

Abstract
Assembly effectors are small molecules that induce inappropriate virus capsid assembly to antiviral effect. To identify attributes of hepatitis B virus (HBV) assembly effectors, assembly reaction products (normal capsid, noncapsid polymer, intermediates, and free dimeric core protein) were quantified in the presence of three experimental effectors: HAP12, HAP13, and AT-130. Effectors bound stoichiometrically to capsid protein polymers, but not free protein. Thermodynamic and kinetic effects, not aberrant assembly, correlate with maximal antiviral activity.
TEXT
Hepatitis B virus (HBV) is a global public health problem. According to World Health Organization estimates, 360 million people suffer from chronic HBV infection, contributing to approximately 600,000 deaths every year (1). HBV-specific antiviral drug development has focused on targeting the viral reverse transcriptase (RT). However, RT inhibitors do not usually clear HBV infection, even after prolonged treatment (2–4). Furthermore, cessation of RT inhibitors can lead to life-threatening viral flares; therefore, they are generally a lifelong therapy (5). An alternative therapeutic target is highly desirable. One attractive target is assembly of HBV's capsid from core protein.
HBV is a DNA virus composed of a protein-studded lipid envelope surrounding an icosahedral nucleoprotein core (6, 7). The protein shell of the core, the capsid, is a T=4 icosahedral complex built from 120 copies of core protein homodimer. The core protein is a 183-amino-acid protein comprised of a 149-residue assembly domain (which includes the dimerization motif) and a C-terminal 34-residue RNA binding domain which is not required for assembly (8). The core protein assembly domain has no human homolog (9). The assembly domain is referred to as Cp149. In the HBV life cycle, like many icosahedral viruses, the capsid has critical roles in virus replication, making it an excellent target for antiviral therapy (10, 11). Cp149 assembly is a function of protein concentration, ionic strength, and temperature (12). A molecule that modulates capsid assembly could interfere with the geometry of core protein interaction, packaging viral nucleic acid, and the stability of newly assembled virions (13–16). A number of HBV assembly effectors have been investigated (17–21). Recently, capsid assembly has also been targeted in other viral systems, including HIV and HCV (22–24).
Two classes of HBV assembly effectors have been discovered in searches for nonnucleoside inhibitors of HBV replication, the heteroaryldihydropyrimidines (HAPs) and phenylpropenamides (25–29). On the basis of observations with purified Cp149, HAPs increase the kinetics of assembly and strengthen dimer-dimer association to stabilize capsids, and at high concentrations, they misdirect assembly (14, 20). On the basis of a crystal structure of the HAP-HBV complex, a series of HAPs with different properties were designed; their effects on the thermodynamics and kinetics of assembly of purified Cp149 were compared with inhibition of virion production in HepG2.2.15 cells (17, 30). The AT-130 and AT-61 phenylpropenamides had the unusual antiviral activity of generating empty cytoplasmic capsids (31). Like HAPs, phenylpropenamides were shown to accelerate assembly and stabilize capsids; however, they do not misdirect assembly (19). HAPs and AT-130 have antiviral activity in cells, although they have distinct effects on assembly products with purified protein. Because searches for assembly effectors are most efficiently based on biochemical screens, here we identify activities of selected HAPs and phenylpropenamides to define the characteristics that are most important for antiviral activities.
To compare assembly effectors, we have generated phase diagrams of assembly as a function of effector and Cp149 dimer concentrations. To obtain a breadth of understanding of different effectors, we examined HAP12, which substantially strengthens pairwise protein-protein association energy and accelerates kinetics of capsid assembly, and HAP13, which has weaker effects on association energy and kinetics (17). To generalize beyond the HAP family, we included AT-130 in our study (19); structures of these molecules are shown in Fig. 1. To examine equilibrium assembly, Cp149 dimer (at 2.5 to 15 μM in 50 mM HEPES) was incubated with assembly effectors (0 μM to 20 μM) for 20 min prior to inducing assembly by addition of NaCl to 150 mM and incubation at 37°C for 24 h. Reaction products were discriminated using 500-Å pore and 1,000-Å pore Agilent BIO SEC 5 size exclusion columns in series (Fig. 2).
