above. The copy numbers obtained were compared to those obtained for the target bacteria alone in water.
Statistical analysis.
To identify correlations between the values for rectal swabs and stool samples, a linear regression analysis was performed with the use of Microsoft Excel (Microsoft Corporation, Redmond, WA) and the Analyze-It version 2.26 software (Analyze-it Software, Ltd., Leeds, United Kingdom). Three outliers out of the 60 samples in the correlation graphs, two in the microbiological (culture) graph and one in the molecular quantification graph were not included in the final statistical analysis. The outliers were not specific to the stool sample or to the swab sample, and the reasons for these outliers are unknown. As they divert from the general behavior of all the other samples, they were excluded from the correlation analysis. Nevertheless, the correlations with these outliers are indicated in the Results section, and they did not change the conclusions. Statistical analysis of the regression plots was done using the JMP IN v 3.2.1 software (SAS Institute Inc.). All tests were two-sided, and a P value of <0.05 was considered significant.
RESULTS
Establishment of a qPCR for the blaKPC gene and the 16S rRNA genes.
In this study, we chose to use quantitative PCR as a molecular biology-based method to compare two sampling methods, rectal swabs versus stool samples, using two singleplex assays. We first evaluated the performance of the two qPCR assays (blaKPC gene and 16S rRNA genes) by following guidelines presented by Espy et al. (27) and Bustin et al. (28).
qPCR evaluation. (i) Specificity of the blaKPC and 16S rRNA gene qPCRs.
The performance of the 16S rRNA gene PCR assay and the newly designed blaKPC qPCR assay was evaluated by PCR on 35 DNA samples purified from target strains (i.e., carrying the blaKPC gene; n = 5) and nontarget strains (not carrying the blaKPC gene; n = 30) (see Table S1 in the supplemental material).
PCR amplifications revealed a single band of the expected size for the blaKPC gene and the 16S rRNA genes, 160 bp and 180 bp, respectively. Using the blaKPC primers, only DNA from bacteria that were known to contain the blaKPC gene (target bacteria) were amplified (data not shown). We further tested the specificity on DNA extracted from rectal swabs of 2 known KPC carriers. Sequencing of the PCR products showed 100% identity to known KPC or 16S rRNA gene sequences (data not shown). These results show that the primers and the assay conditions for the detection of blaKPC and 16S rRNA genes are highly specific.
(ii) Sensitivity of the blaKPC-3 and 16S rRNA gene qPCRs.
We analyzed the sensitivity and precision of the reaction by serial dilutions of known concentrations of plasmids carrying the blaKPC-3 gene or 16S rRNA genes. The amplification was linear over 6 log dilutions for the blaKPC (r2 = 0.996; slope, −3.4299; efficiency = 96%; P < 0.0001; TaqMan qPCR) and over 7 log dilutions for the 16S rRNA gene (r2 = 0.999; slope, −3.350; efficiency = 98%; P < 0.0001; SYBR green-based qPCR). The detection limit was 10 and 40 plasmid molecules/PCR for the blaKPC and 16S rRNA gene assays, respectively. The performance of the assays on purified plasmid DNA (105 copies) over 10 runs indicated that the qPCR was highly stable and precise with a mean threshold cycle (CT) of 18.34 ± 0.13 or 19.081 ± 0.182419 for blaKPC and 16S rRNA gene qPCR, respectively.
(iii) Inhibition.
DNA extracted from fecal specimens often contains PCR inhibitors that can reduce the sensitivity of the qPCRs (15). Therefore, we evaluated the effect of fecal DNA on the sensitivity of the reaction. We found inhibition only in an undiluted fecal DNA sample, whereas further 10-fold serial dilutions showed no inhibition. Spiking of KPC-Kpn or K. oxytoca alone or preprepared in an artificial mix with fecal DNA extracts did not affect the sensitivity of the assay (data not shown).
Enumeration of CRE and blaKPC-carrying populations in rectal swabs versus stool samples in different individuals.
