

Abstract
The antifungal efficacy of voriconazole (VRC) differs among host species, with potent efficacy in humans but less in rodents. We investigated the possible involvement of pregnane X receptor (PXR) and constitutive androstane receptor (CAR) in the species-specific efficacy of VRC through pharmacokinetic analyses using genetically modified mice and primary human hepatocytes. VRC (30 mg/kg) was orally administered to wild-type, Pxr-null, Car-null, and Pxr- and Car-null (Pxr/Car-null) mice for 7 days. Hepatic VRC metabolism was significantly increased by VRC administration, and the elimination rates of plasma VRC were much higher on day 7 than on day 1 in wild-type mice. This autoinduction was also observed in Pxr-null and Car-null mice but not in Pxr/Car-null mice, suggesting coordinated roles of PXR and CAR in the autoinduction of VRC metabolism in mice. Hepatic Cyp3a11 mRNA levels were increased by VRC administration, hepatic metabolic activities for VRC were correlated with CYP3A activities, and the induced VRC metabolism was inhibited by ketoconazole (a CYP3A inhibitor). In primary human hepatocytes, VRC barely increased mRNA levels of CYP3A4 and CYP2B6 (human PXR/CAR target genes) at its therapeutic concentrations. In conclusion, these results suggest that VRC is metabolized mainly by CYP3A11 in mouse livers and that PXR- and CAR-mediated CYP3A11 induction, namely, autoinduction of VRC metabolism, is a primary reason for the ineffectiveness of VRC in mice. A limited ability of VRC to activate human PXR/CAR at its clinical concentration might explain the VRC efficacy in humans. Therefore, the ability to activate PXR/CAR might determine the VRC efficacy in different mammalian species.
INTRODUCTION
Invasive fungal infections, including aspergillosis and candidiasis, continue to pose serious clinical problems in immunocompromised patients. Although improved diagnostics and a new class of antifungals have been developed (1), recent clinical studies have reported persistently higher mortality rates of 30% to 50% and difficult treatment against drug-resistant strains, including resistance to azoles in Aspergillus fumigatus (2, 3). At present, voriconazole (VRC) is widely used as a standard treatment for patients with invasive aspergillosis (4); however, its efficacy differs by host species, with greater efficacy observed in humans but less in rodents due to their rapid metabolism (5). These species differences have hampered efforts to predict the in vivo efficacy of new drug candidates or combination therapies in humans with VRC monotherapy from preclinical studies using mice or rats.
VRC is a second-generation triazole compound which is known to exert a broad and potent range of activities against fungal pathogens, including itraconazole-resistant isolates. VRC is metabolized into pharmacologically inactive metabolites by cytochrome P450 2C9 (CYP2C9), CYP2C19, and CYP3A4 or flavin-containing monooxygenases (FMOs) in humans (6–8). Roffey et al., however, reported that the clearance of VRC in mice and rats was enhanced after multiple administrations, suggesting the autoinduction of metabolism (5). The mechanism of such autoinduction remains unknown.
Nuclear receptors are transcription factors that regulate the expression of a battery of genes in response to various extracellular and intracellular signals. The pregnane X receptor (PXR) and the constitutive androstane receptor (CAR) belong to the same nuclear receptor subfamily (NR1I) and function as xenobiotic sensors (9, 10). Both receptors regulate the expression of many phase I and II drug-metabolizing enzymes and drug transporters to promote the detoxification and elimination of toxic chemicals from the body. PXR is commonly associated with the regulation of CYP3A, which participates in the metabolism of over 50% of clinically used drugs. PXR has a flexible and large binding pocket with a diameter of over 1,100 Å, and it is activated by many structurally unrelated ligands (11). As such, PXR plays an important role in clinical drug-drug interactions. CAR was first identified as a critical transcription factor in phenobarbital-mediated CYP2B induction and has been found to regulate CYP2C, CYP3A, UDP-glucuronosyltransferase 1A1, and multidrug resistance-associated protein 2 as well (12, 13).
Although a number of endogenous and exogenous chemicals, including steroids, bile acids, antibiotics, and anticancer agents, activate PXR and CAR, significant species differences have been reported in the response to these nuclear receptor ligands (9, 10). For example, pregnenolone 16α-carbonitrile (PCN) activates rodent PXR but is a weak activator of human PXR, whereas the antituberculous agent rifampin strongly activates human PXR but is a weak activator of rodent PXR (14). With regard to CAR, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) has been reported to specifically bind to mouse CAR, and 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) has been reported to bind to human CAR (15, 16). Based on these facts, we have speculated that the ligand specificities of PXR and CAR for VRC are associated with the observed species differences in the efficacy of VRC.
To clarify the mechanism of the autoinduction of VRC metabolism in mice, we investigated the possible involvements of PXR and CAR in the ineffectiveness of VRC after repeated doses through pharmacokinetic analyses with genetically modified mice. In addition, we also evaluated the influences of VRC on human CYP levels using primary hepatocytes to examine whether VRC activates human PXR and/or CAR.
MATERIALS AND METHODS
Materials.
VRC was purified from VRC tablets purchased from Pfizer, Inc. (New York, NY), at the Chemistry Research Laboratories of Astellas Pharma Inc. (Ibaraki, Japan). Phenacetin, midazolam, alprazolam, phenobarbital, and omeprazole were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan), and rifampin and EDTA were purchased from Sigma-Aldrich Japan K.K. (Tokyo, Japan). NADPH and acetic acid were obtained from Nacalai Tesque (Kyoto, Japan), and 1′- and 4-hydroxymidazolam were obtained from Toronto Research Chemicals Inc. (North York, ON, Canada). Ketoconazole was purchased from LKT Laboratories, Inc. (St. Paul, MN). All other chemicals and reagents were of analytical grade and purchased from commercial sources.
Animals and treatments.
The development of the Pxr-null, Car-null, and Pxr/Car-null mice has been described elsewhere (17, 18). In the present study, male C57BL/6J (wild-type), Pxr-null, Car-null, and Pxr/Car-null mice were obtained from Harlan Laboratories Inc. (Indianapolis, IN) and TaconicArtemis GmbH (Cologne, Germany), and the animal experiments were carried out at CXR Biosciences Ltd. (Dundee, United Kingdom). The mice were housed in a temperature-controlled environment with a 12-h light-dark cycle and permitted ad libitum access to a standard diet and water. For all experiments, the mice were acclimatized for at least 5 days and used in experiments at an age of 7 to 12 weeks. The animals were handled and cared for in accordance with United Kingdom Home Office guidelines and the regulations of the Astellas Pharma Inc. Animal Ethical Committee.
