

Abstract
The clinical efficacy of a pegylated form of human lambda 1 interferon (IFN-λ1; also referred to herein as lambda) has been demonstrated in patients chronically infected with hepatitis C virus (HCV) representing genotypes 1 through 4. In these proof-of-concept studies, lambda showed an improved safety profile compared to the pegylated form of alfa interferon (referred to herein as alfa). In the study described in this report, an assessment of the in vitro antiviral activity of type III IFNs toward different HCV replicons revealed that the unpegylated recombinant form of IFN-λ1 (rIFN-λ1) exerted the most robust effect, while rIFN-λ3 exhibited greater activity than rIFN-λ2. More importantly, cross-resistance to rIFN-λ1 was not observed in replicon cell lines known to have reduced susceptibility to investigational direct-acting antiviral (DAA) agents targeting the essential HCV nonstructural protein NS3, NS5A, or NS5B. When combined with either rIFN-α, the NS3 protease inhibitor (NS3 PI) asunaprevir (ASV), the NS5A replication complex inhibitor (NS5A RCI) daclatasvir (DCV), or the NS5B polymerase site I inhibitor (NS5B I) BMS-791325, rIFN-λ1 displayed a mixture of additive and synergistic effects. In three-drug combination studies, inclusion of lambda with ASV and DCV also yielded additive to synergistic effects. In line with these observations, it was demonstrated that a regimen that used a combination of rIFN-λ1 with one or two DAAs was superior to an IFN-free regimen in clearing HCV RNA in genotype 1a cell lines representing wild-type and NS3 protease inhibitor-resistant sequences. Overall, these data support further clinical development of lambda as part of alternative combination treatments with DAAs for patients chronically infected with HCV.
INTRODUCTION
Hepatitis C virus (HCV), a positive-strand RNA virus that belongs to the Flaviviridae family, is a major causative agent of chronic liver disease, affecting an estimated 170 million individuals worldwide (1). Until recently, treatment options for chronic HCV infection comprised the combination of the pegylated form of alfa interferon (IFN) (referred to here as alfa) with ribavirin (RBV). This regimen is associated with significant side effects, resulting in high rates of noncompliance, and demonstrates variable efficacy against numerous HCV genotypes. Although various host and viral factors are believed to influence the outcome of infection, different genotypes (GTs) are also associated with variable responses to alfa-based treatment (2–4). More specifically, an increased risk of treatment failure is observed against the most predominant HCV GT, GT1 (subtypes 1a and 1b), which accounts for approximately 60% of global infections and against which an extended duration of therapy (48 to 72 weeks) is required to enhance the response. Successful treatment, referred to as a sustained virological response (SVR), is achieved in only 40 to 50% of patients infected with HCV GT1, whereas higher rates (78 to 86%) have been reported with those infected with HCV GT2 and GT3 (5). In addition, completion of treatment often suffers because of poor adherence by patients due to drug-related adverse events, including psychiatric disorders, flu-like symptoms, and/or hematological abnormalities, such as hemolytic anemia and neutropenia (6). Recently, the addition of a direct-acting antiviral (DAA) targeting the HCV NS3 protease activity (telaprevir and boceprevir) to the alfa-RBV regimen was approved as the new standard of care for the treatment of chronic GT1 infection, a consequence of enhanced SVR rates to about 70 to 75% in patients (7, 8). Unfortunately, the side effects associated with alfa-containing treatments remain. This highlights a medical need for new HCV therapeutic agents that are more effective and tolerable.
Human lambda 1 interferon (IFN-λ1), also known as interleukin-29 (IL-29) and referred to here as lambda, is a recently described human type III IFN which has a close evolutionary relationship to the IL-10 cytokine family and is distantly related to the type I IFNs (9). Two other IFN-λ cytokines simultaneously identified, IFN-λ2 (IL-28A) and IFN-λ3 (IL-28B), share approximately 81% sequence identity with IFN-λ1. The biological characteristics of these cytokines are comparable to those of type I IFNs, such as IFN-α and IFN-β, although sequence homology is low. These various classes of IFNs exert their antiviral activities by inducing the expression of IFN-stimulated genes (ISGs) through activation of the Janus kinases Jak1 and Tyk2 and subsequent phosphorylation of the signal transducer and activator of transcription (STAT) factors STAT1 and STAT2. The complete spectrum of ISGs that mediate an antiviral effect on HCV replication has not yet been defined (10). Additionally, similar to IFN-α/β, expression of IFN-λ is induced upon viral infection or stimulation with double-stranded RNA, and IFN-λ has demonstrated broad antiviral activity in vitro, including inhibition of viral RNA replication in the HCV replicon model (11, 12). However, the receptor complex for type III IFNs is structurally unique, with a cell and tissue distribution more restricted than that of the type I IFN receptor, being minimally expressed on cells of hematopoietic lineage (13). These observations suggest that systemic administration of IFN-λ1 may be associated with fewer side effects than alfa-based treatments. Indeed, proof-of-concept clinical studies with lambda indicated minimal adverse events and hematologic effects, while demonstrating promising antiviral activity in patients with chronic HCV GT2 or GT3 infection (14). Moreover, a series of genome-wide association studies (GWASs) independently reported a strong association between common host genetic polymorphisms in the region of the locus for the IL28B gene (which encodes IFN-λ3), spontaneous viral clearance, and a more favorable outcome to alfa-based treatments in chronic HCV subjects (15, 16). Although a causal immunological mechanism involved in the IL28B genotype remains elusive, a link between HCV clearance and type III IFN regulation underlies the potential importance of lambda in future therapeutic indications for patients refractory to current treatment options.
Lambda has been shown to exert an antiviral response comparable to that exerted by alfa in patients infected with HCV GT1 through GT4 (17). As with alfa, it will be important to determine that lambda can be combined with DAAs and also result in improved SVR rates. We recently described three classes of investigational DAAs in clinical development that target the activity of different HCV proteins: the NS3 protease inhibitor (NS3 PI) asunaprevir (ASV; BMS-650032), the first-in-class NS5A replication complex inhibitor (NS5A RCI) daclatasvir (DCV; BMS-790052), and the NS5B RNA-dependent RNA polymerase nonnucleoside site I inhibitor (NS5B I) BMS-791325 (18–20). The combination of each of these inhibitors with alfa-RBV was shown to be generally well tolerated and demonstrated greater antiviral activity than a placebo regimen of alfa-RBV in treatment-naive patients infected with HCV GT1 (21–23). In this report, we document the antiviral activity of the various recombinant forms of IFN-λ cytokines in a number of HCV cell culture systems representing GT1a, GT1b, and GT2a. In addition, we show that a recombinant IFN-λ1 (rIFN-λ1)-based regimen compares favorably with an rIFN-α-based regimen in reducing the emergence of HCV resistance in replicon cells when combined with various classes of DAAs currently in clinical development. Overall, these data support the inclusion of lambda as part of new treatment options for HCV therapy.
MATERIALS AND METHODS
Compounds.
rIFN-α2b (rIFN-α; Intron A) and the pegylated form of IFN-α2a (alfa; Pegasys) were purchased from Myoderm Medical Supply (Norristown, PA) and Hoffmann-La Roche, Inc. (Nutley, NJ), respectively. Human recombinant forms of IL-29 (rIFN-λ1), IL-28A (rIFN-λ2), IL-28B (rIFN-λ3), and IL-28B K70R (rIFN-λ3-K70R) were synthesized at ZymoGenetics, Inc. (Seattle, WA). The pegylated form of IFN-λ1 (lambda) is a covalent conjugate of rIFN-λ1 (molecular mass, 19.6 kDa) and a 20-kDa linear polyethylene glycol chain. All forms of IFN-λ have been extensively characterized by a number of methods, including amino acid analysis, N-terminal sequencing, size-exclusion chromatography–multiangle light scattering (SEC-MALS), and peptide map analysis and showed equivalent whole mass values (range, 19.6 to 20 kDa). ASV, DCV, and BMS-791325, synthesized by Bristol-Myers Squibb Co. (BMS), have been previously described (18–20).