Fig 1.
Structures of the assembly effectors studied here. Me, methyl.
Fig 2.
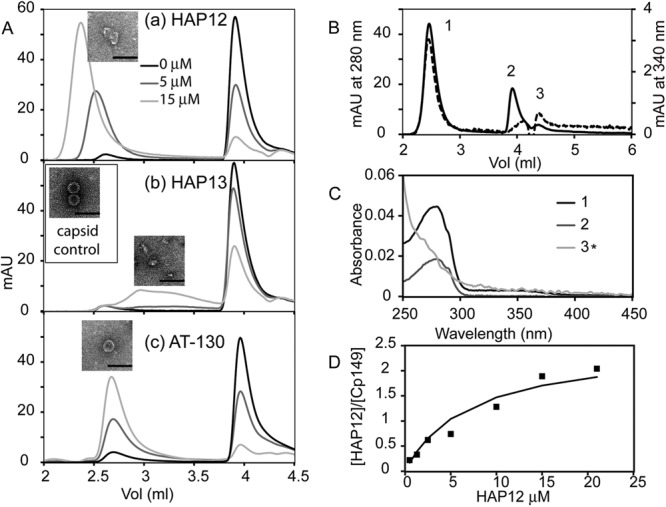
Size exclusion chromatograms of equilibrated 5 μM HBV Cp149 assembly reactions with increasing concentrations of assembly effectors. (A) Cp149 assembly reactions showing assembly behavior as a function of assembly effector, HAP12 (a), HAP13 (b), and AT-130 (c). For each assembly effector, seven concentrations (0, 1.25, 2.5, 5, 10, 15, and 20 μM) were investigated; for clarity, only three concentrations are shown: 0 μM in black, 5 μM in dark gray, and 15 μM in gray. Each experiment was performed three to five times, but only one representative chromatogram is shown. The void volume for the linked 500-Å pore and 1,000-Å pore Agilent BIO SEC 5 size exclusion columns was 1.5 ml. Capsids eluted at 2.6 ml, dimers eluted at 3.9 ml, and small molecules eluted at the end of the column at 4.4 ml. Electron micrographs are insets for the polymer/capsid fractions of assembly reactions with 5 μM Cp149 and 15 μM assembly effector. For a control, a micrograph of 5 μM Cp149 capsid is shown in panel Ab. Bars, 100 nm. mAU, milli absorbance units. (B and C) Chromatogram (B) and UV spectra (C) respective peak fractions of 5 μM Cp149 assembled in the presence of 10 μM HAP12. The elution profiles at 280 nm (solid line, left-hand y axis) and 340 nm (dashed line, right-hand y axis) show peaks for polymer/capsid (1), dimer (2), and HAP 12 (3). (C) UV spectra of peaks 1, 2, and 3 in panel B indicate the presence and absence of HAP12 in samples. 3* is the absorbance spectrum of peak 3 multiplied by five to make it easier for the reader to see. (D) The ratio of HAP12 to Cp149 dimer in the polymer fraction as a function of HAP12 concentration appears to saturate at 2 HAPs per dimer.
The three assembly effectors had distinct effects on capsid assembly. With the strong effector HAP12, the core protein progressively formed large noncapsid polymers, indicated by the decrease in the dimer peak and the shift of the capsid peak toward the void volume (Fig. 2Aa). The continuous shift of the capsid peak was consistent with heterogeneity of assembly products. HAP13, a weak assembly effector, caused intermediates to accumulate without changing the capsid peak, suggesting stabilization of abortive assembly products (Fig. 2Ab). AT-130 induced the formation of more capsid; neither polymer nor intermediate was detected (Fig. 2Ac). Electron micrographs (EM) of the peak center fraction in size exclusion chromatography (SEC) further confirmed the formation of polymers, intermediates, and capsids in the capsid assembly with HAP12, HAP13, and AT-130, respectively.