Wild-type, Pxr-null, Car-null, and Pxr/Car-null mice (n = 4/group) were administered repeated oral doses of either vehicle (50% polyethylene glycol 400 [PEG 400] solution) or VRC suspended in the vehicle at 30 mg/kg body weight daily for 7 consecutive days. On days 1 and 7 of dosing, approximately 40 μl of blood was collected from the tail vein into heparinized microtubes at 2 and 8 h postdose for plasma preparation. On day 8, blood samples were collected 24 h after the final administration, after which the mice were euthanized by exposure to a rising concentration of CO2, and their livers were quickly collected for the preparation of liver microsomes and RNA extraction. Plasma was stored at −70°C until analysis. The livers were excised and frozen in liquid nitrogen before being stored at approximately −70°C.
Determination of plasma concentrations of voriconazole.
Plasma concentrations of VRC were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Briefly, 10 μl of mouse plasma was mixed with 10 μl of 50% acetonitrile, 10 μl of 5 μg/ml phenacetin as an internal standard, and 75 μl of acetonitrile. The mixture was centrifuged at 20,800 × g for 5 min, and 50 μl of the obtained supernatant was combined with 100 μl of 0.1% acetic acid. Five microliters of this sample was then injected into the LC-MS/MS apparatus for analysis. The calibration curves were linear (r > 0.99) over the range of 10 to 5,000 ng/ml.
LC-MS/MS analyses were conducted using an Agilent 1100 high-pressure liquid chromatography (HPLC) system (Agilent Technologies, Inc., Santa Clara, CA) and an API4000 triple tandem mass spectrometer (AB Sciex, Toronto, Canada) with an HTC-PAL autosampler (CTC Analytics AG, Zwingen, Switzerland). The analytes were separated with a TSKgel octyldecylsilane (ODS)-100V column (50 by 2.0 mm, 3 μm) (Tosoh, Tokyo, Japan) at 50°C using 0.1% acetic acid-acetonitrile (65:35 [vol/vol]) as the mobile phase with a flow rate of 0.3 ml/min. Detection was conducted using a mass spectrometer equipped with an electrospray ionization (ESI) source and selected reaction monitoring (SRM) in a positive-ion mode. With regard to parameter settings, the ion spray voltage was set at 5,000 V, and the transfer capillary temperature was set at 450°C. The mass spectrometer was set to monitor the transitions of the precursors to the product ions as follows: m/z 350.1 to 281.1 for VRC and m/z 180.1 to 110.1 for phenacetin. Data were processed using Analyst software version 1. 4. 1 (AB Sciex).
Preparation of liver microsomes.
Liver samples were homogenized in 1.15% KCl and centrifuged at 9,000 × g for 20 min at 4°C, after which the supernatant was centrifuged at 105,000 × g for 60 min at 4°C. The resulting pellets were washed and resuspended in 1.15% KCl and then centrifuged again at 105,000 × g for 60 min at 4°C. The final resulting pellet was then suspended in a small amount of 1.15% KCl. The protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific Inc., Rockford, IL).
VRC metabolism in liver microsomes.
The reaction mixture was composed of 125 μl of 200 mM Na-K phosphate buffer (pH 7.4), 25 μl of 1 mM EDTA, 5 μl of 50 μM VRC (final concentration, 1 μM), 12.5 μl of 10 mg protein/ml mouse liver microsomes, and 57.5 μl of water. After 5 min of preincubation at 37°C, the reaction was initiated by adding 25 μl of 10 mM NADPH in a total volume of 250 μl and allowed to proceed at 37°C for 30 min before it was terminated by adding 500 μl of acetonitrile. After 25 μl of 5 μg/ml phenacetin and 25 μl of 50% acetonitrile had been added, the mixture was centrifuged at 1,870 × g for 10 min at 4°C. A 50-μl aliquot of the supernatant was combined with 100 μl of 0.1% acetic acid, and then 2 μl of this sample was injected into the LC-MS/MS apparatus. For the inhibition assays, VRC was incubated in the absence or presence of 0.02 to 20 μM ketoconazole (a CYP3A inhibitor) with liver microsomes prepared from VRC-treated mice.
Midazolam metabolism in liver microsomes.
The reaction mixture was composed of 100 μl of 200 mM Na-K phosphate buffer (pH 7.4), 20 μl of 1 mM EDTA, 2 μl of 100 μM midazolam (final concentration, 1 μM), 10 μl of 2.5 mg protein/ml mouse liver microsomes, and 48 μl of water. After 5 min of preincubation at 37°C, the reaction was initiated by adding 20 μl of 10 mM NADPH in a total volume of 200 μl and allowed to proceed at 37°C for 5 min. The reaction was terminated by adding 20 μl of formic acid-50% acetonitrile (10:90 [vol/vol]) containing 0.316 μM alprazolam (internal standard). After 20 μl of 50% acetonitrile had been added, the mixture was centrifuged at 1,870 × g and 4°C for 3 min. A 10-μl aliquot of the sample was injected into the LC-MS/MS apparatus. The calibration curves were linear (r > 0.99) over the range of 20 to 4,000 nM for 1′-hydroxymidazolam and 1 to 200 nM for 4-hydroxymidazolam.
LC-MS/MS analyses were conducted using a TSQ Quantam Ultra system (Thermo Fisher Scientific Inc., San Jose, CA). The analytes were separated with a Luna C18(2) column (100 by 2.1 mm, 3 μm) (Phenomenex, Torrance, CA) at 40°C with a flow rate of 0.3 ml/min. The mobile phase consisted of 0.1% acetic acid-acetonitrile (90:10 [vol/vol]) (A) and acetic acid-acetonitrile (0.1:100 [vol/vol]) (B). The gradient condition was linearly increased from 5% to 17% B over 0.5 min and maintained at 17% B for 3.5 min, linearly increased to 80% B over 1.5 min and maintained for 2.5 min, and then finally kept at 5% B for 4.5 min. Detection was conducted using a positive electrospray ionization SRM mode. With regard to parameter settings, the ion spray voltage was set at 4,500 V, and the transfer capillary temperature was set at 350°C. The mass spectrometer was set to monitor the transitions of the precursors to the product ions as follows: m/z 342.0 to 324.0 for 1′-hydroxymidazolam, m/z 342.0 to 325.0 for 4-hydroxymidazolam, and m/z 309.0 to 205.0 for alprazolam. Data were processed using Xcalibur software version 1.4.1 (Thermo Fisher Scientific Inc., San Jose, CA).
