

Abstract
To investigate the pharmacokinetics, safety, and tolerability of GS-9851 (formerly PSI-7851), a new nucleotide analog inhibitor of hepatitis C virus (HCV), we conducted a double-blind, parallel, placebo-controlled, randomized, single-ascending-dose study. Healthy subjects received oral doses of 25 to 800 mg GS-9851. Peak concentrations of GS-9851 in plasma were achieved more rapidly than those of the metabolites GS-566500 (formerly PSI-352707) and GS-331007 (formerly PSI-6206), with time to maximum concentration of drug in plasma (tmax) values of 1.0 to 1.8 h, 1.5 to 3.0 h, and 3.0 to 6.0 h, respectively. The majority of systemic drug exposure was from the nucleoside GS-331007, with maximum concentration of drug in plasma (Cmax) and area under the concentration-time curve to the last measurable concentration (AUC0–t) values at least 7- and 41-fold higher, respectively, than those obtained for GS-9851 after adjusting for differences in molecular weight. The terminal elimination half-life (t1/2) of GS-331007 increased with the dose, achieving a t1/2 of 25.7 h at 800 mg GS-9851. Dose proportionality was not observed for GS-331007. The majority of drug recovered in urine was in the form of GS-331007, with the percentage of this metabolite in urine samples ranging from 57% to 27% with increasing dose. GS-9851 was generally well tolerated, with no maximum tolerated dose identified. In conclusion, GS-9851 and its metabolites demonstrated a favorable pharmacokinetic profile consistent with once-daily dosing, and therefore, further clinical studies evaluating GS-9851 in HCV-infected patients are warranted.
INTRODUCTION
Chronic infection with hepatitis C virus (HCV) is a major cause of chronic liver disease, cirrhosis, and liver cancer worldwide (1). A broadly effective cure has remained elusive (2–5). The new current standard-of-care (SOC) treatment for HCV is a combination of weekly injections of pegylated alpha interferon plus oral daily administration of ribavirin and a protease inhibitor (6). However, this treatment regimen is recommended only for patients infected with HCV genotype 1, against which it has proven to be effective. Other limitations of the SOC are the variable levels of treatment success, with nonresponse rates of approximately 30%, and the frequent side effects that affect patient adherence (6, 7). The well-recognized need for an effective treatment for HCV has driven continuous efforts to develop more effective therapies. Hepatitis C virus RNA-dependent RNA polymerase is a virally encoded molecule that has been identified as a potential target for antiviral agents (8). Essential for viral replication, the enzyme is encoded by the nonstructural protein 5B (NS5B) region of the HCV genome. There is no known structural homologue of RNA polymerase NS5B in the uninfected host cell (8).
GS-9851 (formerly PSI-7851) is a phosphoramidate nucleotide prodrug that potently and selectively inhibits NS5B (9, 10). In vitro, GS-9851 was shown to be a highly effective pan-genotype HCV inhibitor, with the GS-9851 concentration resulting in 90% inhibition (EC90) of HCV replicon determined to be 0.4 μM (9). However, to inhibit NS5B, this prodrug must be first metabolized to the active triphosphate form GS-461203 (formerly PSI-7409). In vitro, studies conducted with primary human hepatocytes and with primary hepatocytes isolated from rat, dog, and monkey, together with a preliminary in vivo study in rats, demonstrated that GS-9851 is first hydrolyzed to the inactive, nonisomeric GS-566500 (formerly PSI-352707) intermediate form (11). GS-566500 is further metabolized to either the inactive nucleoside metabolite GS-331007 (formerly PSI-6206) or an inactive uridine monophosphate, GS-606965 (formerly PSI-7411). Inside the hepatocyte, GS-9851 is converted to GS-606965, which is further phosphorylated to an active triphosphate metabolite, GS-461203. GS-461203 selectively inhibits recombinant NS5B polymerase (Fig. 1) (12). High liver-to-plasma ratios for GS-9851 and relevant metabolites have been observed in preclinical testing using three different species following oral dosing with GS-9851 (13). Thus far, there has been no evidence of cytotoxicity or mitochondrial toxicity at all concentrations tested (up to 100 μM) (9, 13). The observed antiviral activity of GS-9851 appears to be additive with pegylated interferon and ribavirin combinations in vitro (14) and can be considered additive to synergistic when used in combination with NS3 protease inhibitors and other inhibitors of the NS5B (9). Data from preclinical studies demonstrated that the antiviral potency of GS-9851 and its distribution and metabolism profile warrant clinical evaluation.
Fig 1.

Schematic of the proposed metabolic pathway of GS-9851. GS-9851 is a mixture of two diastereoisomers, GS-491241 and GS-7977. Studies in vitro demonstrated that GS-9851 is metabolized in the plasma to the intermediate GS-566500 and to the inactive nucleoside derivative GS-331007 (step not shown). Inside the hepatocyte, both GS-491241 and GS-7977 undergo hydrolysis of the carboxyl ester catalyzed by the hepatically expressed carboxyl esterase 1 (hCE1) and cathepsin A (CatA) to form GS-566500. GS-566500 is further hydrolyzed by the histidine triad nucleotide binding protein 1 (HINT1) to either GS-331007 or a uridine monophosphate, GS-606965 (step not shown). GS-606965 is further phosphorylated to the nucleotide diphosphate GS-607596 (step not shown) and then to the active triphosphate NS5B inhibitor GS-461203.