Cell lines and viral constructs.
Human hepatoma cells Huh-7 and Huh-7.5 were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and l-glutamine (2 mM). HCV subgenomic replicon cell lines representing GT1b (Con1 strain), GT1a (H77c strain), and GT2a (JFH1 strain) wild-type (WT) sequences were generated at BMS as previously described (24). Stable Huh-7 cells containing HCV replicon variants with specific NS3, NS5A, or NS5B amino acid substitutions were established following selection with G418, as previously described (24).
Phenotypic analysis.
To determine the antiviral activity of compounds, HCV replicon cells were incubated (37°C, 5% CO2) in 96-well black-clear assay plates (Costar 3614; BD Biosciences, San Diego, CA) in the presence of 3-fold serial dilutions of test compound in dimethyl sulfoxide (DMSO) for 3 or 4 days. The 50% effective concentration (EC50) was calculated using XLfit software (version 2.0; IDBusiness Solutions, Burlington, MA). For replicons containing a humanized Renilla luciferase reporter gene, HCV RNA replication was measured using a Renilla luciferase assay system (Promega Corporation, Madison, WI) as previously described (25). Briefly, luciferase reagent, dissolved in DMSO, was diluted in medium containing DMEM and 2 mM l-glutamine and added to each plate well at a final concentration of 15 μM. Plates were then incubated for 1 h at 37°C in 5% CO2 prior to reading on a Perkin-Elmer TopCount NXT counter set for luminescence. For replicons lacking a reporter gene, NS3 protease activity was used as an indirect measurement of the amount of HCV replicon RNA present within cells as previously described (25). Following incubation (37°C, 5% CO2) with serial dilutions of test compound, NS3 protease activity was measured using a fluorescence resonance energy transfer (FRET)-labeled peptide substrate (final concentration, 10 μM) diluted in Promega 1× cell lysis buffer (Promega, Madison, WI). Cells were washed with Dulbecco's phosphate-buffered saline (D-PBS; 3×, 150 μl per well), and 25 μl of the above-described buffer containing the FRET-labeled peptide was added per well. Protease activity was measured using a Cytofluor 4000 instrument set to wavelengths of 340 nm (excitation) and 490 nm (emission). Linear rates obtained during 15 cycles in kinetic mode were used in EC50 calculations.
Western immunoblot analysis.
To monitor the capacity of rIFN-λs to induce the JAK-STAT signaling pathway, phosphorylation of STAT1 was monitored in cells upon IFN stimulation. Briefly, equivalent numbers of cells (1 × 105) seeded in 12-well assay plates (Becton, Dickinson, Franklin Lakes, NJ) were treated with rIFN-α or rIFN-λ for 30 min at 37°C in 5% CO2. IFN-treated and untreated replicon cells were then washed once with D-PBS and lysed with 100 μl of radioimmunoprecipitation assay buffer (Sigma) supplemented with protease inhibitors and phosphatase inhibitor cocktails (Sigma). Equal amounts of protein lysates were separated on a 3 to 8% Tris-acetate gel and then transferred onto an Invitrolon polyvinylidene difluoride membrane (Invitrogen, Carlsbad, CA). The membrane was incubated with the indicated primary antibody, followed by incubation with a horseradish peroxidase-conjugated antibody (Bio-Rad Laboratories, Hercules, CA). Proteins were visualized using Kodak BioMax film (Carestream Health, Rochester, NY).
Luminex beads assay.
HCV replicon cells seeded at a density of 10 × 103 cells per well in 96-well black-clear assay plates received 3-fold serial dilutions of test compounds and were incubated at 37°C in 5% CO2 for 15 min. Following incubation, the medium was removed and the cells were washed with 100 μl ice-cold D-PBS and put on ice to stop the reaction. Subsequently, 50 μl Milliplex multianalyte panel (MAP) lysis buffer (Millipore Corporation, Billerica, MA) was added to each well and the plate was then agitated on a microplate platform shaker for 20 min at 300 rpm and 2 to 8°C. Plates were centrifuged at 3,200 rpm and 2 to 8°C for 20 min. Supernatants were collected and transferred to a new microtiter plate for storage at −20°C. The status of STAT1 molecules in the samples was monitored by Luminex technology using a Milliplex phospho-STAT1 assay (Millipore Corporation, Billerica, MA) as previously described (26). Median fluorescence intensity (MFI) values for each test compound concentration, estimated on a Bio-Plex array reader, were exported for subsequent analysis. MFI values were transferred to XLfit software (version 2.0; IDBusiness Solutions, Burlington, MA). EC50s for each curve were estimated, and plots of the dose-response curve were generated for visual comparison.
Drug-drug interaction studies.
Inhibitor combination studies with rIFN-α, rIFN-λ1, lambda, and HCV inhibitors targeting NS3, NS5A, and NS5B were carried out as previously described (19), and the data were analyzed using the combination index (CI) method of Chou (27).
Colony formation assays.
GT1a HCV replicon cells seeded at a density of 6 × 104 cells per well in 6-well assay plates (Becton, Dickinson, Franklin Lakes, NJ) were maintained in growth medium containing 0.5 mg/ml G418. Subsequently, rIFN-λ1 or rIFN-α was added in combination with various concentrations of HCV inhibitors diluted in DMSO at a final concentration of 0.5% (vol/vol). Each compound was provided to the cell cultures at increasing multiples of their respective EC50s (ASV, 6 nM; DCV, 0.050 nM; BMS-791325, 3 nM; rIFN-α, 50 IU/ml; rIFN-λ1, 3 ng/ml) as part of either a mono-, dual-, or triple-treatment regimen. No-compound (untreated) replicon cell cultures were maintained in parallel in growth medium containing an equivalent percent volume (0.5%) of DMSO and served as a control. Following 7 days of incubation, the cells were split 1:4, placed into new 6-well assay plates, and incubated with growth medium containing the appropriate compound concentrations. Thereafter, fresh medium containing the appropriate compound regimens was replenished twice weekly for a total of 25 days. The cells were then fixed with 4% (wt/vol) formaldehyde and stained with 0.2% (wt/vol) crystal violet to determine the frequency of resistance colony formation.
Genotypic analysis.
Total RNA was isolated from selected replicon cells using an RNeasy minikit (Qiagen, Valencia, CA). First-strand cDNA was synthesized from a specific genotype 1a primer with SuperScript II reverse transcriptase reagents (Invitrogen Corporation, Carlsbad, CA). PCR was then performed with Platinum Taq high-fidelity DNA polymerase (Invitrogen Corporation, Carlsbad, CA). The entire NS3- to NS5B-coding region was amplified with genotype 1a primers 5′-TGAATGTCGTGAAGGAAGCAG-3′ (forward) and 5′-GGAGTGTTTACCCCAACCTT-3′ (reverse). Purified amplicons were subjected to sequence analysis, and alignments were performed using Sequencher software (Gene Codes, Ann Arbor, MI).
RESULTS
Antiviral activity of type III IFNs in cell culture.