Hepatocyte culture.
In the present study, cultures of matured primary human hepatocytes were used in accordance with regulatory guidelines (19–21). Cryopreserved primary human hepatocytes from three donors (lot no. Hu4205, Caucasian, male, 43 years old; lot no. Hu4228, Indian, female, 47 years old; lot no. Hu8114, Caucasian, female, 47 years old) were purchased from Life Technologies Corporation (Grand Island, NY) and thawed using cryopreserved hepatocyte recovery medium (CHRM) (Life Technologies Corporation) according to the manufacturer's protocol. The cells were seeded onto 96-well collagen-coated plates (Life Technologies Corporation) at a density of 1.2 × 105 cells/well and maintained in fetal bovine serum-added Williams' E medium (CM3000; Life Technologies Corporation) for 4 to 6 h in an atmosphere of 5% CO2-95% air at 37°C. The medium was then changed to serum-free Williams' E medium (CM4000; Life Technologies Corporation) every 24 h and cultured for approximately 48 h in an atmosphere of 5% CO2-95% air at 37°C. The hepatocytes were then treated daily with serum-free Williams' E medium containing VRC (1 to 300 μM), 100 μM omeprazole (an aryl hydrocarbon receptor activator), 20 μM rifampin (a human PXR activator), 1 mM phenobarbital (a CAR activator), or vehicle (0.1% dimethyl sulfoxide [DMSO]) for 48 h.
RNA extraction and cDNA synthesis.
RNA was extracted from mouse liver or primary human hepatocytes using the RNeasy minikit (Qiagen, Hilden, Germany), and cDNA was generated from a random hexamer using the high-capacity cDNA reverse transcription kit (Life Technologies Corporation).
RT-PCR.
Quantitative real-time (RT)-PCR was conducted using the ABI Prism 7900HT sequence detection system (Life Technologies Corporation) with TaqMan gene expression assays (Life Technologies Corporation). The primer and probe sets used were Mm00487224_m1 for Cyp1a2, Mm00456591_m1 for Cyp2b10, Mm00725580_s1 for Cyp2c29, Mm00833845_m1 for Cyp2c37, Mm00472168_m1 for Cyp2c55, Mm00731567_m1 for Cyp3a11, Mm00515795_m1 for Fmo1, Mm00490159_m1 for Fmo2, Mm01306348_m1 for Fmo3, Mm00515805_m1 for Fmo5, 4352341E for β-Actin, Hs00167927_m1 for CYP1A2, Hs03044636_m1 for CYP2B6, Hs00430021_m1 for CYP3A4, and 4326315 for β-ACTIN. mRNA levels were normalized against those of β-ACTIN (human) or β-Actin (mouse).
Statistic analysis.
Data in the tables and figures are shown as the means and standard deviations (SD), except for Fig. 3, where individual data points are plotted. Statistical analysis was performed by unpaired Student's t test using PRISM 5.0 software (GraphPad Software Inc., San Diego, CA). The correlation coefficient (Pearson's r) was determined using PRISM 5.0 software (GraphPad Software, Inc.). Kinetic parameters (maximum effect [Emax], 50% effective concentration [EC50], and 50% inhibitory concentration [IC50]) were determined using WinNonlin 6.1 software (Pharsight Corporation, Mountain View, CA).
Fig 3.
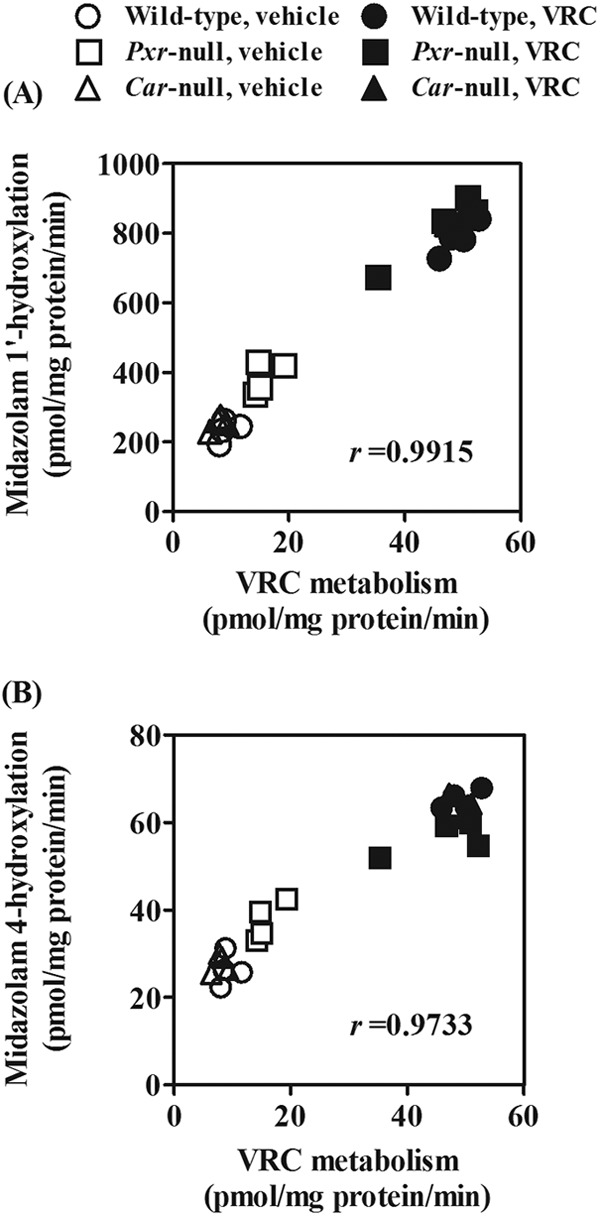
Correlations of metabolic activities for VRC with CYP3A activities. Metabolic activities for VRC and midazolam were determined as the elimination rates of VRC and conversion rates of midazolam into 1′-hydroxylated (A) and 4-hydroxylated (B) metabolites in liver microsomes, respectively. Liver microsomes prepared from vehicle- or VRC-treated wild-type, Pxr-null, and Car-null mice were used. Twenty-four individual data points are plotted.
RESULTS
VRC concentrations in mouse plasma.