This study involved the first administration of GS-9851 to healthy human subjects. The aim of the study was to determine whether plasma concentrations of GS-9851 resulting from administration of single oral doses of 25 mg to 800 mg were safe and tolerable in humans and to characterize the pharmacokinetic profile of GS-9851 and its metabolites. The rate and extent of absorption of GS-9851 administered as a solution and as a capsule were also investigated.
GS-9851 is a mixture comprised of two chemically identical isomers, GS-7977 (formerly PSI-7977) and GS-491241 (formerly PSI-7976). Originally, GS-9851 was selected for development because it was not possible to efficiently separate the diastereoisomers at that point in time and both had antiviral activity. However, a method of separation was eventually developed, and GS-7977 was selected for continued development. Nonetheless, an initial evaluation of GS-9851 was performed and comprised two studies: the one we describe here and a multiple-ascending-dose study of patients infected with HCV, described in our accompanying article (16). Since GS-9851, GS-7977, and GS-491241 share the same metabolic pathway (11), the results obtained for GS-9851 can be translated to GS-7977.
(This work was presented in part at the 60th Annual Meeting of the American Association for the Study of Liver Diseases, Boston, MA, November 2009.)
MATERIALS AND METHODS
Study population.
Forty-two healthy male and female subjects of ages between 18 and 55 years, with body mass indices of 19 to 30 kg/m2, were enrolled. Female subjects were required to be of non-childbearing potential or to take protocol-specified contraceptive measures. Subjects were excluded if they tested serologically positive for hepatitis B, HCV, or human immunodeficiency virus. Concurrent drugs known to affect the elimination of serum creatinine or substrates/inhibitors of renal tubular secretion were excluded within 60 days prior to the first dose of the study drug, and use of medication associated with QT interval prolongation was not permitted within 30 days prior to dosing or during the study. Subjects unwilling to abstain from alcohol, caffeine- or xanthine-containing products, strenuous exercise, grapefruit, or grapefruit products for 72 h prior to dosing were ineligible. Subjects unwilling to refrain from smoking or the use of tobacco or nicotine-containing products for 3 months prior to the screening visit or during the study were also ineligible.
The study (protocol number P7851-1101) was conducted at Covance Clinical Research Unit, Inc. (Madison, WI) following protocol approval by the Independent Investigational Review Board, Inc. (Plantation, FL). All subjects provided signed and dated informed consent prior to screening.
Study design.
The study utilized a double-blind, randomized, placebo-controlled, ascending-dose design. The study involved administration of single oral ascending doses of GS-9851: 25 mg, 50 mg, 100 mg, 200 mg, 400 mg, and 800 mg. The initial starting dose was selected in accordance with FDA guidance based upon the no-observed-effect levels in nonclinical species. Subjects were allocated to one of four groups that consisted of 10 subjects who were randomized to receive either an active dose or a placebo during each dosing session. Subjects underwent dosing at the following dose levels: 25 mg capsule and 200 mg capsule or placebo (group 1), 50 mg capsule and 50 mg solution or placebo (group 2), 100 mg capsule and 400 mg capsule or placebo (group 3), or 800 mg capsule alone or placebo (group 4). At each dose level, 10 subjects were randomized, with 8 receiving GS-9851 and 2 receiving placebo. Groups were dosed in ascending dose order with a washout period of at least 1 week between dosing sessions; subjects allocated to the 800-mg treatment group underwent only a single dosing session.
Subjects attended a screening visit within 28 days prior to receiving the first dose of study drug. They were admitted to the clinical study unit the day before dosing for baseline assessments that included measurement of vital signs, electrocardiogram (ECG) and clinical laboratory tests. The day after admission, subjects received their randomized treatment and underwent a period of observation and assessment for 5 days. After the washout period, subjects returned to the unit to receive their second randomized dose of study medication, with an additional 5-day inpatient assessment. Subjects returned for a follow-up visit 7 to 10 days after their last dose of study medication.
Dosing and sample collection.
GS-9851 (manufactured by Samchully, Inc., Seoul, South Korea, on behalf of Pharmasset, Inc.) or placebo was administered after a 10-h fast. Subjects who received capsules were instructed to take their study medication with at least 240 ml of water. Subjects who received the solution dose consumed a total volume of 240 ml of water (including the dosing solution). During a 3-h period around the dosing time (1 h predose until 2 h postdose), no water was allowed, with the exception of that taken with the study drug. On the completion of each dose level, pharmacokinetic and safety data were examined by the sponsor and investigator. If no clinically significant issues were found, escalation to the next dose level was permitted. Blood samples were drawn for pharmacokinetic analysis predose (within 15 min prior to dosing) and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 48, 72, and 96 h after dosing. An aliquot of urine was collected predose; after dosing, all urine was collected and pooled over the following time intervals: 0 to 6, 6 to 12, 12 to 24, 24 to 36, 36 to 48, 48 to 72, and 72 to 96 h for analysis of concentrations of GS-9851 and metabolites.
Pharmacokinetic analysis.