Since IFN-λ1 is one of three closely related type III IFNs previously reported to exert inhibitory activity against HCV, the cell potency of these cytokines was evaluated in parallel in a number of HCV subgenomic replicon reporter assays. Across these cell-based systems, rIFN-λ1 displayed comparable activity against subgenomic replicons representing GT1a, GT1b, and GT2a, with EC50s ranging from 3 to 5 ng/ml (Table 1). Furthermore, rIFN-λ1 exerted greater antiviral activity than rIFN-λ3 (EC50 range, 10 to 14 ng/ml), which was more potent than rIFN-λ2 (EC50 range, 17 to 22 ng/ml) in all HCV replication cell systems. Initial GWASs identified a nonsynonymous polymorphism tightly associated with SVR rates within the coding region of the IL28B gene that encodes a lysine for an arginine substitution at position 70 (K70R) (28). The impact of the K70R substitution on the biological activity of IFN-λ3, however, has yet to be defined. Results shown in Table 1 indicate that the antiviral potency of rIFN-λ3-K70R was similar to that of the parental form of rIFN-λ3; comparable EC50s were obtained when tested in HCV GT1a, GT1b, and GT2a replicon assays. Furthermore, the greater antiviral activity seen with rIFN-λ1 compared to that seen with rIFN-λ2 and rIFN-λ3 correlated with the capacity of each form to mediate the induction of the JAK-STAT signaling pathway. As shown in Fig. 1A, phosphorylation of STAT1 in replicon cells was induced more readily upon stimulation with 10 ng/ml of rIFN-λ1, while it required 100 ng/ml of rIFN-λ2 or rIFN-λ3. Using the more sensitive Luminex bead array technology, we showed that all three cytokines were highly active in stimulating STAT1 phosphorylation in a dose-dependent manner (Fig. 1B). Analysis of the curve fits was used to derive EC50s, and results confirmed that rIFN-λ1 (18 ng/ml) is more potent than rIFN-λ2 (55 ng/ml) and rIFN-λ3 (45 ng/ml) in inducing STAT1 phosphorylation. Moreover, lambda revealed comparable pan-genotypic antiviral activity, with EC50s varying from 4 to 13 ng/ml in different HCV replication assays representing GT1a, GT1b, and GT2a, albeit it was less potent (4- to 6-fold) than alfa (see Table S1 in the supplemental material). These data indicate that the antiviral activity of lambda is not limited to a single viral genotype, as would be expected on the basis of the presumed overlapping of its mechanism of action to that of alfa. Finally, as summarized in Table 2, no cross-resistance to rIFN-λ1 was observed in replicon cell lines harboring substitutions known to confer resistance to NS3 PIs, NS5A RCIs, or various classes of NS5B polymerase inhibitors (24, 29). When tested in these replicon variants, rIFN-λ1 yielded EC50s ranging from 2 to 7 ng/ml, consistent with the cell potencies reported against the parental WT replicons.
Table 1.
Cell potencies of IFN-λ cytokines against various HCV subgenomic replicons
| HCV GT (replicon cell line) | Compound EC50a (ng/ml) |
|||
|---|---|---|---|---|
| rIFN-λ1 | rIFN-λ2 | rIFN-λ3 | rIFN-λ3-K70R | |
| 1a (H77) | 3.4 ± 0.4 | 19 ± 5 | 11 ± 5 | 9.6 ± 1.2 |
| 1b (Con1) | 3.3 ± 0.5 | 11 ± 2 | 6.2 ± 2 | 7.3 ± 3 |
| 2a (JFH1) | 3.7 ± 1.1 | 22 ± 7 | 14 ± 4 | 8.8 ± 0.5 |
Data indicate the mean EC50 value ± standard deviation from at least three independent experiments.
Fig 1.

rIFN-λ1 induces JAK-STAT signaling more readily than rIFN-λ2 or rIFN-λ3. (A) Replicon cells were treated for 30 min with various concentrations of rIFN-λ1, rIFN-λ2, or rIFN-λ3. Cell lysates were prepared, and equal amounts of proteins were used to examine the levels of STAT1 phosphorylation using an antibody directed against phospho-STAT1 (pSTAT1; Tyr701). A STAT1 antibody was used as a loading control to ensure that equivalent amounts of protein extracts were analyzed among the samples. (B) Phosphorylation of STAT1 in replicon cells was evaluated upon stimulation with rIFN-λ1, rIFN-λ2, or rIFN-λ3 using the Luminex bead-based assay. MFI values were exported and transferred to XLfit. Log transformation of the concentration data was performed, and the resulting curve fits are reported for each IFN. Data are the average of 3 independent experiments. Error bars show the standard deviations.
Table 2.
Cell potency of rIFN-λ1 cytokines against various HCV subgenomic replicon variants
| HCV GT (replicon cell line) | Compound EC50a |
|||
|---|---|---|---|---|
| rIFN-λ1 | ASV | DCV | NS5B I | |
| 1a (H77) | 4.6 ± 1.5 | 9.4 ± 3.2 | 0.055 ± 0.011 | 4.6 ± 1.5 |
| 1a-NS3-R155K | 1.6 ± 0.6 | 359 ± 63 | 0.073 ± 0.005 | 1.6 ± 0.6 |
| 1a-NS5A-Q30E | 7.0 ± 0.7 | 7.4 ± 0.7 | 447 ± 10 | 7.0 ± 0.7 |
| 1a-NS5A-L31V | 5.7 ± 0.6 | 5.9 ± 0.8 | 121 ± 4 | 5.7 ± 0.6 |
| 1a-NS5A-Y93H | 4.4 ± 1.0 | 7.2 ± 0.5 | 246 ± 3 | 4.4 ± 1.0 |
| 1b (Con1) | 3.3 ± 0.5 | ND | ND | 5.5 ± 1.2 |
| 1b-NS5B-P495L | 2.7 ± 0.7 | ND | ND | 2648 ± 378 |
Data are in ng/ml for rIFN-λ1 and nM for ASV, DCV, and NS5B I. ND, not determined. Data indicate the mean EC50 value ± standard deviation from at least three independent experiments.
In vitro drug-drug interaction studies between IFN-λ1 and various classes of DAAs.
Recent clinical data highlighted the rapid enrichment of preexisting or emerging viral variants with reduced susceptibility to investigational DAAs upon monotherapy in HCV-infected patients (30, 31). Despite the impressive decline in viral load reported during these clinical studies, the high rate of viral resistance underscored the importance of combination therapy to achieve sustained virological responses. Therefore, we next investigated the antiviral activity of rIFN-λ1 in combination with various HCV replication inhibitors using the GT1b subgenomic HCV replicon cell system to determine whether the in vitro drug-drug interactions were additive, antagonistic, or synergistic. Specific combinations included rIFN-λ1 mixed with rIFN-α or various DAAs at different ratios. Serial dilutions of a mixture of four HCV antivirals (rIFN-α, ASV [NS3 PI], DCV [NS5A RCI], and BMS-791325 [NS5B I]) were tested in two-drug combinations with rIFN-λ1. The degree of antagonism, additivity, or synergy was determined over a range of drug concentrations, and combination-response curves were fit to assess the antiviral effects of the drug treatment combinations. The combination effects were analyzed for departure of the results from additivity at the estimated EC50 level (Table 3) for each combination using the method described by Chou (27). Results from several independent experiments are summarized in Table 3. No enhanced cytotoxicity was reported for any of the combination agents tested and analyzed in parallel using a CellTiter blue assay (data not shown). Combination of rIFN-λ1 with rIFN-α resulted in CIs predominantly reflective of additivity at the 50% effective level and/or synergy at the 75% or 90% effective level. When combined with ASV, additivity was observed at the 50% effective level, while mixed additivity and synergy were observed at the 75% and 90% effective levels. Similarly, combination of rIFN-λ1 with DCV yielded a mixture of synergistic and additive effects in these experiments. Finally, additive effect levels were solely observed during combinations of rIFN-λ1 with BMS-791325. More importantly, no drug antagonism was observed at any effective doses in the various combination treatments, presumably reflecting the mechanistically distinct modes of action of IFN-λ1 and DAAs. Furthermore, three-drug combinations of lambda with ASV and DCV were tested for Loewe's additivity (see Table S2 in the supplemental material). The data from these experiments showed additivity at the 50% effective dose level and a mixture of additivity and synergy at the 75% and 90% effective dose levels. Overall, these results indicate that the combination of lambda with ASV and DCV displays mixed additivity and synergistic effects and are in agreement with data obtained with rIFN-λ1 (data not shown). Thus, lambda holds promise as part of a combination regimen involving different classes of DAAs.