After the qPCRs were established, we used the assay to assess the CRE and blaKPC-carrying populations in stool samples and rectal swabs of 37 known KPC carriers. We determined the amount of CRE and TAB (22) and the amount of blaKPC and 16S rRNA genes (with the newly developed qPCR assays) and then calculated the ratios of CRE/TAB or blaKPC/16S rRNA genes in each of the samples. In the culture-based method, the CRE/TAB showed positive linear correlation between stool samples and rectal swabs (Fig. 1A; r2 = 0.367, P < 0.0002). A higher positive linear correlation was observed between the blaKPC/16S rRNA genes in stool samples versus rectal swabs (Fig. 1B; r2 = 0.8397, P < 0.0001). When the data were analyzed using log transformation, we observed a higher correlation between the stool samples and rectal swabs by the molecular quantification method than by the culture-based method (P < 0.0001 and P = 0.0022, respectively; Fig. 1C and D). Finally, we examined the correlation between the values of the culture-based method and the molecular quantification method (Fig. 2A and B). The two methods had a correlation value of close to 1; y = 0.9882x for stool (r2 = −0.862, P = 0.1664; Fig. 2A) and y = 1.1602x for rectal swab (r2 = −0.275, P = 0.0461; Fig. 2B) with a significant P value for the rectal swab samples.
Fig 1.

(A) Correlation between the values of CRE/TAB of stool samples versus rectal swabs (as percentages) as obtained by the bacteriological (culture-based) method (two outliers [two filled circles] were not included in the analysis shown [see “Statistical analysis” in Materials and Methods]; r2 = 0.045 when the two points are included). (B) Correlation between the values of blaKPC/16S rRNA genes of stool samples versus rectal swabs (as percentages) as obtained by the molecular quantification method (one extreme outlier [one filled circle] was not included in the analysis showed [see “Statistical analysis” in Materials and Methods]; r2 = 0.397 when the point is included). The broken lines indicate 95% confidence intervals (95% CI). (C) Correlation between the log values of CRE/TAB of stool samples versus rectal swabs as obtained by the bacteriological method (the two outliers excluded from panel A were included). (D) Correlation between the log values of blaKPC/16S rRNA genes of stool samples versus rectal swabs as obtained by the molecular quantification method (the one extreme point excluded from panel B was included).
Fig 2.
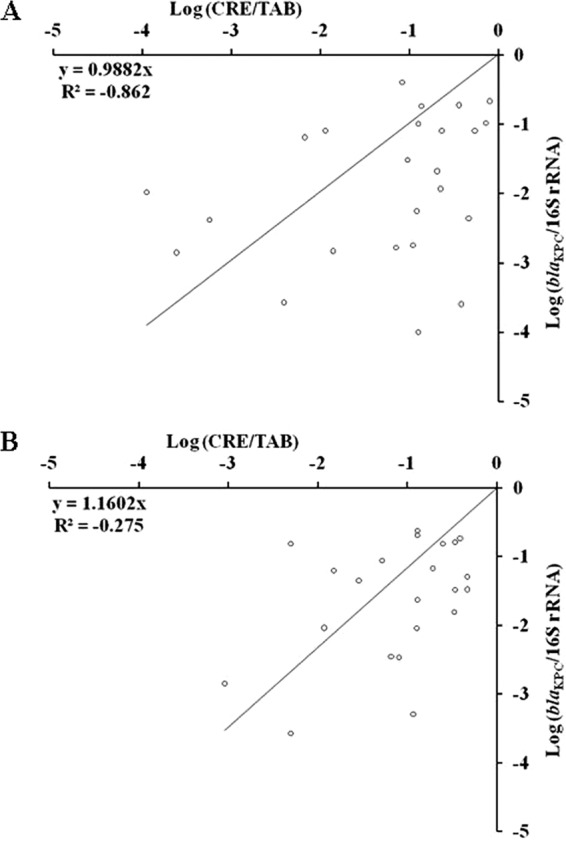
(A) Correlation between the values for the culture-based method (log CRE/TAB) versus the molecular quantification method (log blaKPC/16S rRNA genes) for stool samples. (B) Correlation between the values of the culture-based method (log CRE/TAB) versus the molecular quantification method (log blaKPC/16S rRNA genes) for rectal swabs.
As shown in Table 2, we noticed that in the culture-based analysis, the stool ratios were lower than the rectal swab values in 13/30 (43%) samples, while in 17/30 (57%) samples, they were higher. However, with the use of qPCR-based method, the stool ratios were lower in 15/29 samples (52%), higher in 13/29 samples (45%), and equal in 1/29 samples (3%; Table 2). These results indicate that rectal swabs are as efficient and accurate as stool samples in detecting CRE by either method.
Table 2.