The plasma concentrations of the prodrug GS-9851 and of the metabolites GS-566500, GS-606965, and GS-331007 were determined by a validated methodology that included liquid chromatography with tandem mass spectroscopy (LC-MS/MS). The assay was validated by QPS, LLC, Newark, DE. The lower limit of quantitation (LLQ) of the assay was 5 ng/ml for GS-9851 and 10 ng/ml for GS-566500, GS-606965, and GS-331007. The linear range of the plasma assay was 5 to 5,000 ng/ml. Urine samples were analyzed using a validated LC-MS/MS method (QPS, LLC, Newark, DE) to determine concentrations of GS-9851, GS-566500, and GS-331007. The LLQ of the assay was 10 ng/ml for each compound. The linear range of the urine assay was 10 to 10,000 ng/ml. In plasma, the intraday and interday precision ranges (percent coefficient of variation [% CV]) were, respectively, 1.8 to 13.8 and 4.1 to 9.3 for GS-9851, 1.5 to 11.7 and 4.4 to 10.1 for GS-566500, and 1.5 to 8.1 and 2.2 to 6.2 for GS-331007. In urine, the intraday and interday precision ranges (% CV) were, respectively, 1.7 to 12.9 and 3.1 to 11.9 for GS-9851, 1.5 to 10.1 and 3.2 to 9.6 for GS-566500, 1.8 to 16.1 and 2.1 to 15.5 for GS-331007.
For each subject, plasma pharmacokinetic parameters for GS-9851, GS-566500, and GS-331007 were determined by noncompartmental analysis of plasma concentration-time data using the software program WinNonlin Professional, version 5.2 (Pharmasight Corporation, Mountain View, CA). The following pharmacokinetic parameters were calculated: maximum concentration of drug in plasma (Cmax), time of maximum drug concentration (tmax), area under the plasma drug concentration-time curve to the last measurable concentration (AUC0–t), half-life (t1/2), area under the plasma concentration-time curve from time zero and extrapolated to infinity (AUC0–∞), renal clearance (CLrenal), and percentage of study drug or its metabolites excreted in urine (urine %; molecular weight adjusted to GS-9851 for metabolites). Molecular-weight-adjusted metabolite-to-parent Cmax and AUC ratios were also calculated.
Safety and tolerability assessments.
Adverse-event data were recorded from first dose until the follow-up visit. For each event, the information collected included onset, duration, intensity, and potential causal relationship with the study drug as assessed by the investigator. Vital signs, including blood pressure, heart rate, and respiratory rate, were measured predose, at 4, 24, 48, 72, and 96 h postdose, and at follow-up. Twelve-lead electrocardiograms were recorded predose (in triplicate, separated by approximately 1 min) on the first dosing day and at 1, 2, 3, 4, 6, 8, 12, 24, 48, 72, and 96 h postdose. Telemetry was evaluated continuously from 12 h predose until 48 h postdose. A 10-s printed telemetry strip was taken at baseline within 30 min prior to dosing. Any abnormal findings in telemetry were confirmed by 12-lead ECG. Clinical laboratory samples were obtained at the screening visit, admission day (a day prior to initiation of dosing of study drug), in the mornings of day 2 and day 4 postdose, and at the follow-up visit. Clinical laboratory changes were graded according to criteria specified in the protocol using the DAIDS Table for Grading the Severity of Adult and Pediatric Adverse Events (15).
Statistical analysis.
Standard summary statistics were determined for all pharmacokinetic parameters.
Dose proportionality of Cmax, AUC0–∞, and AUC0–t were determined for GS-9851, GS-566500, and GS-331007, if appropriate. Dose proportionality was determined by utilizing a power model and a supportive analysis of variance (ANOVA) using log-transformed pharmacokinetic data for each dose normalized to a 100-mg reference dose.
Relative bioavailability of the solution formulation to the capsule formulation for selected pharmacokinetic parameters at one dose level (AUC0–∞, AUC0–t, and Cmax) was assessed by ANOVA. Upon log transformation, estimates of the difference between capsule and solution were made, together with 90% confidence intervals (CIs). These differences and CIs were transformed back to the original scale to provide an estimate of the relative bioavailability of the solution.
RESULTS
Subject disposition.
Forty-two healthy subjects were enrolled in the study. Most subjects were male (69%) and Caucasian (88%), with a mean (range) age of 29.5 years (19 to 51 years). In two dosing sessions, 33 subjects received GS-9851 and 9 received placebo (Fig. 2). Five subjects (three after GS-9851 and two after placebo) successfully completed the first dose of the planned two-dose administration but were found to no longer meet the inclusion criteria prior to receiving the second dose of study medication and were excluded from further dosing by the sponsor. The three subjects who received GS-9851 were discontinued for non-clinically significant telemetry abnormalities unrelated to the study drug on day −1 in period 2 (GS-9851 at 25 mg; n = 1), preexisting hematuria and a febrile illness prior to period 2 (GS-9851 at 50 mg; n = 1), and asymptomatic premature ventricular contractions later discovered to be preexisting (GS-9851 at 100 mg; n = 1). The two subjects who received placebo were discontinued because of an isolated four-beat run of nonsustained monomorphic ventricular tachycardia (n = 1) and a heart murmur (n = 1) found during check-in for period 2.
Fig 2.

CONSORT diagram of subject disposition.
Safety and tolerability.
GS-9851 was generally well tolerated, with no dose-limiting toxicities and no maximum tolerated dose identified. There was no increase in the frequency or severity of adverse events with dose escalation of GS-9851 from 25 mg to 800 mg. No adverse events attributed to the study drug were rated by the investigator as serious or severe. One subject (receiving GS-9851 at 800 mg) reported an episode of dizziness that was considered moderate in intensity. Sixteen (48%) subjects who received GS-9851 at any dose and four (44%) who received a placebo experienced at least one adverse event (Table 1). The most frequently reported adverse event was erythema (n = 16) at the telemetry and/or ECG patch sites. Six (18%) subjects who received GS-9851 experienced adverse events that were judged to be drug related by the investigator. The most frequently reported of these events were dizziness, headache, pain in the upper abdomen, and nausea (two subjects each). All adverse events considered by the investigator to be related to the study drug resolved without intervention. Clinical laboratory evaluations showed that there were no trends detected when assessing changes from the baseline in clinical chemistry and hematology values over time following study drug administration. Treatment-emergent grade 1 or grade 2 laboratory changes were infrequent and sporadic and did not appear to be dose related. There were no grade 3 or grade 4 laboratory abnormalities reported. No abnormalities in vital signs or 12-lead ECG parameters were considered sufficiently notable to be reported as adverse events.