Table 3.
Antiviral activity of rIFN-λ1 in combination with various classes of HCV replication inhibitors
| Drug combination (estimated EC50s) | Ratio of indicated inhibitor to rIFN-λ1 | CI (95% confidence interval) |
Overall result | ||
|---|---|---|---|---|---|
| 50% effective level | 75% effective level | 90% effective level | |||
| rIFN-α–rIFN-λ1 (18 IU/ml-0.3 ng/ml) | 9:1 | 0.94 (0.78, 1.09) | 0.89 (0.68, 1.10) | 0.85 (0.54, 1.16) | Additivity/synergy |
| 18:5 | 0.93 (0.78, 1.08) | 0.82 (0.64, 1.00) | 0.73 (0.47, 0.99) | ||
| 45:2 | 0.82 (0.70, 0.95) | 0.64 (0.50, 0.78) | 0.51 (0.33, 0.68) | ||
| ASV–rIFN-λ1 (1.3 nM-0.4 ng/ml) | 7:5 | 0.98 (0.82, 1.15) | 0.82 (0.63, 1.01) | 0.72 (0.46, 0.99) | Additivity/synergy |
| 14:5 | 0.97 (0.81, 1.14) | 0.85 (0.64, 1.05) | 0.76 (0.48, 1.05) | ||
| 7:2 | 0.88 (0.74, 1.02) | 0.75 (0.58, 0.92) | 0.67 (0.43, 0.91) | ||
| DCV–rIFN-λ1 (5 nM-3.9 ng/ml) | 1:310 | 0.99 (0.94, 1.04) | 0.93 (0.87, 0.99) | 0.88 (0.78, 0.98) | Additivity/synergy |
| 1:775 | 0.93 (0.87, 0.98) | 0.92 (0.84, 0.99) | 0.92 (0.81, 1.03) | ||
| 1:124 | 1.06 (1.00, 1.12) | 1.06 (0.97, 1.16) | 1.07 (0.93, 1.22) | ||
| NS5B I–rIFN-λ1 (3.6 nM-9.8 ng/ml) | 100:93 | 1.07 (0.98, 1.17) | 1.12 (0.99, 1.25) | 1.18 (0.97, 1.39) | Additivity |
| 40:93 | 1.08 (0.99, 1.17) | 1.05 (0.93, 1.17) | 1.03 (0.86, 1.20) | ||
| 250:93 | 0.91 (0.83, 1.00) | 1.03 (0.90, 1.17) | 1.18 (0.93, 1.42) | ||
Clearance of HCV replicons during IFN-based combination treatments.
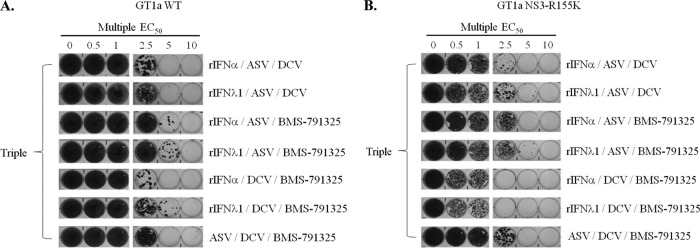
Elimination of HCV RNA and resistance development following selection under rIFN-α- or rIFN-λ1-based regimens in combination with various classes of DAAs were evaluated in the GT1a subgenomic HCV replicon. As shown in Fig. 2A, single treatment with ASV or DCV failed to reduce the frequency of replicon replication in cells, as seen by the appearance of intact monolayers following selective pressure up to 30 times the EC50 of each compound. In contrast, inclusion of rIFN-λ1 or rIFN-α in combination regimens with ASV or DCV greatly enhanced the elimination of replicons from cells in a dose-dependent manner. Furthermore, these regimens showed a greater capacity in reducing colony formation than a combination treatment of ASV and DCV in an IFN-free regimen. Interestingly, the reduction in colony formation appeared to be more efficient with rIFN-α than with rIFN-λ1 during dual therapy with ASV or DCV; however, the colony formation profiles of both IFN-based regimens were comparable when applied as part of a triple combination with ASV. Indeed, clearance of HCV replicons was achieved following triple combination treatment at 5 times EC50 multiples of ASV and DCV with either rIFN-λ1 or rIFN-α (15 times total EC50 multiples).
Fig 2.

Combination treatment of rIFN-λ1 with a DAA reduces the emergence of drug-resistant replicon colonies. Huh-7 cells that stably maintain HCV subgenomic replicons representing either a WT GT1a (A) or an engineered 1a-NS3-R155K variant (B) were incubated with identical treatment regimens including one, two, or three HCV inhibitors at 1, 2.5, 5, 10, or 30 times the EC50s estimated against the WT replicon. After a 25-day selection period in the presence of G418, surviving replicon colonies were fixed and stained with crystal violet. Results of a representative example of ≥2 independent experiments is shown.
The two novel NS3 PIs telaprevir and boceprevir were recently approved for use in combination treatments with alfa and RBV in patients chronically infected with HCV GT1. In clinical studies, the emergence of NS3-R155K has frequently been associated with resistance to telaprevir and boceprevir in patients failing therapies (32). The importance of viral resistance profiling in this new era of DAA-based therapy remains to be elucidated and will require detailed preclinical characterization. Therefore, we evaluated the capacity of rIFN-λ1 to reduce replicon colony survival under conditions similar to those described earlier using a GT1a HCV replicon cell line harboring the NS3-R155K substitution (1a-NS3-R155K). In these experiments (Fig. 2B), 1a-NS3-R155K replicon cells were treated with monotherapy or dual- and triple-combination regimens by applying a pressure (EC50s determined against the WT replicon) similar to that employed in the GT1a WT replicon selection. GT1a replicons carrying NS3-R155K conferred reduced susceptibility (∼30- to 50-fold) to ASV (Table 2). Consequently, combination regimens of ASV with rIFN-λ1 or rIFN-α had minimal effects in reducing the frequency of colony formation compared to the effects of the single-agent treatment. In contrast, combination regimens of DCV with rIFN-λ1 or rIFN-α were equally efficient in reducing the formation of replicon colonies. More importantly, clearance of the 1a-NS3-R155K replicon variant was achieved in cells at low EC50 multiples (15 times total EC50 multiples) following triple-combination treatments of ASV and DCV with either rIFN-α or rIFN-λ1. Interestingly, the presence of ASV in this triple-combination regimen resulted in a greater reduction of colony formation than that in the dual DCV–rIFN-α or DCV–rIFN-λ1 regimens (60 times total EC50 multiples). This phenotype may reflect a synergistic or an additive effect of ASV with these DAAs, as previously reported (33), even when applied at a concentration ∼50 times below the estimated EC50 against the NS3-R155K variant. Overall, these data suggest that preexisting or emerging NS3 protease-resistant variants could be suppressed with the use of lambda in combination with other specifically targeted DAAs.
Genotypic and phenotypic analysis of emerging DAA-resistant HCV replicons following IFN-based combination treatments.