Samples with different values for stool sample and rectal swab sampling by two methodsa
| Analysis method | Total no. of samples analyzed | No. of samples (%) |
||
|---|---|---|---|---|
| Stool value = rectal swab value | Stool value > rectal swab value | Stool value < rectal swab value | ||
| Culture | 30 | 17 (57) | 13 (43) | |
| Molecular quantification | 29 | 1 (3) | 13 (45) | 15 (52) |
The CRE/TAB ratio was the value for the culture method, and the number of copies of the blaKPC gene/number of copies of the 16S rDNA gene was the value for the molecular quantification method.
Enumeration of blaKPC populations over time.
blaKPC-encoding populations in stool samples and rectal swabs were monitored longitudinally in a single KPC carrier, over 1 month (at 5 time points) using the qPCR method. We calculated the ratios of blaKPC/16S rRNA genes (see above) at each time point. The results obtained for stool specimens and rectal swabs were highly similar at all time points (Fig. 3).
Fig 3.
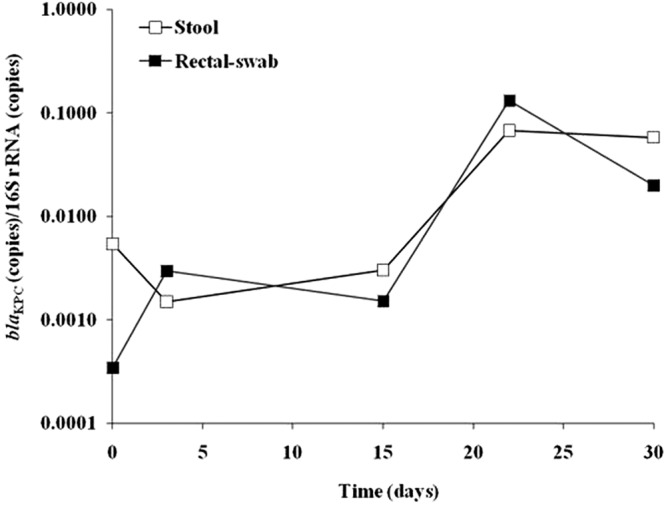
Tracking the bacterial population during a period of 1 month in stool samples and rectal swabs, which were taken from one KPC-CRE carrier patient, using quantitative PCR for blaKPC and 16S rRNA genes. The y axis shows the ratio of blaKPC (number of copies)/16S rRNA genes (number of copies).
DISCUSSION
Quantitative studies of colon flora are of great interest; however, performing a quantitative study is complicated due to the need to obtain a stool specimen and to determine its weight (dry or wet weight) for normalization of the data. Rectal swabs have been considered inappropriate for quantifying the gut flora because the total amount of stool represented on each swab is unknown; thus, the lack of a common denominator makes comparison between different samples impossible.
Here we chose an alternative approach for quantifying resistant bacteria in the gut flora. Rather than measuring the amount of the target organisms per unit of stool weight, we determined the concentration of target organism relative to other members of the gut flora. We hypothesized that by using this approach, we would be able to use rectal swabs rather than stool specimens to perform quantitative analysis of the gut flora. To test our hypothesis, we chose to study the KPC-producing CRE.
Our results show that rectal swabs are an appropriate alternative to stool specimens for quantifying KPC-producing CRE load using our newly designed blaKPC TaqMan assay combined with the 16S rRNA gene assay. In our proposed methodology, calculating the ratio of carbapenem-resistant bacterial CFU or carbapenem resistance gene copies to an appropriate reference, representing other gut flora, enables determination of the relative CRE load. We found that the results obtained for rectal swabs had better similarity to the results for stool samples when using molecular quantification rather than the culture-based method.
We used two quantification methods to determine bacterial populations in the human colon. There are several limitations to these methods. First, the molecular quantification method accounts for all gut bacteria, while the culture-based method does not account for anaerobic, unculturable, and nonviable bacteria. Second, we assumed that there is only one copy of the blaKPC gene. Third, we assumed that the resistance to carbapenem is related to the blaKPC gene, but it might be that other mechanisms, such as IMP, VIM, SPM, and others (29), are also involved. Last, relying on the 16S rRNA gene for bacterial quantification suffers from bias, as bacteria can harbor different numbers of copies of this gene in their genome (30–32).
In summary, the results of our study demonstrate that 16S rRNA genes are a good alternative to stool weight to assess the relative concentration of a target bacterium to other reference strains. In addition, we show that rectal swabs are as good as stool samples in quantifying CRE relative to other bacterial populations. This notion was strengthened with the comparison of the culture-based method versus molecular quantification method, which showed a significant P value for rectal swabs (P = 0.0461) but not for stool samples (P = 0.1664). We believe that this method offers a practical reliable alternative, especially in studies with a large number of individuals.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Y. Paitan (Meir Medical Center, Israel) and O. Peleg for technical assistance and to L. Temkin for editing the language of the manuscript.