Table 1.
Summary of most frequent adverse events (≥2 subjects who received active treatment)
| Adverse event | No. (%) of subjects with adverse event with placebo or dosea |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo (9) | GS-9851 |
||||||||
| All active (33)b | 25 mg, Cap (6) | 50 mg, Cap (8) | 50 mg, Sol (7) | 100 mg, Cap (8) | 200 mg, Cap (8) | 400 mg, Cap (7) | 800 mg, Cap (8) | ||
| Any event | 4 (44) | 16 (48) | 3 (50) | 3 (38) | 1 (14) | 6 (75) | 3 (38) | 3 (43) | 1 (13) |
| Any event judged drug related | 1 (11) | 6 (30) | 3 (50) | 1 (13) | 0 | 0 (75) | 2 (38) | 1 (14) | 1 (25) |
| Erythema | 4 (44) | 12 (36) | 1 (17) | 2 (25) | 1 (14) | 2 (25) | 3 (38) | 2 (29) | 0 |
| Pruritus | 1 (11) | 4 (12) | 1 (17) | 0 | 0 | 0 | 1 (13) | 0 | 0 |
| Dizziness | 0 | 2 (6) | 0 | 0 | 0 | 0 | 1 (13) | 0 | 1 (13) |
| Headache | 0 | 2 (6) | 2 (33) | 0 | 0 | 0 | 0 | 0 | 0 |
| Abdominal pain | 0 | 2 (6) | 0 | 0 | 0 | 0 | 0 | 1 (14) | 1 (13) |
| Nausea | 0 | 2 (6) | 0 | 1 (13) | 0 | 0 | 1 (13) | 0 | 0 |
The number of subjects tested is given in parentheses after the dose. Cap, capsule; Sol, solution.
All subjects dosed with active drug.
Pharmacokinetic results.
GS-606965 was not detected in the plasma of any subject at doses up to 400 mg; however, it was detected at one time point (2 h) in a single individual following a single 800-mg dose of GS-9851 (10.8 ng/ml). There were concentration-time data sufficient to calculate pharmacokinetic parameters for three analytes of interest, GS-9851, GS-566500, and GS-331007. Plots of median plasma concentration-time profiles on semilogarithmic scales for GS-9851 and metabolites GS-566500 and GS-331007 are shown in Fig. 3. GS-331007 accounted for the majority of the systemic drug exposure, whereas exposure to GS-9851 and GS-566500 was generally low. GS-9851 and GS-566500 concentrations fell rapidly to below the limit of detection of the assay. GS-331007 concentrations were detectable for at least 24 h following administration of powder-in-capsule from 100 mg to 800 mg GS-9851.
Fig 3.
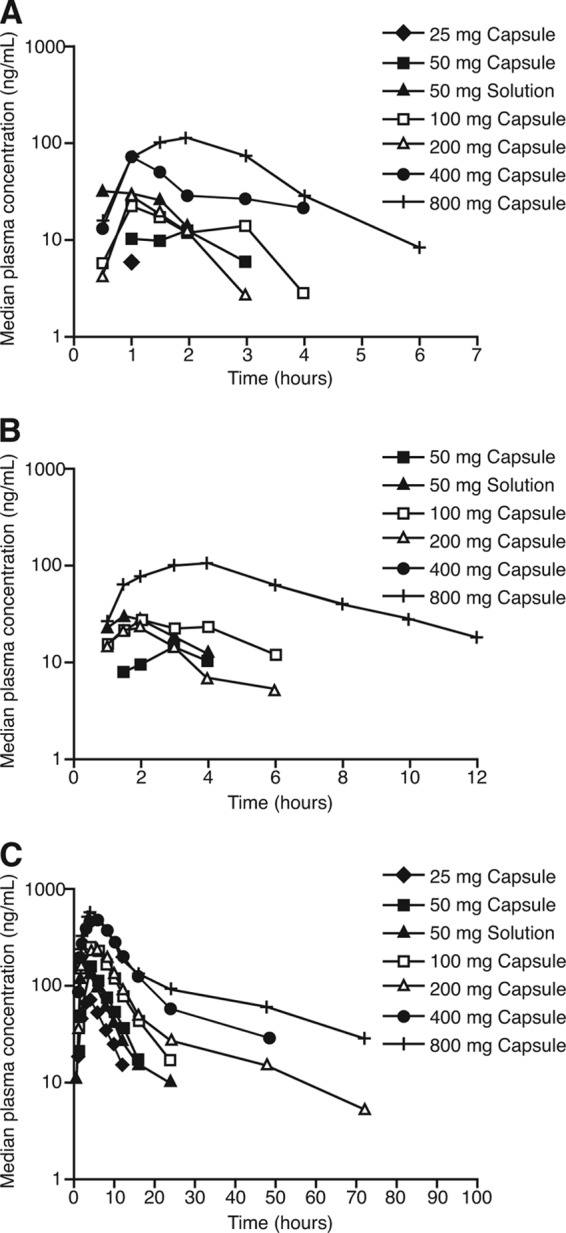
Logarithmic plots of median plasma concentration-time profiles for GS-9851 (A), GS-566500 (B), or GS-331007 (C).