VRC was orally administered to wild-type, Pxr-null, Car-null, and Pxr/Car-null mice at 30 mg/kg/day for 7 days. VRC concentrations in mouse plasma at 2 and 8 h after administration were determined on days 1 and 7. On day 1, almost comparable pharmacokinetics were observed in the wild-type, Pxr-null, Car-null, and Pxr/Car-null mice with plasma VRC concentrations of 17 to 21 μg/ml at 2 h and 10 to 15 μg/ml at 8 h (Fig. 1A to D). VRC concentrations on day 7 were lower than those on day 1 not only in wild-type mice (13 and 3 μg/ml at 2 and 8 h, respectively) but also in Pxr-null mice (9 and 1 μg/ml at 2 and 8 h, respectively) and Car-null mice (11 and 4 μg/ml at 2 and 8 h, respectively) (Fig. 1A to C). Moreover, the elimination rates of VRC were increased after repeated administration in wild-type, Pxr-null, and Car-null mice, suggesting the autoinduction of metabolism. In contrast, the plasma concentrations of VRC in Pxr/Car-null mice were increased on day 7 (55 and 47 μg/ml at 2 and 8 h, respectively), and the elimination rates were unchanged after repeated VRC administration (Fig. 1D).
Fig 1.
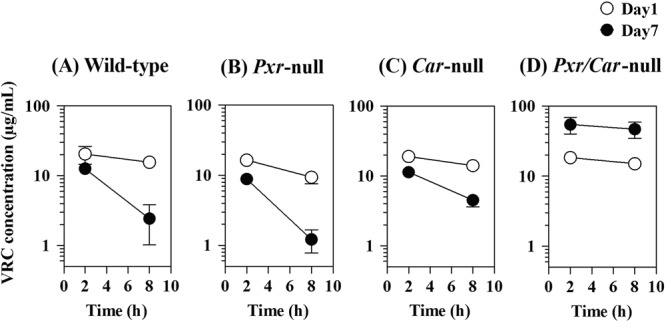
Plasma VRC concentrations in wild-type, Pxr-null, Car-null, and Pxr/Car-null mice on days 1 and 7 following repeated VRC administration. VRC was orally administered to wild-type, Pxr-null, Car-null, and Pxr/Car-null mice at 30 mg/kg for 7 days. Blood samples were collected at 2 and 8 h postdose on days 1 and 7. Plasma concentrations of VRC were determined by LC-MS/MS. Data are shown as means ± SD (n = 4).
Hepatic metabolism of VRC in mice.
To investigate the autoinduction of VRC metabolism, liver microsomes were prepared from vehicle- or VRC-treated mice, and the metabolic activities for VRC were determined in the presence of NADPH. Microsomal VRC metabolism was significantly increased 5.4-, 2.9-, and 6.3-fold after multiple administrations of VRC in wild-type, Pxr-null, and Car-null mice, respectively, indicating the autoinduction of VRC metabolism (Fig. 2A). In contrast, activities in the vehicle- or VRC-treated Pxr/Car-null mice were 15.5 and 6.2 pmol/mg protein/min, respectively, indicating no autoinduction of VRC metabolism in Pxr/Car-null mice (Fig. 2B).
Fig 2.
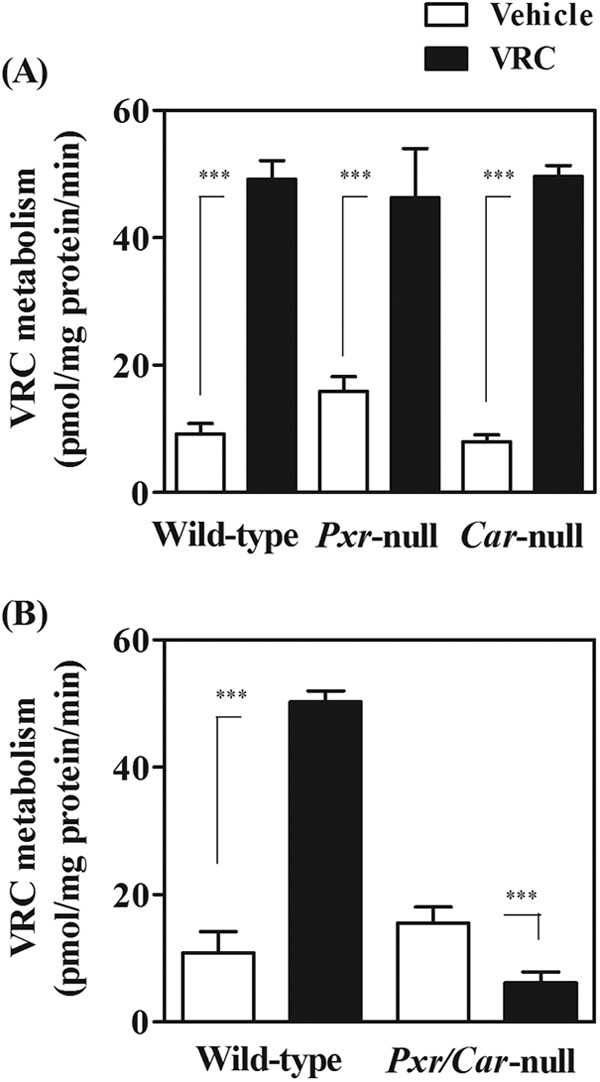
Metabolic activities for VRC in liver microsomes of wild-type, Pxr-null, Car-null, and Pxr/Car-null mice. Liver microsomes were prepared from vehicle- or VRC-treated mice. VRC (1 μM) was incubated with liver microsomes in the presence of NADPH at 37°C for 30 min. Elimination rates of VRC were determined by measuring the amounts of remaining VRC. Data are shown as means + SD (n = 4). ***, P < 0.001.
We also found that metabolic activities to VRC in wild-type, Pxr-null, and Car-null mice were well correlated with CYP3A marker activities, midazolam 1′-hydroxylation (r = 0.9915, P < 0.0001) (Fig. 3A), and 4-hydroxylation (r = 0.9733, P < 0.0001) (Fig. 3B). In addition, the induced VRC metabolism in wild-type mice was almost completely inhibited by the CYP3A inhibitor ketoconazole (IC50, 0.065 μM) (Fig. 4). Ketoconazole (2 μM) also inhibited the induced VRC metabolism in the liver microsomes prepared from Pxr-null and Car-null mice (data not shown).
Fig 4.