This work was supported by the European Commission FP7: SATURN-Impact of Specific Antibiotic Therapies on the Prevalence of Human Host Resistant Bacteria research grant 241796 and by the European Commission FP7 AIDA project (preserving old antibiotics for the future: assessment of clinical efficacy by a pharmacokinetic/pharmacodynamic approach to optimize effectiveness and reduce resistance for off-patent antibiotics) research grant 278348.
Footnotes
Published ahead of print 7 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01275-12.
REFERENCES
- 1. Queenan AM, Bush K. 2007. Carbapenemases: the versatile beta-lactamases. Clin. Microbiol. Rev. 20:440–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nordmann P, Cuzon G, Naas T. 2009. The real threat of Klebsiella pneumoniae carbapenemase producing bacteria. Lancet Infect. Dis. 9:228–236 [DOI] [PubMed] [Google Scholar]
- 3. Schwaber MJ, Carmeli Y. 2008. Carbapenem-resistant Enterobacteriaceae: a potential threat. JAMA 300:2911–2913 [DOI] [PubMed] [Google Scholar]
- 4. Carmeli Y, Akova M, Cornaglia G, Daikos GL, Garau J, Harbarth S, Rossolini GM, Souli M, Giamarellou H. 2010. Controlling the spread of carbapenemase-producing Gram-negatives: therapeutic approach and infection control. Clin. Microbiol. Infect. 16:102–111 [DOI] [PubMed] [Google Scholar]
- 5. Lin LH, Tsai CY, Hung MH, Fang YT, Ling QD. 2011. Rectal swab sampling followed by an enrichment culture-based real-time PCR assay to detect Salmonella enterocolitis in children. Clin. Microbiol. Infect. 17:1421–1425 [DOI] [PubMed] [Google Scholar]
- 6. Papaconstantinou HT, Thomas JS. 2007. Bacterial colitis. Clin. Colon Rectal Surg. 20:18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nordmann P, Gniadkowski M, Giske CG, Poirel L, Woodford N, Miriagou V, European Network on Carbapenemases 2012. Identification and screening of carbapenemase-producing Enterobacteriaceae. Clin. Microbiol. Infect. 18:432–438 [DOI] [PubMed] [Google Scholar]
- 8. Anderson B, Nicholas S, Sprague B, Campos J, Short B, Singh N. 2008. Molecular and descriptive epidemiology of multidrug-resistant Enterobacteriaceae in hospitalized infants. Infect. Control Hosp. Epidemiol. 29:250–255 [DOI] [PubMed] [Google Scholar]
- 9. Carrër A, Lassel L, Fortineau N, Mansouri M, Anguel N, Richard C, Nordmann P. 2009. Outbreak of CTX-M-15-producing Klebsiella pneumoniae in the intensive care unit of a French hospital. Microb. Drug Resist. 15:47–54 [DOI] [PubMed] [Google Scholar]
- 10. Friedmann R, Raveh D, Zartzer E, Rudensky B, Broide E, Attias D, Yinnon AM. 2009. Prospective evaluation of colonization with extended-spectrum beta-lactamase (ESBL)-producing Enterobacteriaceae among patients at hospital admission and of subsequent colonization with ESBL-producing Enterobacteriaceae among patients during hospitalization. Infect. Control Hosp. Epidemiol. 30:534–542 [DOI] [PubMed] [Google Scholar]
- 11. Lautenbach E, Harris AD, Perencevich EN, Nachamkin I, Tolomeo P, Metlay JP. 2005. Test characteristics of perirectal and rectal swab compared to stool sample for detection of fluoroquinolone-resistant Escherichia coli in the gastrointestinal tract. Antimicrob. Agents Chemother. 49:798–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaplan RL, Goodman LJ, Barrett JE, Trenholme GM, Landau W. 1982. Comparison of rectal swabs and stool cultures in detecting Campylobacter fetus subsp. jejuni. J. Clin. Microbiol. 15:959–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pusterla N, Mapes S, Johnson C, Slovis N, Page A, Gebhart C. 2010. Comparison of feces versus rectal swabs for the molecular detection of Lawsonia intracellularis in foals with equine proliferative enteropathy. J. Vet. Diagn. Invest. 22:741–744 [DOI] [PubMed] [Google Scholar]