Derived pharmacokinetic parameters for the three molecules are summarized in Table 2, Table 3, and Table 4. Absorption of GS-9851 was relatively rapid, with Cmax at 1.0 to 1.8 h across the dose range studied (Table 2). GS-9851 elimination was likewise rapid, with a t1/2 of approximately 1 h. GS-566500 reached Cmax at 1.5 to 3.0 h postdose and was cleared with an elimination t1/2 of 2.0 to 3.0 h (Table 3). GS-331007 achieved Cmax at 3.0 to 6.0 h postdose, and values of t1/2 increased with dose: t1/2 was 3.4 h at the 25-mg dose of GS-9851 and 25.7 h at the 800-mg dose (Table 4). The AUC0–t value for GS-331007 was at least 41-fold higher than the value obtained for GS-9851 after adjusting for differences in molecular weight. This observation was consistent with the rapid conversion of GS-9851 into its metabolites and the prolonged t1/2 of GS-331007 relative to that of GS-9851. The majority of the drug recovered in the urine was recovered as GS-331007. After adjustment for molecular weight differences between prodrug and metabolites, the percentage of GS-331007 recovered in the urine ranged from 57% to 27% across the dose range studied. Renal clearance values for GS-331007 remained relatively constant, at approximately 0.2 liters/min, across the ascending-dose groups.
Table 2.
Derived pharmacokinetic parameters for GS-9851
| Parameter | Mean (SD) value for doseb |
||||||
|---|---|---|---|---|---|---|---|
| 25 mg, capsule (6) | 50 mg, capsule (8) | 50 mg, solution (7) | 100 mg, capsule (8) | 200 mg, capsule (8) | 400 mg, capsule (7) | 800 mg, capsule (8) | |
| Cmax (ng/ml) | 7.6 (2.0) | 18.5 (9.4) | 38.4 (11.9) | 31.9 (16.0) | 54.6 (40.5) | 69.2 (24.0) | 317.5 (356.8) |
| tmax (h) | 1.0 [0.5–2.0] | 1.0 [0.5–2.0] | 1.0 [0.5–1.0] | 1.8 [1.0–8.0] | 1.0 [1.0–1.5] | 1.0 [1.0–3.0] | 1.8 [1.0–4.0] |
| AUC0–t (h · ng/ml) | 7.3 (0.5) | 32.5 (17.8) | 52.7 (20.0) | 71.5 (54.7) | 79.0 (64.0) | 158.9 (67.2) | 541.7 (416.1) |
| AUC0–∞ (h · ng/ml) | 87.8 (9.3) | 92.3 (53.4) | 931.0 (873.7) | ||||
| t1/2 (h) | 0.8 (0.1) | 1.6 (1.3) | 1.1 (0.3) | ||||
| CLrenal (liters/h) | 0.14 (0.01) | 0.18 (0.07) | 0.16 (0.02) | ||||
| Urine %a | 0.43 (0.16) | 0.58 (0.34) | 1.13 (0.36) | 0.68 (0.32) | 0.56 (0.51) | 0.40 (0.12) | 0.67 (0.57) |
Urine %: percentage of study drug or its metabolites excreted in urine, adjusted to molecular weight.
No. of subjects tested is given in parentheses after the dose. Data for tmax are presented as median [range].
Table 3.
Derived pharmacokinetic parameters for GS-566500
| Parameter | Mean (SD) value for doseb |
||||||
|---|---|---|---|---|---|---|---|
| 25 mg, capsule (6) | 50 mg, capsule (8) | 50 mg, solution (7) | 100 mg, capsule (8) | 200 mg, capsule (8) | 400 mg, capsule (7) | 800 mg, capsule (8) | |
| Cmax (ng/ml) | 21.0 (4.9) | 29.6 (5.7) | 34.4 (11.9) | 38.4 (30.4) | 59.7 (13.4) | 175.3 (160.8) | |
| tmax (h) | 3.0 [1.5–3.0] | 1.5 [1.5–2.0] | 2.0 [1.0–4.0] | 2.0 [1.0–2.0] | 1.5 [1.5–3.0] | 3.0 [2.0–4.0] | |
| AUC0–t (h · ng/ml) | 53.2 (23.7) | 76.9 (23.2) | 102.8 (42.8) | 133.2 (127.3) | 271.9 (75.4) | 799.8 (586.0) | |
| AUC0–∞ (h · ng/ml) | 149.1 (–) | 161.8 (53.0) | 294.7 (79.6) | 338.6 (87.8) | 926.8 (601.1) | ||
| t1/2 (h) | 1.7 | 2.0 (0.2) | 1.9 (0.1) | 2.4 (1.0) | 2.8 (0.5) | 3.2 (0.9) | |
| CLrenal (liters/h) | 0.09 | 0.11 (0.02) | 0.13 (0.02) | 0.10 (0.02) | 0.11 (0.02) | ||
| Urine %a | 0.78 (0.34) | 1.00 (0.48) | 1.76 (0.43) | 1.10 (0.33) | 0.75 (0.63) | 0.62 (0.17) | 0.88 (0.65) |
Urine %, percentage of study drug or its metabolites excreted in urine, adjusted to molecular weight.
No. of subjects tested is given in parentheses after the dose. Data for tmax are presented as median [range].
Table 4.