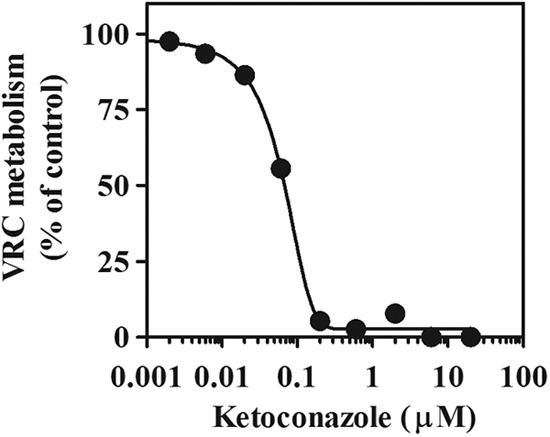
Inhibition of the induced VRC metabolism in mouse liver microsomes by ketoconazole. Liver microsomes prepared from VRC-treated wild-type mice were pooled and used. Metabolic activities to VRC were determined in the absence or presence of 0.002 to 20 μM ketoconazole. Data are shown as the means from duplicate experiments.
Effects of VRC on Cyp and Fmo mRNA levels in mouse liver.
To confirm the involvement of CYP3A in the autoinduction of VRC metabolism, we investigated whether VRC treatment induced hepatic CYP3A expression through the activation of PXR, CAR, or both. Hepatic mRNA levels of Cyp3a11, a major Cyp3a transcript in mouse livers, were increased approximately 11-, 10-, and 10-fold by repeated VRC administration in wild-type, Pxr-null, and Car-null mice, respectively (Fig. 5A). In contrast, Cyp3a11 mRNA levels were not altered by VRC administration in Pxr/Car-null mice (Fig. 5B).
Fig 5.
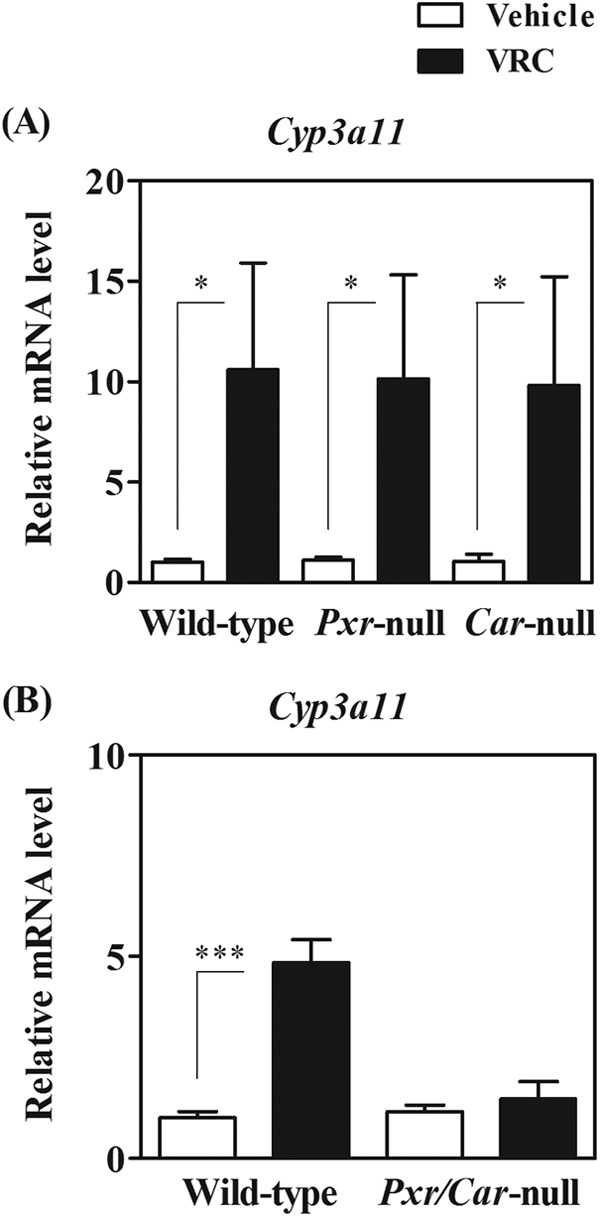
Hepatic Cyp3a11 mRNA levels in vehicle- or VRC-treated wild-type, Pxr-null, Car-null, and Pxr/Car-null mice. VRC was orally administered to wild-type, Pxr-null, and Car-null mice (A) and Pxr/Car-null mice (B) at 30 mg/kg for 7 days. Hepatic Cyp3a11 mRNA levels were determined by real-time PCR and normalized to those of β-actin. Relative mRNA levels are shown as means + SD (n = 4). *, P < 0.05; ***, P < 0.001.
To ascertain whether VRC activates PXR/CAR, mRNA levels of other PXR/CAR target CYP genes such as Cyp2b10 and Cyp2c were investigated (see Tables S1 and S2 in the supplemental material). Repeated VRC administration also significantly increased Cyp2b10, Cyp2c29, Cyp2c37, and Cyp2c55 mRNA levels in the wild-type, Pxr-null, and Car-null mice. In contrast, Cyp2b10, Cyp2c29, Cyp2c37, Cyp2c55, and Cyp3a11 mRNA levels were not increased by VRC treatment in Pxr/Car-null mice. mRNA levels of Cyp1a2, which is mainly regulated by an aryl hydrocarbon receptor, were not induced more than 2-fold by VRC treatment in any of the mice. Fmo mRNA levels in mouse livers were also measured in both wild-type and Pxr/Car-null mice, but those of Fmo1, Fmo2, Fmo3, and Fmo5 were not increased more than 2-fold by VRC administration in these mice (see Table S3 in the supplemental material).
Effects of VRC on CYP mRNA levels in primary human hepatocytes.
The influence of VRC on human CYP levels was studied in primary hepatocytes, and the induction potential of VRC was compared with those of clinical CYP inducers, such as rifampin (a PXR activator and CYP3A/2B inducer), phenobarbital (a CAR activator and CYP3A/2B inducer), and omeprazole (an aryl hydrocarbon activator and CYP1A2 inducer).