To determine whether the selected colonies were resistant to the respective inhibitors, total RNA was extracted from cells treated for 25 days, and population-based sequencing analysis of the entire HCV genome encompassing the NS3- to the NS5B-coding region was performed to identify potential amino acid substitutions. As summarized in Table 4, replicons that contain previously known resistance-associated changes were isolated under selective pressure of 10 times the EC50s with either ASV or DCV. A mutation in the NS3 protease domain encoding residue R155 (R155K) was identified in replicon cell populations selected under ASV pressure, as previously reported (24). Similarly, mutants identified in the NS5A region following DCV selection encoded M28T, Q30R, or Y93C, which have been previously reported (29). None of the replicon cell lines selected under rIFN-α or rIFN-λ1 pressure showed identical genomic sequences, with multiple point mutations emerging throughout the NS3-, NS5A-, and NS5B-coding regions of the various HCV genomes. This is in accordance with in vitro studies indicating that an altered host-related JAK-STAT signaling pathway rather than viral factors contributes to lambda resistance in the HCV replicon cell system (J. Friborg et al., unpublished data). In dual-treatment regimens, sequencing analysis revealed the emergence of NS3-R155K when ASV was combined with rIFN-λ1, while DCV combined with either rIFN-α or rIFN-λ1 led to the selection of NS5A-M28T, -Q30R, and -L31M. Finally, replicon cell populations under selective pressure with either rIFN-α- or rIFN-λ1 in combination with ASV and DCV showed similar genotypic profiles, leading to substitution predominantly at NS5A-Q30 (Q30R or Q30H). Other amino acid substitutions in the NS3 domain (Q89R, G176R, and T177A) were identified less frequently during the combination treatments and occurred at positions previously described to be cell culture-adaptive variants or growth-compensatory substitutions (25, 33). The NS3 substitutions I132V and Y134H have not previously been detected during selection of ASV resistance, although the former has been associated with clinical resistance to telaprevir (34). Neither substitution conferred resistance to ASV when reintroduced into the GT1a WT replicon (data not shown).
Table 4.
Population sequencing analysis of GT1a WT replicon cells selected following treatment with rIFN-λ1 or rIFN-α in combination with DAAs
| Regimen | Treatment (EC50 multiple) | Amino acid change at the indicated position |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NS3 |
NS5A |
||||||||||||
| Q80 | Q89 | I132 | Y134 | R155 | G176 | T177 | K24 | M28 | Q30 | L31 | Y93 | ||
| Mono | Untreated | ||||||||||||
| ASV (10) | R/K | ||||||||||||
| DCV (10) | Q/R | M/T | Q/H | Y/C | |||||||||
| rIFN-α (10) | Q/R | ||||||||||||
| rIFN-λ1 (10) | |||||||||||||
| Dual | ASV+ rIFN-α (2.5) | Q/R | T/A | ||||||||||
| ASV + rIFN-α (5) | |||||||||||||
| ASV + rIFN-λ1 (2.5) | Q/R | I/V | |||||||||||
| ASV + rIFN-λ1 (5) | H/Y | ||||||||||||
| ASV + rIFN-λ1 (10) | Q/R | R/K | G/R | ||||||||||
| DCV + rIFN-α (2.5) | M/T | Q/R | L/M | ||||||||||
| DCV + rIFN-λ1 (2.5) | K/R | M/T | Q/R | ||||||||||
| ASV + DCV (2.5) | T/A | K/R | M/T | Q/R | |||||||||
| ASV + DCV (5) | H/Y | M/T | Q/R | ||||||||||
| ASV + DCV (10) | M/T | ||||||||||||
| Triple | ASV + DCV + rIFN-α (2.5) | Q/R | K/Q | Q/R | |||||||||
| ASV + DCV + rIFN-λ1 (2.5) | Q/R | V | H | ||||||||||
Sequencing analysis of the 1a-NS3-R155K-selected replicons revealed a resistance development pathway similar to the pathways for the resistance breakthrough profiles of GT1a drug-resistant replicons. Not surprisingly, by applying ASV at a selective pressure approximately 50 times below the EC50s determined against the NS3-R155K variant, no other resistance substitutions emerged during the course of the various NS3 PI-based treatments (Table 5). Conversely, the 1a-NS3-R155K replicon selected using 10 times DCV EC50 multiples led to the detection of an NS5A mutation encoding Q30H. The Q30H substitution emerged during combination treatment with DCV and rIFN-α at lower EC50 multiples, while a Y93F substitution emerged at higher EC50 multiple (5 times). The NS5A-M28T and -Y93H substitutions emerged when rIFN-λ1 was combined with DCV.
Table 5.
Population sequencing analysis of GT1a NS3-R155K replicon cells selected following treatment with of rIFN-λ1or rIFN-α in combination with DAAs
| Regimen | Treatment (EC50 multiple) | Amino acid change at the indicated position |
|||||
|---|---|---|---|---|---|---|---|
| NS3 R155 | NS5A |
||||||
| K24 | M28 | Q30 | L31 | Y93 | |||
| Mono | Untreated | K | K/T | ||||
| ASV (10) | K | ||||||
| DCV (10) | K | K/T | Q/H | ||||
| rIFN-α (10) | K | ||||||
| rIFN-λ1 (10) | K | ||||||
| Dual | ASV + rIFN-α (2.5) | K | K/T | ||||
| ASV + rIFN-α (5) | K | ||||||
| ASV + rIFN-λ1 (2.5) | K | K/T | |||||
| ASV + rIFN-λ1 (5) | K | ||||||
| ASV + rIFN-λ1 (10) | K | ||||||
| ASV + rIFN-λ1 (30) | K | ||||||
| DCV + rIFN-α (2.5) | K | K/T/E | H/Q | ||||
| DCV + rIFN-α (5) | K | F/Y | |||||
| DCV + rIFN-λ1 (5) | K | M/T | Y/H | ||||
| ASV + DCV (2.5) | K | K/T | Q/H/R | ||||
| ASV + DCV (5) | K | K/T | Q/H/R | L/M | |||
| ASV + DCV (10) | K | Q/H/R | L/M | ||||
| ASV + DCV (30) | K | H/Q | |||||
| Triple | ASV + DCV + rIFN-α (1) | K | |||||
| ASV + DCV + rIFN-λ1 (1) | K | ||||||
| ASV + DCV + rIFN-λ1 (2.5) | K | K/T | |||||
To confirm the contributions of these mutations to the reduced susceptibility to the respective inhibitors, phenotypic assays were performed on the breakthrough GT1a replicon cell populations. As shown in Fig. 3, the reduced susceptibility of the ASV-resistant replicon cell populations varied from 11- to 20-fold relative to the susceptibility of the parental control replicon, depending on the drug selection combination pressure. In contrast, the susceptibility to DCV was not affected in these selected replicon cell populations. The replicon cell populations emerging from the various DCV-based regimens demonstrated modest levels of resistance against DCV ranging from 12- to 943-fold over the control parental level, while showing no cross-resistance to ASV. Both ASV- and DCV-resistant cell populations remained susceptible to the NS5B I BMS-791325, rIFN-α, or rIFN-λ1 (data not shown). These data thus provide direct evidence that the resistance phenotype was specific to mutations identified during sequencing analysis and known to affect ASV or DCV susceptibility.
Fig 3.
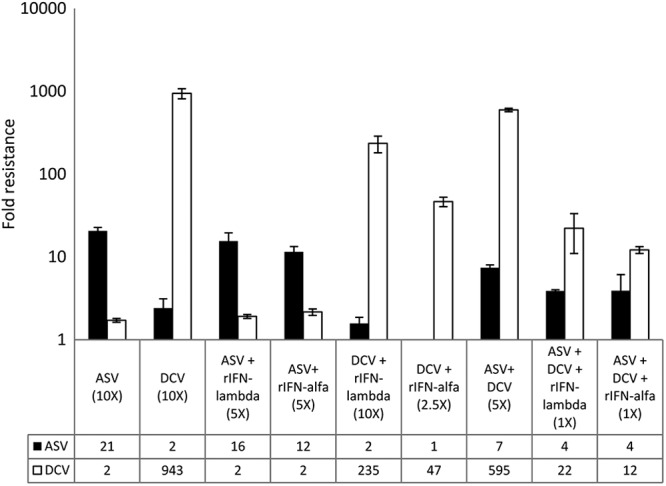
Phenotypic profiles of GT1a WT replicon cell populations selected following IFN-based combination treatments. Cell populations were assayed for sensitivity to ASV and DCV as described in the Material and Methods section. Fold resistance was plotted and represents the inhibitor EC50 against drug-resistant cells divided by the EC50 against DMSO-treated replicon control cells. Error bars represent standard deviations generated from three independent experiments.