Derived pharmacokinetic parameters for GS-331007
| Parameter | Mean (SD) value for dosec |
||||||
|---|---|---|---|---|---|---|---|
| 25 mg, capsule (6) | 50 mg, capsule (8) | 50 mg, solution (7) | 100 mg, capsule (8) | 200 mg, capsule (8) | 400 mg, capsule (7) | 800 mg, capsule (8) | |
| Cmax (ng/ml) | 74.9 (14.2) | 144.0 (46.7) | 159.5 (29.9) | 273.8 (28.6) | 299.3 (94.5) | 545.4 (108.5) | 724.3 (185.4) |
| tmax (h) | 4.0 [2.0–4.0] | 3.5 [3.0–4.0] | 3.0 [2.0–4.0] | 4.0 [3.0–6.0] | 4.0 [3.0–8.0] | 6.0 [4.0–6.0] | 4.0 [3.0–6.0] |
| AUC0–t (h · ng/ml) | 490.0 (107.5) | 1124.5 (461.8) | 1213.6 (317.7) | 2324.6 (585.6) | 3246.3 (906.7) | 6702.2 (976.6) | 9489.5 (2484.8) |
| AUC0–∞ (h · ng/ml) | 561.1 (107.5) | 1245.6 (515.2) | 1361.0 (405.9) | 2507.4 (650.6) | 4200.9 (1119.7) | 7242.3 (1097.6) | 10065.2 (2756.5) |
| t1/2 (h) | 3.4 (0.6) | 5.1 (1.8) | 6.7 (2.8) | 7.5 (3.4) | 19.1 (13.3) | 18.0 (8.0) | 25.7 (6.2) |
| GS-331007/GS-9851 AUC0–t ratioa | 160.1 (33.2) | 102.1 (53.6) | 54.1 (24.5) | 96.0 (58.5) | 146.7 (94.6) | 97.2 (35.8) | 44.0 (16.2) |
| GS-331007/GS-9851 Cmax ratioa | 21.1 (6.3) | 17.3 (5.6) | 9.2 (3.4) | 22.9 (14.3) | 15.6 (7.5) | 18.2 (8.0) | 8.0 (4.9) |
| CLrenal (liters/h) | 0.21 (0.04) | 0.19 (0.08) | 0.17 (0.05) | 0.18 (0.04) | 0.22 (0.07) | 0.16 (0.04) | 0.19 (0.04) |
| Urine %b | 56.5 (6.6) | 51.0 (12.4) | 54.4 (7.0) | 53.3 (8.0) | 37.3 (13.5) | 35.3 (7.0) | 27.3 (8.2) |
GS-331007/GS-9851 AUC0–t and Cmax ratios were calculated following conversion of GS-331007 into nanogram equivalents of GS-9851 · h ml and nanogram equivalents of GS-9851/ml, respectively.
Urine %, percentage of study drug or its metabolites excreted in urine, adjusted to molecular weight.
No. of subjects tested is given in parentheses after the dose. Data for tmax are presented as median [range].
Dose proportionality analysis.
Power model analysis demonstrated that GS-9851 or metabolites did not exhibit dose proportionality across the dose range studied (25 mg to 800 mg GS-9851). Wide 90% CIs indicated a high level of data variability. Dose proportionality was also assessed using a pairwise ANOVA (Table 5). GS-9851 did not consistently exhibit dose proportionality as assessed by comparison with the 100-mg GS-9851 dose group (Table 5). Values of AUC for GS-9851 generally increased in a less than proportional manner for doses higher than 100 mg GS-9851. The results obtained for the metabolite GS-566500 showed a similar pattern. For GS-331007, AUC values increased in a close to dose-proportional fashion up to the 100-mg GS-9851 dose level; for higher doses, increases in the AUC were less than dose proportional, with the greatest deviation from linearity at the highest dose (800 mg).
Table 5.
Analysis of variance of results of dose proportionality for GS-9851, GS-566500, and GS-331007 (all capsule formulations)
| Parameter | Comparison | ANOVA result for drug |
|||||
|---|---|---|---|---|---|---|---|
| GS-9851 |
GS-566500 |
GS-331007 |
|||||
| Ratio | 90% CI | Ratio | 90% CI | Ratio | 90% CI | ||
| AUC0–∞ (h · ng/ml) | 25 mg/100 mg | 0.91 | 0.68, 1.22 | ||||
| 50 mg/100 mg | 0.92 | 0.70, 1.21 | |||||
| 200 mg/100 mg | 0.91 | 0.48, 1.76 | 0.84 | 0.58, 1.22 | |||
| 400 mg/100 mg | 0.52 | 0.29, 0.94 | 0.74 | 0.56, 0.98 | |||
| 800 mg/100 mg | 0.64 | 0.36, 1.14 | 0.50 | 0.37, 0.67 | |||
| AUC0–t (h · ng/ml) | 25 mg/100 mg | 0.52 | 0.24, 1.11 | 0.85 | 0.63, 1.15 | ||
| 50 mg/100 mg | 1.00 | 0.56, 1.79 | 1.01 | 0.51, 1.98 | 0.89 | 0.68, 1.18 | |
| 200 mg/100 mg | 0.48 | 0.27, 0.85 | 0.40 | 0.22, 0.75 | 0.69 | 0.53, 0.91 | |
| 400 mg/100 mg | 0.64 | 0.36, 1.15 | 0.70 | 0.38, 1.29 | 0.73 | 0.55, 0.98 | |
| 800 mg/100 mg | 1.01 | 0.57, 1.77 | 0.89 | 0.49, 1.61 | 0.51 | 0.39, 0.67 | |
| Cmax (ng/ml) | 25 mg/100 mg | 1.06 | 0.61, 1.84 | 1.08 | 0.85, 1.38 | ||
| 50 mg/100 mg | 1.19 | 0.72, 1.98 | 1.26 | 0.74, 2.12 | 0.99 | 0.79, 1.24 | |
| 200 mg/100 mg | 0.76 | 0.46, 1.26 | 0.45 | 0.28, 0.71 | 0.53 | 0.42, 0.66 | |
| 400 mg/100 mg | 0.58 | 0.34, 0.97 | 0.45 | 0.28, 0.72 | 0.49 | 0.39, 0.62 | |
| 800 mg/100 mg | 0.99 | 0.59, 1.64 | 0.53 | 0.33, 0.84 | 0.32 | 0.26, 0.40 | |
Relative bioavailability.