VRC did not increase CYP3A4 mRNA levels more than 3-fold in hepatocytes from two donors (Hu4228 and Hu8114) (Fig. 6A). In only one lot of hepatocytes (Hu4205) did VRC upregulate CYP3A4 mRNA levels in a concentration-dependent manner, although the increase in CYP3A4 mRNA levels by VRC was less than 3-fold at 10 to 30 μM (the maximum plasma concentration in VRC-treated patients) and maximally 6.3-fold at 300 μM compared to those of the vehicle control (Fig. 6A). The extent of increase was much smaller than that with rifampin (17-fold) or phenobarbital (11-fold). The relative increase of CYP3A4 mRNA levels after VRC treatment at the clinical maximum concentration of drug in serum (Cmax) was less than 20% of that of the positive control (rifampin). The Emax and EC50 of VRC for CYP3A4 mRNA levels in Hu4205 hepatocytes were calculated as 7.1-fold and 129 μM, respectively, and the EC50 was at least 4-fold higher than the clinical Cmax (10 to 30 μM) in the patients (22). VRC (1 to 300 μM) did not increase CYP2B6 and CYP1A2 mRNA levels more than 3-fold compared to that of the vehicle control (Fig. 6B and C). In contrast, rifampin and phenobarbital increased CYP2B6 mRNA levels 6- to 14-fold and 6- to 11-fold, respectively. Omeprazole increased CYP1A2 mRNA levels 19- to 40-fold. VRC and rifampin did not increase CYP2C19 mRNA levels in any of the hepatocyte lots (data not shown).
Fig 6.
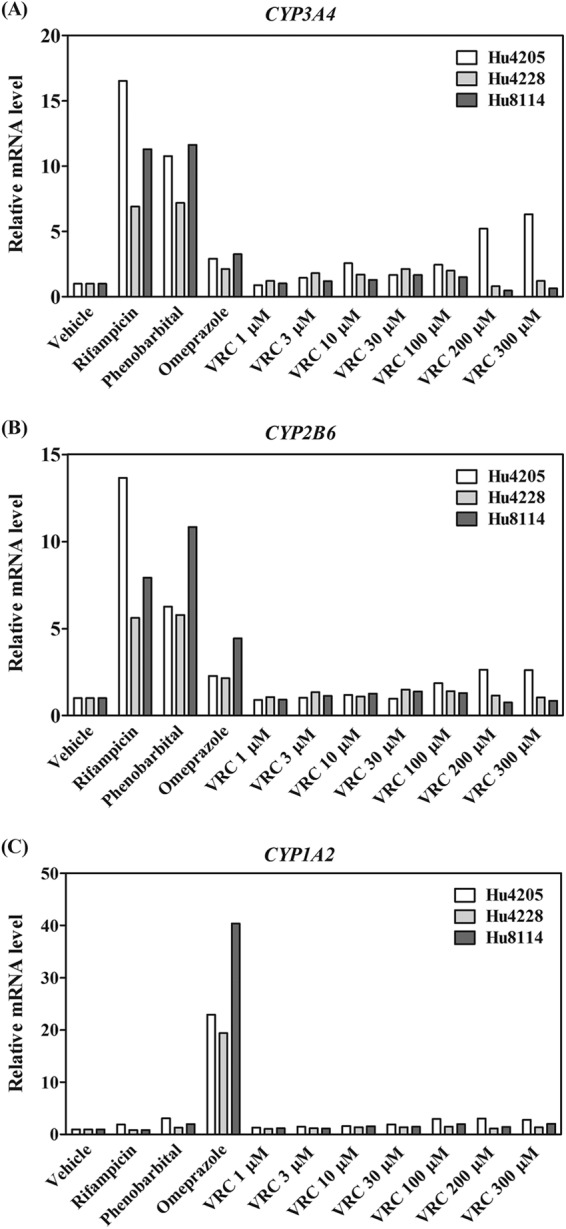
Influence of VRC treatment on CYP3A4, CYP2B6, and CYP1A2 mRNA levels in primary human hepatocytes. Human hepatocytes (Hu4205, Hu4228, and Hu8114) were treated with vehicle (0.1% DMSO), VRC (1 to 300 μM), or positive control rifampin, phenobarbital, or omeprazole. CYP mRNA levels were determined by real-time PCR and normalized to those of β-ACTIN. Relative mRNA levels are shown as the means from triplicate experiments.
DISCUSSION
The present study demonstrates that (i) repeated administration of VRC causes its autoinduction of metabolism in mice, (ii) both PXR and CAR are involved in the VRC autoinduction, (iii) the increase in Cyp3a expression by VRC administration is mainly involved in the autoinduction in mice, and (iv) VRC treatment is less effective in human hepatocytes in terms of CYP3A induction than in mice in vivo.
The clearance of VRC in mice was increased after repeated VRC administration, which is consistent with a previous study (5). Furthermore, hepatic VRC metabolism was increased by VRC treatment in mice. Given that VRC is mainly eliminated by metabolism in rodents as well as humans (5, 6), the decrease in plasma concentrations of VRC in mice after repeated administration is probably attributable to the induced metabolism in the liver. Intriguingly, this decrease after repeated administration was also observed in Pxr-null and Car-null mice. In contrast, the plasma concentration was increased in Pxr/Car-null mice after repeated doses. These results suggest that both PXR and CAR are coordinately involved in the autoinduction of VRC metabolism in mice. Because the antifungal efficacy of azole agents, including VRC, is related to the area under the curve/MIC ratio (AUC/MIC) (23, 24), PXR- and CAR-mediated autoinduction might be a primary reason for the ineffectiveness of VRC in mice.
Several findings support the likely role of CYP3A in the autoinduction of VRC metabolism. First, hepatic metabolic activities for VRC were closely correlated with those for midazolam (CYP3A marker activity) in vehicle- and VRC-treated (wild-type, Pxr-null, and Car-null) mice. Second, hepatic Cyp3a11 mRNA levels and VRC-metabolizing activities were upregulated by VRC treatment. Third, ketoconazole (a representative CYP3A inhibitor) almost completely inhibited the induced VRC metabolism.
In addition to CYP3A, CYP2C9, CYP2C19, and FMOs have been reported to catalyze the N-oxidation of VRC in humans (6, 7, 22). In the present study, mRNA levels of genes for other CYP forms, including Cyp2b10, Cyp2c29, Cyp2c37, and Cyp2c55, were increased by VRC treatment in wild-type mice, raising the possibility that these enzymes are partially involved in the autoinduction of VRC in mice. It has been reported that coadministration with grapefruit juice increases serum VRC concentrations in mice, prolonging survival in mouse models of fungal infection with Aspergillus flavus, Candida tropicalis, Candida krusei, Fusarium, Paecilomyces lilacinus, or Blastomyces dermatitidis (25–30). Because furanocoumarin derivatives in grapefruit juice are known to inhibit CYP3A activities (31, 32), these facts corroborate the idea that CYP3A is mainly involved in VRC metabolism in mouse livers. Moreover, our present findings provide a rationale for the use of grapefruit juice as a CYP3A inhibitor to repress VRC metabolism in murine models to study the efficacy of VRC in vivo.