Because assembly effectors stabilize Cp149-Cp149 interactions, it was likely that they bind tightly to capsid and noncapsid polymers but not necessarily to dimers. Analysis of the UV spectrum of the elution profile was complicated by overlapping absorbance of chromophores and light-scattering artifacts due to large polymers, such as capsid (32). For example, a chromatogram of the assembly of 5 μM Cp149 with 10 μM HAP12 (Fig. 2B) included absorbance of HAP and protein at 280 nm, HAP at 340 nm, and light scattering (which increases at shorter wavelengths) throughout. Analysis of the UV spectra (Fig. 2C) of chromatographic fractions showed that the polymer/capsid had a typical protein peak with an added shoulder from 300 nm to 400 nm corresponding to HAP12, whereas the dimer peak had negligible absorbance in this region. To accurately interpret spectra, we calculated corrections for light scatter and determined the concentrations of Cp149 and assembly effector (32). HAP12 saturated the capsid/polymer complex at a ratio of two HAPs per dimer (Fig. 2D). This agreed with our HAP-capsid crystal structure which identified one HAP site per monomer (though only some quasiequivalent sites were filled in the context of an HBV capsid crystal structure) (30).
We determined the concentration of Cp149 in polymer, intermediate, capsid, and dimer peaks. To do this, we assumed that chromatographic peaks were well described as a sum of Gaussian peaks and fit them using the Gaussian function implemented in the program Origin 8.5 (OriginLab). From this quantification, we generated a three-dimensional (3D) phase diagram for each effector (Fig. 3A to C). In contrast to standard phase diagrams, no single “pure” phase exists in the assembly reaction due to the pseudocritical concentration nature of capsid assembly (33). The phase diagrams show that all three effectors resulted in a dose-dependent decrease of free dimer in the assembly reaction, but different capsid assembly patterns. In the absence of small-molecule effectors, assembly is Cp149 concentration dependent. When HAP12 was introduced into the assembly reactions, the polymers aggressively increased coupled with decreases in capsids and dimers. HAP13 led to intermediates but only at very high concentrations. AT-130 only had capsid and dimer phases. All three assembly effectors disturbed normal capsid assembly.
Fig 3.

(A to C) 3D phase diagrams of assembly reactions for Cp149 and assembly effectors: HAP12 (A), HAP13 (B), and AT-130 (C). Capsids are shown in green, polymers or intermediates are shown in red, and dimers are shown in blue; the concentrations of each species are also indicated by the intensity of the color. (D to F) 2D contour views of the phase diagrams for HAP12 (D), HAP13 (E), and AT-130 (F). Contours are colored when they represent over 50% of the Cp149 mass. Contour lines (with corresponding percentages) of each product are also shown, colored as in panels A, B, and C. The antiviral activity in terms of the concentration of assembly effector needed to suppress virus production HepG2.2.15 cells by 50%, EC50, is shown by the black horizontal line (17, 25).
The correlation between the phase diagram and antiviral effect is most easily demonstrated when the phase diagram is compressed into two dimensions. Since the diagrams have no “pure” phases, regions were designated as representing the polymer, capsid, or dimer phase when over 50% of the total mass corresponded to that form of the protein. The concentration of effector needed to suppress virion production in culture by 50%, EC50, is represented by a horizontal black line (Fig. 3D and E) (17, 25). This analysis revealed that antiviral activity was not related to the formation of polymer or intermediate structure: the EC50 was far below the polymer or intermediate phase. Instead, the antiviral activity of each effector seemed to follow the same trend as its energetic effect on capsid assembly (Table 1; 17, 7, 3). Normal capsid assembly is characterized by slow nucleation rate and weak pairwise dimer-dimer association energy. Therefore, changing either the strength or rate of association will affect the capsid production. Previously, we defined a kinetic index (Kindex), which is the negative log of the rate of appearance of capsids, to characterize assembly effectors (17). HAP12 has the strongest antiviral activity and also has the greatest kinetic and thermodynamic effects on capsid assembly; its extremely low EC50 would seem to correlate with its kinetic effect (17). Though HAP13 increased the rate of assembly more than AT-130, its effect on dimer-dimer association was weaker. Therefore, our data indicate that both kinetics and thermodynamics are critical to preventing formation of virions, but aberrant assembly is not required.