The increase in relative bioavailability based upon Cmax for the 50-mg-solution GS-9851 versus 50-mg-capsule GS-9851 was 121% for GS-9851, 42% for GS-566500, and 16% for GS-331007 (Table 6). An increase of 17% in the value of AUC0–∞ was observed for GS-331007 for the 50-mg solution versus the 50-mg capsule; AUC0–∞ could not be calculated for GS-9851 and GS-566500. Values of AUC0–t revealed an increase of 74% and 54% for GS-9851 and GS-566500, respectively, for the 50-mg solution versus the 50-mg capsule. In summary, GS-9851 in solution form provided higher parent and metabolite exposures with less variability than the drug in capsule form.
Table 6.
Analysis of variance of results of bioavailability for GS-9851, GS-566500, and GS-331007 (50-mg solution versus 50-mg capsules)
| Parameter | ANOVA result for drug |
|||||
|---|---|---|---|---|---|---|
| GS-9851 |
GS-566500 |
GS-331007 |
||||
| Ratio | 90% CI | Ratio | 90% CI | Ratio | 90% CI | |
| AUC0–∞ (h · ng/ml) | 1.17 | 0.77, 1.76 | ||||
| AUC0–t (h · ng/ml) | 1.74 | 1.10, 2.77 | 1.54 | 0.97, 2.45 | 1.17 | 0.77, 1.76 |
| Cmax (ng/ml) | 2.21 | 1.52, 3.20 | 1.42 | 1.12, 1.80 | 1.16 | 0.86, 1.57 |
DISCUSSION
In this first-time-in-human study, GS-9851 (nucleotide prodrug) was rapidly absorbed and converted to GS-331007. GS-331007 exhibited a tmax of approximately 3.0 to 6.0 h and demonstrated multiphasic elimination with a maximum t1/2 of 25.7 h at the 800-mg dose. Given the fact that drug-related material is detectable beyond 24 h at some doses, the high liver-to-plasma drug ratios observed in species used for nonclinical testing, and the in vitro half-life of GS-461203 (9, 13), the pharmacokinetic profiles of GS-9851 and the metabolites GS-566500 and GS-331007 should be compatible with once-daily dosing when given to patients at sufficient dose levels. In addition, GS-9851 showed a favorable safety and tolerability profile in healthy adult subjects following single ascending doses from 25 mg to 800 mg.
The pharmacokinetic profile obtained for the prodrug GS-9851 and its analytes revealed that systemic exposure to GS-9851 and GS-566500 was low, with the majority of systemic drug exposure measured as GS-331007. This observation is consistent with rapid and efficient metabolism of GS-9851 to GS-331007 in the blood and likely rapid hepatic uptake of the prodrug. The pharmacokinetic profile obtained for GS-9851 is consistent with the preclinical data demonstrating that only GS-9851 can enter the hepatocyte and undergo conversion to GS-461203 and any metabolite of GS-9851 in the blood will ultimately lead to the inactive GS-331007. The metabolite GS-606965 was not detected in the plasma of any subject at doses up to 400 mg GS-9851, and thus it was not possible to provide a pharmacokinetic profile for GS-606965. Although the study was not powered for a formal analysis, dose proportionality (Cmax and AUC0–t) was observed for GS-331007 up to a dose of 100 mg GS-9851 but not for the 200-mg-to-800-mg GS-9851 dose range. GS-331007 exhibited less than proportional exposure increases with increasing doses of GS-9851. Urinary recovery of GS-331007 was reduced as the prodrug dose increased above 200 mg.
The rate and extent of absorption of GS-9851 were also investigated when it was administered as a solution formulation or as a capsule formulation in healthy subjects. Administration of GS-9851 as a solution resulted in higher Cmax and AUC0–t values for the prodrug than those seen with the powder-in-capsule formulation. These differences appear to indicate an incomplete dissolution of the powder-in-capsule and/or an increased gut metabolism of the prodrug when it is administered as a capsule. Since the solution produced only slightly higher systemic exposure of GS-331007, the capsule formulation was an acceptable dosage form for this single-ascending-dose study. In addition, for the capsule dosages, stable metabolic ratios in the 25-mg-to-400-mg GS-9851 dose range strongly suggest that the exposure of GS-331007 was relatively consistent with the exposure of the prodrug. However, for the 800-mg GS-9851 dose level, there was less GS-331007 exposure relative to the prodrug. A possible explanation for this observation could be that the formation of GS-331007 became saturated. However, the results obtained for dose proportionality and metabolic ratios may be a consequence of the small number of subjects in each dose group, which is reflected in wide CIs surrounding those parameters.