The murine model in which VRC is coadministered with grapefruit juice has also been used in the comparison of the in vivo efficacy of isavuconazole, itraconazole, fluconazole, and amphotericin B with that of VRC (27, 29, 30). Based on the present findings, the simultaneous knockdown or inhibition of both PXR and CAR might be another useful approach to improving the antifungal efficacy of VRC with effective exposure in mice, where the autoinduction of VRC metabolism might be completely blocked.
Hepatic Cyp3a11 mRNA levels were upregulated 9- to 11-fold by VRC treatment in wild-type, Pxr-null, and Car-null mice but not in Pxr/Car-null mice. These results suggest that both PXR and CAR are involved in VRC-mediated Cyp3a11 induction and that VRC is a common activator for murine PXR and CAR. In addition, VRC treatment increased mRNA levels of other PXR/CAR target CYP genes such as Cyp2b10 and Cyp2c55 in wild-type, Pxr-null, and Car-null mice, with Cyp2b10 being more CAR dependent and Cyp2c55 being more PXR dependent. Given that PXR and CAR share only approximately 40% amino acid identity in their ligand binding domains, VRC might be unique in activating both murine PXR and CAR. Moreover, the present results suggest that the contributions of VRC-activated PXR and CAR are different among their target genes.
Primary human hepatocytes contain human nuclear receptors, native human target genes and their promoters, as well as human drug-metabolizing enzymes and transporters. In addition, the induction data obtained with primary human hepatocytes were reported to be well correlated with the clinical observations (33, 34). Thus, cultured primary human hepatocytes are recognized as the most accepted in vitro system for assessing the potential of a drug candidate to induce human CYP expression (19–21). The present study using primary human hepatocytes revealed that the maximum inducibility of VRC against CYP3A4 or CYP2B6 (the target genes of human PXR and CAR) was lower than that of rifampin (a human PXR activator) or phenobarbital (a human CAR activator). In addition, the potential of VRC to increase CYP3A4 or CYP2B6 around the maximum concentration in human plasma (about 10 to 30 μM) was negligible, and CYP2C19 was not induced by VRC treatment in the hepatocytes used. These results suggest that VRC is unable to activate human PXR and CAR to enhance its own metabolism in clinical situations. In fact, the autoinduction of VRC metabolism has not been reported in humans; clinical pharmacokinetic studies have reported that VRC causes drug-drug interaction based on CYP3A inhibition but not CYP induction (35, 36). There seems to be a species difference between mice and humans in the ability of VRC to activate PXR and CAR.
The mRNA levels of CYP3A4 and CYP2B6 in Hu4205 hepatocytes were increased, in contrast to those of Hu4228 and Hu8114 cells, when high concentrations of VRC (200 to 300 μM) were used. Madan et al. reported a large interindividual variability in the response of primary human hepatocytes to CYP inducers (37). Although the reason for this variability is currently unclear, many factors might be involved in the interindividual variability, such as basal expression levels of CYPs, body mass index, and liver diseases (such as fatty liver and viral infections) or the ages of the donors. Nonetheless, given that the VRC concentration at which CYP3A4 mRNA levels were increased in the Hu4205 lot was much higher than the clinically observed VRC concentration (22), no CYP3A4 induction would occur in the livers of VRC-treated subjects.
In conclusion, we have demonstrated the coordinated roles of PXR and CAR in the autoinduction of CYP3A-mediated VRC metabolism in mice. Since the antifungal efficacy of azole agents is tightly associated with the AUC/MIC, PXR- and CAR-mediated autoinduction might be a primary reason for the ineffectiveness of VRC in mice. In contrast, our results demonstrate that the autoinduction of VRC metabolism is unlikely to occur in humans, ensuring its potent antifungal activity with effective exposure in humans. Since the ability to activate PXR and CAR might be a major determinant of the efficacy of VRC in different mammalian species, knockdown or inhibition of both PXR and CAR is proposed as a useful approach for improving the efficacy of VRC in mice.
Supplementary Material
ACKNOWLEDGMENTS
We especially appreciate the advice and expertise of Hidetaka Kamimura (ADME & Tox. Research Institute, Sekisui Medical Co., Ltd.). We also sincerely thank Ayako Mera (Drug Metabolism Research Laboratories, Astellas Pharma Inc.), Marie Bowers, and Eddie Dolyle (CXR Biosciences) for their experimental assistance.
We declare no conflicts of interest.
Footnotes
Published ahead of print 28 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01900-12.