Table 1.
Antiviral activity and thermodynamic and kinetic effects of HAP12, HAP13, and AT-130
| Assembly effector | EC50 (μM) (mean ± SEM) | ΔΔGcont (kcal/mol) (mean ± SEM) | Kindexa (mean ± SEM) |
|---|---|---|---|
| HAP12b | 0.012 ± 0.002 | −1.92 ± 0.07 | −3.69 ± 0.05 |
| AT-130c | 2.40 ± 0.92 | −0.99 ± 0.18 | −2.08 ± 0.07 |
| HAP13b | 6.10 ± 0.50 | −0.69 ± 0.11 | −2.41 ± 0.03 |
The kinetic index was determined by light scattering experiments as follows: Kindex = −log(slope/concentration of assembly effector), where slope is the steepest slope of the light scattering (LS) trace in arbitrary units, and the assembly effector concentration is micromolar.
Data for HAP12 and HAP13 assembly energetics and EC50 are from reference 17. EC50 was the concentration of the compound required to reduce the concentration of HBV DNA produced in HepG2.2.15 cell culture medium by 50%. DNA concentrations were determined by quantitative Southern blotting.
In conclusion, the antiviral activity of the assembly effectors studied here requires an interplay of the thermodynamics and kinetics induced by the effector. A good assembly effector must be able to initiate capsid assembly fast enough to “escape” the normal capsid assembly path, as well as stabilize intermediates to support further assembly to deplete available core protein.
ACKNOWLEDGMENT
This work was supported by a grant from the National Institutes of Health (R01-AI067417) to A.Z.
Footnotes
Published ahead of print 3 December 2012
REFERENCES
- 1. WHO 2004. WHO position on the use of hepatitis B vaccines. Wkly Epidemiol. Rec. 28:255–263 [Google Scholar]
- 2. Matthews GV, Seaberg E, Dore GJ, Bowden S, Lewin SR, Sasadeusz J, Marks P, Goodman Z, Philp FH, Tang Y, Locarnini S, Thio CL. 2009. Combination HBV therapy is linked to greater HBV DNA suppression in a cohort of lamivudine-experienced HIV/HBV coinfected individuals. AIDS 23:1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nguyen T, Locarnini S. 2009. Hepatitis: monitoring drug therapy for hepatitis B–a global challenge? Nat. Rev. Gastroenterol. Hepatol. 6:565–567 [DOI] [PubMed] [Google Scholar]
- 4. Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–1608 [DOI] [PubMed] [Google Scholar]
- 5. Committee on the Prevention and Control of Viral Hepatitis Infection, Institute of Medicine 2010. Hepatitis and liver cancer: a national strategy for prevention and control of hepatitis B and C. The National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- 6. Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235–249 [DOI] [PubMed] [Google Scholar]
- 7. Seeger C, Zoulim F, Mason WS. 2007. Hepadnaviruses, p 2977–3029 In Knipe DM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 8. Nassal M. 1992. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 66:4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ricco R, Kanduc D. 2010. Hepatitis B virus and Homo sapiens proteomewide analysis: a profusion of viral peptide overlaps in neuron-specific human proteins. Biologics 4:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steven AC, Conway JF, Cheng N, Watts NR, Belnap DM, Harris A, Stahl SJ, Wingfield PT. 2005. Structure, assembly, and antigenicity of hepatitis B virus capsid proteins. Adv. Virus Res. 64:125–164 [DOI] [PubMed] [Google Scholar]
- 12. Ceres P, Stray SJ, Zlotnick A. 2004. Hepatitis B virus capsid assembly is enhanced by naturally occurring mutation F97L. J. Virol. 78:9538–9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prevelige PEJ. 1998. Inhibiting virus-capsid assembly by altering the polymerisation pathway. Trends Biotechnol. 16:61–65 [DOI] [PubMed] [Google Scholar]