The dual DAA–rIFN-λ1 regimen compares favorably to a triple DAA regimen in clearance of HCV DAA-resistant replicons.
Recently, results from a phase 2a trial indicated that 90% of HCV GT1 prior null responders to alfa-RBV treatment achieved SVR at 24 weeks (SVR24) after treatment with a regimen of ASV and DCV in combination with alfa-RBV (35). Therefore, we next examined in parallel the efficacy of rIFN-α or rIFN-λ1 when combined with two DAAs (ASV-DCV, ASV–BMS-791325, or DCV–BMS-791325) for its capacity to eliminate GT1a replicons. An IFN-free regimen that comprised all three DAAs was used as a control to assess the impact of rIFN-α or rIFN-λ1 in these regimens. Similar to the results shown in Fig. 1, clearance of the GT1a replicon was achieved following triple-combination treatment at 5 times EC50 multiples of ASV and DCV combined with rIFN-λ1 or rIFN-α (Fig. 4A). For most of the IFN-based regimens tested, both rIFN-λ1 and rIFN-α demonstrated comparable capacities in reducing colony formation at low EC50 multiples. The only exception was a combination of DCV–BMS-791325 with rIFN-λ1, which was unable to clear replicons at 5 times EC50 multiples, in contrast to the rIFN-α-based regimen. This is consistent with the increased frequency of emerging DAA-resistant colonies noted in the GT1a WT replicon system following dual-combination treatment with DCV and rIFN-λ1 compared to the frequency for the rIFN-α-based regimen. Interestingly, combinations of ASV plus DCV with either rIFN-λ1 or rIFN-α eliminated HCV replicons from cells with an efficiency comparable to that for an IFN-free triple-combination-based DAA treatment (ASV–DCV–BMS-791325).
Fig 4.
Combination of rIFN-λ1 with various classes of DAAs efficiently reduces replication of the 1a-NS3-R155K replicon variant. Huh-7 cells that stably maintain subgenomic replicons representing a GT1a WT (A) or 1a-NS3-R155K (B) carrying the neomycin resistance gene were treated in parallel with various combination regimens of three HCV inhibitors at 0.5, 1, 2.5, 5, or 10 times EC50 multiple. After a 25-day selection period in the presence of G418, surviving replicon colonies were fixed and stained with crystal violet. A representative example of two independent experiments is shown for each selection.
Finally, under identical selective pressure, the colony formation profiles for the 1a-NS3-R155K variant following treatments with the various triple-combination regimens did not differ from the profiles for the GT1a WT (Fig. 4B). Clearly, the combined regimen of DCV–BMS-791325 with rIFN-λ1 not only compared favorably to the rIFN-α-based regimen but also showed greater efficiency than the GT1a WT replicon in suppressing replication of the 1a-NS3-R155K replicon. Clearance of the NS3 variant was achieved at 2.5 times EC50 multiples with this regimen (7.5 times total EC50 multiples), reflecting the lack of cross-resistance to the compounds tested and potentially a higher genetic barrier to resistance with the addition of an NS5B nonnucleoside inhibitor, as previously suggested (36).
DISCUSSION
Numerous studies have documented the biological characteristics of type III IFNs since their discovery and depicted an overlapping profile with type I IFNs in respect of their antiviral properties (37). Both types of cytokines can ultimately inhibit HCV replication in a dose-dependent manner through the common activation of the JAK-STAT signaling pathway (12, 38). However, within the induced signaling cascade, there appears to be subtle differences between IFN-α and IFN-λ that are not fully understood. Even though several microarray analyses have shown that the repertoire of ISGs induced in vitro by type I and type III IFNs is essentially similar, the kinetics and magnitude of signal transduction differ between IFN-α and IFN-λ. The overall pattern in ISG expression in response to IFN-α tends to peak early, followed by a rapid decline, whereas IFN-λ triggers a weaker but more sustained increase in ISG expression (12). These fundamental differences may be attributed in part to the cellular distribution and expression levels of their respective cognate receptor complexes. Unlike IFN-α, the unique receptor complex for IFN-λ shows a more restricted expression, being highly detected in human hepatocytes and minimally expressed in hematopoietic cells. These distinct characteristics have made IFN-λ an ideal candidate for development as therapy against HCV infection with the potential of having reduced adverse events normally associated with alfa-based treatments. In this study, we extended the biological assessment of IFN-λ1 by demonstrating that a recombinant form of this cytokine (rIFN-λ1) has robust antiviral activities in various HCV replication cell systems representing GT1 and GT2. Interestingly, rIFN-λ1 demonstrated greater antiviral activity than both rIFN-λ2 and rIFN-λ3, while rIFN-λ3 constantly exhibited greater antiviral activity than rIFN-λ2 in all replicon cell systems. Potency rankings were supported by the stronger induction of STAT1 phosphorylation in cells in response to rIFN-λ1 than to rIFN-λ2 and rIFN-λ3. More importantly, the cell potencies of all three IFN-λs against various replicon variants with reduced susceptibility to investigational DAAs did not differ. The lack of cross-resistance between rIFN-λ1 and other HCV inhibitors may be crucial for future treatment strategies, as the potential development of viral resistance will become a major concern in this new era of DAA-based therapy.
Several lines of evidence have indicated a potential physiological role for type III IFN production in host responses to HCV infection. Early GWASs performed by Ge et al. have linked a single nucleotide polymorphism (SNP) surrounding the IL28B gene encoding IFN-λ3 to the spontaneous clearance and a successful alfa treatment outcome of HCV infection (28). The rs12979860 SNP identified in this study is located upstream of the IL28B locus. Patients homozygous for the major allele (the favorable CC genotype) were shown to have SVR rates more than 2-fold higher than those with the minor risk allele (the unfavorable TT or CT genotype) following alfa-based therapy. Subsequent GWASs have independently reported the identification of other SNPs associated with treatment failure. The majority of SNPs identified are not located within the regulatory elements or the coding region of IL28B itself but may be involved in the regulation of the functionality of the IFN-λ3 cytokine. The only SNP that is in the coding region of IL28B encodes a K70R variant which was associated with treatment failure. To this date, it has yet to be defined if the IFN-λ3-associated polymorphisms affect the immune responses or exert specific antiviral effects in HCV patients. Our data indicate that the SNP encoding the K70R variant did not alter the antiviral activity of IFN-λ3 against HCV. The potency of the recombinant form of IFN-λ3-K70R was comparable to that of the WT cytokine across various HCV replicon systems (Table 1). In turn, this finding supports the hypothesis that IL28B SNPs may affect the levels of IFN-λ3 endogenous expression upon viral infection. Interestingly, recent studies have shown that HCV infection is capable of inducing the production of all forms of type III IFNs in primary human hepatocyte-cell systems and in the chimpanzee animal model, while showing minimal effects on IFN-α/β mRNA (39–41). The level of endogenous IFN-λ1 induced upon infection was shown to be sufficient to control in part the HCV replication in a virus-based model. This observation raises the intriguing possibility of a relationship between the various IL28B genotypes and HCV clearance. Therefore, more refined quantitative studies for the detection of IFN-λ3 expression in infected subjects should provide valuable insight into the mechanism of regulation of IL28B SNPs.