Values of t1/2 for GS-331007 increased with increasing doses of GS-9851. At doses of GS-9851 at 200 mg and above, values for t1/2 ranged from 18.0 to 25.7 h. The longer t1/2 associated with higher doses reflects the terminal elimination t1/2, which was not observed at the lower dose as the plasma concentration of GS-331007 fell below the LLQ. The t1/2 measured in the lower doses was reflective of the distribution t1/2. The 18.0- to 25.7-h terminal elimination t1/2 observed at the higher dose indicates that GS-9851 could potentially be administered on a once-daily basis, which would be beneficial to patients as simple dosing regimens can lead to higher levels of adherence.
GS-9851 showed a good tolerability profile, since all adverse events reported were of mild intensity, except for one episode of dizziness that was considered moderate. Events judged possibly related to the study drug by the investigator resolved without intervention. There was no dose-limiting toxicity and no increase in the frequency or intensity of events with increasing doses of GS-9851. No clinically significant trends in treatment-emergent laboratory abnormalities, vital signs, or 12-lead ECG assessments were observed.
In conclusion, the results obtained in this single-ascending-dose study demonstrate that GS-9851 is a generally safe and well-tolerated compound with a pharmacokinetic profile consistent with once-daily dosing. The findings of this study in healthy subjects indicate that further studies of GS-9851 are warranted for patients with HCV infection.
ACKNOWLEDGMENTS
We thank the subjects and staff who participated in the study.
This work was funded by Pharmasset, Inc., Princeton, New Jersey. J.D., M.C., M.M.B., and W.T.S. were employees of Pharmasset. S.D.F. was paid by Pharmasset to conduct this study. Justin Cook and Severina Moreira from Niche Science and Technology Ltd. (Richmond-upon-Thames, United Kingdom) provided writing and editorial support during the preparation of the manuscript and were paid for these services by Pharmasset.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Lauer GM, Walker BD. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345:41–52 [DOI] [PubMed] [Google Scholar]
- 2. Birerdinc A, Younossi ZM. 2010. Emerging therapies for hepatitis C virus. Expert Opin. Emerg. Drugs 15:535–544 [DOI] [PubMed] [Google Scholar]
- 3. Rong L, Perelson AS. 2010. Treatment of hepatitis C virus infection with interferon and small molecule direct antivirals: viral kinetics and modelling. Crit. Rev. Immunol. 30:131–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schinazi RF, Bassit L, Gavegnano C. 2010. HCV drug discovery aimed at viral eradication. J. Viral Hepat. 17:77–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wyles DL. 2010. Moving beyond interferon alfa: investigational drugs for hepatitis C virus infection. Top. HIV Med. 18:132–136 [PubMed] [Google Scholar]
- 6. Hofmann WP, Zeuzem S. 2011. A new standard of care for the treatment of chronic HCV infection. Nat. Rev. Gastroenterol. Hepatol. 8:257–264 [DOI] [PubMed] [Google Scholar]
- 7. Negro P. 2010. Adverse effects of drugs in the treatment of viral hepatitis. Best Pract. Res. Clin. Gastroenterol. 24:183–192 [DOI] [PubMed] [Google Scholar]
- 8. Walker PM, Hong Z. 2002. HCV RNA-dependent RNA polymerase as a target for antiviral development. Curr. Opin. Pharmacol. 2:534–540 [DOI] [PubMed] [Google Scholar]
- 9. Lam AM, Murakami E, Espiritu C, Steuer HM, Niu C, Keilman M, Bao H, Zennou V, Bourne N, Julander JG, Morrey JD, Smee DF, Frick DN, Heck JA, Wang P, Nagarathnam D, Ross BS, Sofia MJ, Otto MJ, Furman PA. 2010. PSI-7851, a pronucleotide of β-D-2′-deoxy-2′-fluoro-2′-C-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis C virus replication. Antimicrob. Agents Chemother. 54:3187–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a β-D-20-deoxy-20-r-fluoro-20-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53:7202–7218 [DOI] [PubMed] [Google Scholar]
- 11. Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA. 2010. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 285:34337–34347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murakami E, Niu C, Bao H, Micolochick Steuer HM, Whitaker T, Nachman T, Sofia MA, Wang P, Otto MJ, Furman PA. 2008. The mechanism of action of β-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine involves a second metabolic pathway leading to β-D-2′-deoxy-2′-fluoro-2′-C-methyluridine 5′-triphosphate, a potent inhibitor of the HCV RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 52:458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furman PA, Wang P, Niu C, Bao D, Symonds S, Nagarathnam Steuer HM, Rachakonda S, Ross BS, Otto MJ, Sofia MJ.Abstr. 59th Annu. Meet. Am. Assoc. Study Liver Dis., abstr. 1901.2008. [Google Scholar]
- 14. Lawitz E, Lalezari JP, Rodriguez-Torres M, Kowdley KV, Nelson D, DeJesus E, McHutchison JG, Cornpropst MT, De La Rosa A, Symonds WT, Berrey MM. 2010. Clinical synergy of an anti-HCV nucleotide analog with SOC: viral kinetics of PSI-7977 with SOC, abstr 1861. Abstr. 61st Annu. Meet. Am. Assoc. Study Liver Dis [Google Scholar]
- 15. Lawitz E, Rodriguez-Torres M, Denning JM, Albanis E, Cornpropst M, Berrey MM, Symonds WT. 2013. Pharmacokinetics, pharmacodynamics, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor, following multiple ascending doses in patients with chronic hepatitis C infection. Antimicrob. Agents Chemother. 57:1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. National Institute of Allergy and Infectious Diseases December 2004, posting date Division of AIDS table for grading the severity of adult and pediatric adverse events. http://www.hptn.org/web%20documents/hptn046/ssp/appendices/appendixe-toxicitytables_daids_ae_gradingtable_finaldec2004.pdf