- 14. Zlotnick A, Ceres P, Singh S, Johnson JM. 2002. A small molecule inhibits and misdirects assembly of hepatitis B virus capsids. J. Virol. 76:4848–4854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zlotnick A, Mukhopadhyay S. 2011. Virus assembly, allostery and antivirals. Trends Microbiol. 19:14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zlotnick A, Stray SJ. 2003. How does your virus grow? Understanding and interfering with virus assembly. Trends Biotechnol. 21:536–542 [DOI] [PubMed] [Google Scholar]
- 17. Bourne C, Lee S, Venkataiah B, Lee A, Korba B, Finn MG, Zlotnick A. 2008. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J. Virol. 82:10262–10270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hacker HJ, Deres K, Mildenberger M, Schroder CH. 2003. Antivirals interacting with hepatitis B virus core protein and core mutations may misdirect capsid assembly in a similar fashion. Biochem. Pharmacol. 66:2273–2279 [DOI] [PubMed] [Google Scholar]
- 19. Katen SP, Chirapu SR, Finn MG, Zlotnick A. 2010. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem. Biol. 5:1125–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. U. S. A. 102:8138–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stray SJ, Zlotnick A. 2006. BAY 41-4109 has multiple effects on hepatitis B virus capsid assembly. J. Mol. Recognit. 19:542–548 [DOI] [PubMed] [Google Scholar]
- 22. Adamson CS, Freed EO. 2010. Novel approaches to inhibiting HIV-1 replication. Antiviral Res. 85:119–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kota S, Takahashi V, Ni F, Snyder JK, Strosberg AD. 2012. Direct binding of a hepatitis C virus inhibitor to the viral capsid protein. PLoS One 7:e32207 doi:10.1371/journal.pone.0032207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nangola S, Urvoas A, Valerio-Lepiniec M, Khamaikawin W, Sakkhachornphop S, Hong SS, Boulanger P, Minard P, Tayapiwatana C. 2012. Antiviral activity of recombinant ankyrin targeted to the capsid domain of HIV-1 Gag polyprotein. Retrovirology 9:17 doi:10.1186/1742-4690-9-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delaney WE, IV, Edwards R, Colledge D, Shaw T, Furman P, Painter G, Locarnini S. 2002. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 46:3057–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. 2003. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 299:893–896 [DOI] [PubMed] [Google Scholar]
- 27. King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. 1998. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)β-l-2′,3′-dideoxy-3′-thiacytidine. Antimicrob. Agents Chemother. 42:3179–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perni RB, Conway SC, Ladner SK, Zaifert K, Otto MJ, King RW. 2000. Phenylpropenamide derivatives as inhibitors of hepatitis B virus replication. Bioorg. Med. Chem. Lett. 10:2687–2690 [DOI] [PubMed] [Google Scholar]
- 29. Stoltefuss J, Goldmann S, Krämer T, Schlemmer K-H, Niewöhner U, Paessens A, Lottmann S, Deres K, Weber O. 1 October 1999. New dihydropyrimidine derivatives and their corresponding mesomers useful as antiviral agents. WO patent 9954326
- 30. Bourne C, Finn MG, Zlotnick A. 2006. Global structural changes in hepatitis B capsids induced by the assembly effector HAP1. J. Virol. 80:11055–11061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. 2007. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res. 76:168–177 [DOI] [PubMed] [Google Scholar]
- 32. Porterfield JZ, Zlotnick A. 2010. A simple and general method for determining the protein and nucleic acid content of viruses by UV absorbance. Virology 407:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katen SP, Zlotnick A. 2009. Thermodynamics of virus capsid assembly. Methods Enzymol. 455:395–417 [DOI] [PMC free article] [PubMed] [Google Scholar]