Results from the phase 2 EMERGE trial investigating lambda as a new therapeutic agent against chronic infection with HCV isolates representing GT1 to GT4 were recently reported (14). Compared with a standard form of alfa-RBV, a lambda-based regimen showed similar to slightly better SVR24 rates in patients infected with GT2 or GT3 but, more importantly, was associated with significantly fewer IFN-related adverse events. Furthermore, week 12 on-treatment interim results in patients infected with GT1 and GT4 indicate more robust initial declines in HCV RNA with lambda than with alfa. Herein, we demonstrated for the first time that the inclusion of rIFN-λ1 in combination treatment with various classes of DAAs (an NS3 PI, an NS5A RCI, or an NS5B I) can significantly enhance clearance of HCV GT1a replicons with an efficiency comparable to that of rIFN-α-based regimens. In drug-drug interaction studies, lambda and rIFN-λ1 acted in an additive to synergistic manner when combined with investigational DAAs. In longer-term colony formation experiments, rIFN-λ1 combined with ASV or DCV greatly reduced resistance development compared to that with a single-agent treatment. Furthermore, GT1a replicons (either WT or the NS3-R155K variant) were completely eliminated following prolonged exposure to a triple-combination regimen of rIFN-λ1 with ASV and DCV at low EC50 multiples (Fig. 2). In contrast, a dual-combination-based IFN-sparing regimen combining ASV with DCV was unable to clear replicons in cells at high EC50 multiples (60 times total DAA EC50 multiples), further indicating that this combination does not provide a higher barrier to resistance than each drug used alone. An in vitro replicon model does not represent the most physiologically relevant system for studying the role of host genetic variation and the innate immune response during the course of HCV infection. Nevertheless, the comparable abilities of rIFN-λ1- and rIFN-α-based regimens to clear HCV replicons are important, in light of results from a recent clinical trial investigating the efficacy of combining ASV and DCV with or without alfa-RBV. When administered to a predominantly GT1a-null-responder population, alfa-RBV combined with ASV and DCV suppressed the emergence of resistance; 90% of patients achieved SVR24, whereas 36% of patients achieved SVR24 following treatment with the IFN-free dual-DAA regimen (35). Dual-DAA regimens have the potential to eradicate HCV infection in prior null responders; however, administration of an IFN combined with DAAs for 24 weeks may increase the cure rate in hard-to-treat GT1a-infected patients. Therefore, given its promising safety profile and proven efficacy in clinical studies, lambda has the potential to become an attractive alternative to alfa in IFN-based combination treatments.
Due to the large number of virions produced on a daily basis and the error-prone nature of the HCV-encoded RNA-dependent RNA polymerase, it has been predicted that infected patients harbor preexisting polymorphisms potentially resistant to DAAs (42, 43) The emergence of variants with reduced susceptibility to DAAs depends on the prevalence of preexisting variants and their replication fitness. The first-wave NS3 PIs are efficacious against GT1 infection; however, failure rates of >60% have been reported, depending on the patient population being treated. Since cross-resistance is extensive among the different NS3 PIs (42), future combination regimens should be tailored to prevent the emergence of cross-resistant strains. To overcome this issue, numerous alfa-based treatment approaches are currently being investigated in clinical settings. In our studies, we explored the combination of two DAAs, each with low genetic barriers to resistance (i.e., second-wave NS3 PIs and NS5A RCIs or an NS5B site I inhibitor with an NS5A RCI). We observed a higher efficiency of clearance of 1a-NS3-R155K replicons than the GT1a WT replicon when cells were treated with a triple combination of rIFN-λ1 with DCV and the NS5B site I inhibitor BMS-791325. This finding may be relevant, as various salvage treatments will need to be identified for patients who acquired resistance to the first wave of NS3 PIs. Moreover, our genotypic analyses of the resistant GT1a replicons indicated that many of the variants emerging in the presence of rIFN-λ1-based treatments combined with ASV and/or DCV have been previously identified in vitro or in clinical studies (24, 29, 35, 44). These included NS3-R155K and NS5A-M28T, -Q30H, -Q30R, and -Y93C, which individually were previously shown to confer low to modest levels of resistance in vitro to their respective targeted inhibitors. In accordance with that finding, phenotypic profiling of the GT1a drug-resistant replicons largely validated the population sequencing analysis, with reduced susceptibility to ASV or DCV being associated with the emergence of specific targeted substitutions. Consequently, while rIFN-λ1 works cooperatively with the various DAAs in reducing the emergence of variants, it does not appear to alter their pattern of resistance development.
In summary, we demonstrated that lambda is an HCV inhibitor with the potential for pan-genotypic activity and a nonoverlapping profile of resistance to DAAs. More importantly, lambda showed an additive to synergistic effect in vitro on HCV replication when combined with various classes of DAAs. Accordingly, lambda has the capacity to inhibit the emergence of drug-resistant variants when combined with these DAAs, consistent with a role in future combination therapies. Furthermore, our results suggest that tailored lambda-based regimens could also be effective in patients who failed NS3 PI-based therapies due to resistance development. Taken together, these data provide a rationale and guidance for future clinical evaluation of combination regimens with lambda.
Supplementary Material
ACKNOWLEDGMENTS
We thank Baiqing Lin for technical assistance. We thank Juan-Carlos Lopez-Talavera and Mark Cockett for their support and leadership.
Footnotes
Published ahead of print 28 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02239-12.
REFERENCES
- 1. Munir S, Saleem S, Idrees M, Tariq A, Butt S, Rauff B, Hussain A, Badar S, Naudhani M, Fatima Z, Ali M, Ali L, Akram M, Aftab M, Khubaib B, Awan Z. 2010. Hepatitis C treatment: current and future perspectives. Virol. J. 7:296 doi:10.1186/1743-422X-7-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975–982 [DOI] [PubMed] [Google Scholar]
- 3. McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, Nyberg LM, Lee WM, Ghalib RH, Schiff ER, Galati JS, Bacon BR, Davis MN, Mukhopadhyay P, Koury K, Noviello S, Pedicone LD, Brass CA, Albrecht JK, Sulkowski MS. 2009. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N. Engl. J. Med. 361:580–593 [DOI] [PubMed] [Google Scholar]
- 4. Asselah T, Estrabaud E, Bieche I, Lapalus M, De Muynck S, Vidaud M, Saadoun D, Soumelis V, Marcellin P. 2010. Hepatitis C: viral and host factors associated with non-response to pegylated interferon plus ribavirin. Liver Int. 30:1259–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoofnagle JH, Seeff LB. 2006. Peginterferon and ribavirin for chronic hepatitis C. N. Engl. J. Med. 355:2444–2451 [DOI] [PubMed] [Google Scholar]
- 6. Negro F. 2010. Adverse effects of drugs in the treatment of viral hepatitis. Best Pract. Res. Clin. Gastroenterol. 24:183–192 [DOI] [PubMed] [Google Scholar]
- 7. Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 8. Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. 2003. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4:69–77 [DOI] [PubMed] [Google Scholar]
- 10. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donnelly RP, Kotenko SV. 2010. Interferon-lambda: a new addition to an old family. J. Interferon Cytokine Res. 30:555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. 2006. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131:1887–1898 [DOI] [PubMed] [Google Scholar]