- 14. Stamper PD, Cai M, Lema C, Eskey K, Carroll KC. 2007. Comparison of the BD GeneOhm VanR assay to culture for identification of vancomycin-resistant enterococci in rectal and stool specimens. J. Clin. Microbiol. 45:3360–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chaban B, Musil KM, Himsworth CG, Hill JE. 2009. Development of cpn60-based real-time quantitative PCR assays for the detection of 14 Campylobacter species and application to screening of canine fecal samples. Appl. Environ. Microbiol. 75:3055–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kotton CN, Lankowski AJ, Hohmann EL. 2006. Comparison of rectal swabs with fecal cultures for detection of Salmonella typhimurium in adult volunteers. Diagn. Microbiol. Infect. Dis. 56:123–126 [DOI] [PubMed] [Google Scholar]
- 17. D'Agata EM, Gautam S, Green WK, Tang YW. 2002. High rate of false-negative results of the rectal swab culture method in detection of gastrointestinal colonization with vancomycin-resistant enterococci. Clin. Infect. Dis. 34:167–172 [DOI] [PubMed] [Google Scholar]
- 18. Funk JA, Davies PR, Nichols MA. 2000. The effect of fecal sample weight on detection of Salmonella enterica in swine feces. J. Vet. Diagn. Invest. 12:412–418 [DOI] [PubMed] [Google Scholar]
- 19. Murphy C, Reid-Smith RJ, Prescott JF, Bonnett BN, Poppe C, Boerlin P, Weese JS, Janecko N, McEwen SA. 2009. Occurrence of antimicrobial resistant bacteria in healthy dogs and cats presented to private veterinary hospitals in southern Ontario: a preliminary study. Can. Vet. J. 50:1047–1053 [PMC free article] [PubMed] [Google Scholar]
- 20. Padiglione AA, Wolfe R, Grabsch EA, Olden D, Pearson S, Franklin C, Spelman D, Mayall B, Johnson PD, Grayson ML. 2003. Risk factors for new detection of vancomycin-resistant enterococci in acute-care hospitals that employ strict infection control procedures. Antimicrob. Agents Chemother. 47:2492–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ott SJ, Musfeldt M, Ullmann U, Hampe J, Schreiber S. 2004. Quantification of intestinal bacterial populations by real-time PCR with a universal primer set and minor groove binder probes: a global approach to the enteric flora. J. Clin. Microbiol. 42:2566–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schechner V, Straus-Robinson K, Schwartz D, Pfeffer I, Tarabeia J, Moskovich R, Chmelnitsky I, Schwaber MJ, Carmeli Y, Navon-Venezia S. 2009. Evaluation of PCR-based testing for surveillance of KPC-producing carbapenem-resistant members of the Enterobacteriaceae family. J. Clin. Microbiol. 47:3261–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nikkari S, Lopez FA, Lepp PW, Cieslak PR, Ladd-Wilson S, Passaro D, Danila R, Relman DA. 2002. Broad-range bacterial detection and the analysis of unexplained death and critical illness. Emerg. Infect. Dis. 8:188–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keinänen-Toivola MM, Revetta RP, Santo Domingo JW. 2006. Identification of active bacterial communities in a model drinking water biofilm system using 16S rRNA-based clone libraries. FEMS Microbiol. Lett. 257:182–188 [DOI] [PubMed] [Google Scholar]
- 25. Hindiyeh M, Smollen G, Grossman Z, Ram D, Davidson Y, Mileguir F, Vax M, Ben David D, Tal I, Rahav G, Shamiss A, Mendelson E, Keller N. 2008. Rapid detection of blaKPC carbapenemase genes by real-time PCR. J. Clin. Microbiol. 46:2879–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JD, Wengenack NL, Rosenblatt JE, Cockerill FR, III, Smith TF. 2006. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin. Microbiol. Rev. 19:165–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622 [DOI] [PubMed] [Google Scholar]
- 29. Hawkey PM, Jones AM. 2009. The changing epidemiology of resistance. J. Antimicrob. Chemother. 64(Suppl 1):i3–i10 [DOI] [PubMed] [Google Scholar]
- 30. Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. 2004. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186:2629–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farrelly V, Rainey FA, Stackebrandt E. 1995. Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Appl. Environ. Microbiol. 61:2798–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klappenbach JA, Dunbar JM, Schmidt TM. 2000. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66:1328–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.