- 13. Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, Yao L, Liu H, Barahmand-pour F, Sivakumar P, Chan C, Birks C, Foster D, Clegg CH, Wietzke-Braun P, Mihm S, Klucher KM. 2006. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 44:896–906 [DOI] [PubMed] [Google Scholar]
- 14. Zeuzem SA, Bacon B, Box T, Charlton M, Diago M, Dieterich REMD, Everson G, Fallon M, Ferenci P, Flisiak JGR, Ghalib R, Gitlin N, Gladysz A, Gordon SGS, Hassanein T, Jacobson I, Jeffers KKL, Lawitz E, Lee SS, Leggett B, Lueth DNS, Pockros P, Rodriguez-Torres M, Rustgi LSV, Sherman M, Shiffman M, Sola R, Sulkowski HVM, Vierling J, Yoffe B, Ishak L, Fontana DXD, Gray T, Horga A, Hillson J, Lopez-Talavera AMJC. 2012. Peginterferon lambda-1A (lambda) compared with peginterferon alfa-2A (alfa) in treatment-naive patients with HCV genotypes 2 or 3: first SVR24 results from EMERGE phase IIb. J. Hepatol. 56:S5–S6 [Google Scholar]
- 15. Balagopal A, Thomas DL, Thio CL. 2010. IL28B and the control of hepatitis C virus infection. Gastroenterology 139:1865–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dahari H, Guedj J, Perelson AS, Layden TJ. 2011. Hepatitis C viral kinetics in the era of direct acting antiviral agents and IL28B. Curr. Hepat. Rep. 10:214–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muir AJ, Shiffman ML, Zaman A, Yoffe B, de la Torre A, Flamm SL, Steven G, Stuart C, Morotta P, Vierling J, Lopez-Talavera JC, Byrnes-Blake KA, Fontana D, Freeman JA, Gray T, Hausman D, Hunder N, Lawitz E. 2010. Phase 1b study of pegylated interferon lambda 1 with or without RBV in patients with chronic genotype 1 hepatitis C virus infection. Hepatology 52:822–832 [DOI] [PubMed] [Google Scholar]
- 18. Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, Chen C, Lee MS, Mosure K, Sun LQ, Sinz M, Meanwell NA, Colonno RJ, Knipe J, Scola P. 2012. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob. Agents Chemother. 56:5387–5396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kadow JF, Gentles RG, Ding M, Bender JA, Bergstrom C, Grant-Young K, Hewawasam P, Hudyma TW, Martin S, Nickel A, Regueiro-Ren A, Tu Y, Yang Z, Yeung KS, Zheng X, Chen BC, Chao S, Sun JH, Li JM, Smith AD, Wu DR, Beno B, Hanumegowda U, Knipe J, Parker DD, Zhuo X, Lemm JA, Liu M, Pelosi L, Rigat K, Voss S, Wang Y, Wang YK, Colonno RC, Gao M, Roberts SB, Meanwell NA. 2012. Discovery of BMS-791325, an allosteric NS5B replicase inhibitor for the treatment of hepatitis C, abstr. MEDI-23. Abstr. 243rd ACS Natl. Meet. Expo., San Diego, CA [Google Scholar]
- 21. Bronowicki JP, Pol S, Thuluvath P, Larrey D, Martorell CT, Rustgi VK, Morris DW, Younes Z, Fried MW, Bourliere M, Hezode C, Reddy R, Massoud O, Abrams GA, Ratziu V, He B, Eley T, Ahmad A, Thiry A, Llamoso C, Mendez P, Hughes E. 2012. Asunaprevir (ASV; BMS-650032), an NS3 protease inhibitor, in combination with peginterferon and ribavirin in treatment-naive patients with genotype 1 chronic hepatitis C infection. J. Hepatol. 56(Suppl 2):S431–S432 [Google Scholar]
- 22. Tatum H, Thuluvath P, Lawitz E, Martorell CT, Demicco M, Cohen S, Rustgi V, Ravendhran N, Ghalib R, Hanson J, Zamparo J, Yang R, Hughes E, Cooney E. 2012. A phase 2A study of BMS-791325, an NS5B polymerase inhibitor, with peginterferon alfa-2a and ribavirin in treatment-naive patients with genotype 1 chronic hepatitis C Infection. J. Hepatol. 56(Suppl 2):S460 [Google Scholar]
- 23. Pol S, Ghalib RH, Rustgi VK, Martorell C, Everson GT, Tatum HA, Hézode C, Lim JK, Bronowicki J-P, Abrams GA, Bräu N, Morris DW, Thuluvath PJ, Reindollar RW, Yin PD, Diva U, Hindes R, McPhee F, Hernandez D, Wind-Rotolo M, Hughes EA, Schnittman S. 2012. Daclatasvir for previously untreated chronic hepatitis C genotype-1 infection: a randomised, parallel-group, double-blind, placebo-controlled, dose-finding, phase 2a trial. Lancet Infect. Dis. 12:671–677 [DOI] [PubMed] [Google Scholar]
- 24. McPhee F, Friborg J, Levine S, Chen C, Falk P, Yu F, Hernandez D, Lee M, Chaniewski S, Sheaffer AK, Pasquinelli C. 2012. Resistance analysis of the hepatitis C virus NS3 protease inhibitor asunaprevir. Antimicrob. Agents Chemother. 56:3670–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sheaffer AK, Lee MS, Hernandez D, Chaniewski S, Yu F, Falk P, Friborg J, Zhai G, McPhee F. 2011. Development of a chimeric replicon system for phenotypic analysis of NS3 protease sequences from HCV clinical isolates. Antivir. Ther. 16:705–718 [DOI] [PubMed] [Google Scholar]
- 26. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. 2007. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 81:7749–7758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58:621–681 [DOI] [PubMed] [Google Scholar]
- 28. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, Sulkowski M, McHutchison JG, Goldstein DB. 2009. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401 [DOI] [PubMed] [Google Scholar]
- 29. Wang C, Huang H, Valera L, Sun JH, O'Boyle DR, II, Nower PT, Jia L, Qiu D, Huang X, Altaf A, Gao M, Fridell RA. 2012. Hepatitis C virus RNA elimination and development of resistance in replicon cells treated with BMS-790052. Antimicrob. Agents Chemother. 56:1350–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Muh U, Welker M, Wincheringer D, Zhou Y, Chu HM, Lin C, Weegink C, Reesink H, Zeuzem S, Kwong AD. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777 [DOI] [PubMed] [Google Scholar]
- 31. Susser S, Vermehren J, Forestier N, Welker MW, Grigorian N, Fuller C, Perner D, Zeuzem S, Sarrazin C. 2011. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J. Clin. Virol. 52:321–327 [DOI] [PubMed] [Google Scholar]
- 32. Thompson AJ, Locarnini SA, Beard MR. 2011. Resistance to anti-HCV protease inhibitors. Curr. Opin. Virol. 1:599–606 [DOI] [PubMed] [Google Scholar]
- 33. Pelosi LA, Voss S, Liu M, Gao M, Lemm JA. 2012. Effect on hepatitis C virus replication of combinations of direct-acting antivirals, including NS5A inhibitor daclatasvir. Antimicrob. Agents Chemother. 56:5230–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vertex Pharmaceuticals 2011. IncivekTM (telaprevir) package insert. Vertex Pharmaceuticals, Cambridge, MA [Google Scholar]
- 35. Lok AS, Gardiner DF, Lawitz E, Martorell C, Everson GT, Ghalib R, Reindollar R, Rustgi V, McPhee F, Wind-Rotolo M, Persson A, Zhu K, Dimitrova DI, Eley T, Guo T, Grasela DM, Pasquinelli C. 2012. Preliminary study of two antiviral agents for hepatitis C genotype 1. N. Engl. J. Med. 366:216–224 [DOI] [PubMed] [Google Scholar]
- 36. Vermehren J, Sarrazin C. 2011. New HCV therapies on the horizon. Clin. Microbiol. Infect. 17:122–134 [DOI] [PubMed] [Google Scholar]
- 37. Witte K, Witte E, Sabat R, Wolk K. 2010. IL-28A, IL-28B, and IL-29: promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev. 21:237–251 [DOI] [PubMed] [Google Scholar]
- 38. Zhang L, Jilg N, Shao RX, Lin W, Fusco DN, Zhao H, Goto K, Peng LF, Chen WC, Chung RT. 2011. IL28B inhibits hepatitis C virus replication through the JAK-STAT pathway. J. Hepatol. 55:289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, Dustin LB. 2011. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology 54:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. 2012. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 142:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park H, Serti E, Eke O, Muchmore B, Prokunina-Olsson L, Capone S, Folgori A, Rehermann B. 2012. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology 56:2060–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462 [DOI] [PubMed] [Google Scholar]
- 43. Rong L, Ribeiro RM, Perelson AS. 2012. Modeling quasispecies and drug resistance in hepatitis C patients treated with a protease inhibitor. Bull. Math. Biol. 74:1789–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fridell RA, Wang C, Sun JH, O'Boyle DR, II, Nower P, Valera L, Qiu D, Roberts S, Huang X, Kienzle B, Bifano M, Nettles RE, Gao M. 2011. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology 54:1924–1935 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.