REFERENCES
- 1. Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH. 2010. An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov. 9:719–727 [DOI] [PubMed] [Google Scholar]
- 2. Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, Mellado E, Donders AR, Melchers WJ, Verweij PE. 2008. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med. 5:e219 doi:10.1371/journal.pmed.0050219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howard SJ, Cerar D, Anderson MJ, Albarrag A, Fisher MC, Pasqualotto AC, Laverdiere M, Arendrup MC, Perlin DS, Denning DW. 2009. Frequency and evolution of azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg. Infect. Dis. 15:1068–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scott LJ, Simpson D. 2007. Voriconazole: a review of its use in the management of invasive fungal infections. Drugs 67:269–298 [DOI] [PubMed] [Google Scholar]
- 5. Roffey SJ, Cole S, Comby P, Gibson D, Jezequel SG, Nedderman AN, Smith DA, Walker DK, Wood N. 2003. The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human. Drug Metab. Dispos. 31:731–741 [DOI] [PubMed] [Google Scholar]
- 6. Hyland R, Jones BC, Smith DA. 2003. Identification of the cytochrome P450 enzymes involved in the N-oxidation of voriconazole. Drug Metab. Dispos. 31:540–547 [DOI] [PubMed] [Google Scholar]
- 7. Yanni SB, Annaert PP, Augustijns P, Bridges A, Gao Y, Benjamin DK, Jr, Thakker DR. 2008. Role of flavin-containing monooxygenase in oxidative metabolism of voriconazole by human liver microsomes. Drug Metab. Dispos. 36:1119–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murayama N, Imai N, Nakane T, Shimizu M, Yamazaki H. 2007. Roles of CYP3A4 and CYP2C19 in methyl hydroxylated and N-oxidized metabolite formation from voriconazole, a new anti-fungal agent, in human liver microsomes. Biochem. Pharmacol. 73:2020–2026 [DOI] [PubMed] [Google Scholar]
- 9. Timsit YE, Negishi M. 2007. CAR and PXR: the xenobiotic-sensing receptors. Steroids 72:231–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. di Masi A, Marinis ED, Ascenzi P, Marino M. 2009. Nuclear receptors CAR and PXR: molecular, functional, and biomedical aspects. Mol. Aspects Med. 30:297–343 [DOI] [PubMed] [Google Scholar]
- 11. Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. 2005. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol. Endocrinol. 19:1125–1134 [DOI] [PubMed] [Google Scholar]
- 12. Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, Moore DD. 2003. Induction of bilirubin clearance by the constitutive androstane receptor (CAR). Proc. Natl. Acad. Sci. U. S. A. 100:4156–4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshinari K, Ohno H, Benoki S, Yamazoe Y. 2012. Constitutive androstane receptor transactivates the hepatic expression of mouse Dhcr24 and human DHCR24 encoding a cholesterogenic enzyme 24-dehydrocholesterol reductase. Toxicol. Lett. 208:185–191 [DOI] [PubMed] [Google Scholar]
- 14. Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. 2000. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol. Endocrinol. 14:27–39 [DOI] [PubMed] [Google Scholar]
- 15. Tzameli I, Pissios P, Schuetz EG, Moore DD. 2000. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell. Biol. 20:2951–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, Moore JT. 2003. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 278:17277–17283 [DOI] [PubMed] [Google Scholar]
- 17. Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. 2001. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. U. S. A. 98:3369–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scheer N, Ross J, Rode A, Zevnik B, Niehaves S, Faust N, Wolf CR. 2008. A novel panel of mouse models to evaluate the role of human pregnane X receptor and constitutive androstane receptor in drug response. J. Clin. Invest. 118:3228–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chu V, Einolf HJ, Evers R, Kumar G, Moore D, Ripp S, Silva J, Sinha V, Sinz M, Skerjanec A. 2009. In vitro and in vivo induction of cytochrome p450: a survey of the current practices and recommendations: a pharmaceutical research and manufacturers of America perspective. Drug Metab. Dispos. 37:1339–1354 [DOI] [PubMed] [Google Scholar]
- 20. Food and Drug Administration 2012. Guidance for industry drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations: draft guidance. Food and Drug Administration, Silver Spring, MD: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf [Google Scholar]
- 21. European Medicines Agency 2012. Guideline on the investigation of drug interactions. European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf [Google Scholar]
- 22. Weiss J, Ten Hoevel MM, Burhenne J, Walter-Sack I, Hoffmann MM, Rengelshausen J, Haefeli WE, Mikus G. 2009. CYP2C19 genotype is a major factor contributing to the highly variable pharmacokinetics of voriconazole. J. Clin. Pharmacol. 49:196–204 [DOI] [PubMed] [Google Scholar]
- 23. Andes D, Marchillo K, Stamstad T, Conklin R. 2003. In vivo pharmacokinetics and pharmacodynamics of a new triazole, voriconazole, in a murine candidiasis model. Antimicrob. Agents Chemother. 47:3165–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mavridou E, Bruggemann RJ, Melchers WJ, Verweij PE, Mouton JW. 2010. Impact of cyp51A mutations on the pharmacokinetic and pharmacodynamic properties of voriconazole in a murine model of disseminated aspergillosis. Antimicrob. Agents Chemother. 54:4758–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sugar AM, Liu XP. 2000. Effect of grapefruit juice on serum voriconazole concentrations in the mouse. Med. Mycol. 38:209–212 [DOI] [PubMed] [Google Scholar]
- 26. Sugar AM, Liu XP. 2001. Efficacy of voriconazole in treatment of murine pulmonary blastomycosis. Antimicrob. Agents Chemother. 45:601–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodriguez MM, Pastor FJ, Serena C, Guarro J. 2010. Efficacy of voriconazole in a murine model of invasive paecilomycosis. Int. J. Antimicrob. Agents 35:362–365 [DOI] [PubMed] [Google Scholar]
- 28. Graybill JR, Najvar LK, Gonzalez GM, Hernandez S, Bocanegra R. 2003. Improving the mouse model for studying the efficacy of voriconazole. J. Antimicrob. Chemother. 51:1373–1376 [DOI] [PubMed] [Google Scholar]
- 29. Majithiya J, Sharp A, Parmar A, Denning DW, Warn PA. 2009. Efficacy of isavuconazole, voriconazole and fluconazole in temporarily neutropenic murine models of disseminated Candida tropicalis and Candida krusei. J. Antimicrob. Chemother. 63:161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Warn PA, Sharp A, Mosquera J, Spickermann J, Schmitt-Hoffmann A, Heep M, Denning DW. 2006. Comparative in vivo activity of BAL4815, the active component of the prodrug BAL8557, in a neutropenic murine model of disseminated Aspergillus flavus. J. Antimicrob. Chemother. 58:1198–1207 [DOI] [PubMed] [Google Scholar]
- 31. Guo LQ, Yamazoe Y. 2004. Inhibition of cytochrome P450 by furanocoumarins in grapefruit juice and herbal medicines. Acta Pharmacol. Sin. 25:129–136 [PubMed] [Google Scholar]
- 32. Guo LQ, Fukuda K, Ohta T, Yamazoe Y. 2000. Role of furanocoumarin derivatives on grapefruit juice-mediated inhibition of human CYP3A activity. Drug Metab. Dispos. 28:766–771 [PubMed] [Google Scholar]
- 33. Fahmi OA, Kish M, Boldt S, Obach RS. 2010. Cytochrome P450 3A4 mRNA is a more reliable marker than CYP3A4 activity for detecting pregnane X receptor-activated induction of drug-metabolizing enzymes. Drug Metab. Dispos. 38:1605–1611 [DOI] [PubMed] [Google Scholar]
- 34. Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, Dickins M, Phipps A, Darekar A, Hyland R, Obach RS. 2009. Comparison of different algorithms for predicting clinical drug-drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab. Dispos. 37:1658–1666 [DOI] [PubMed] [Google Scholar]
- 35. Saari TI, Laine K, Leino K, Valtonen M, Neuvonen PJ, Olkkola KT. 2006. Effect of voriconazole on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Clin. Pharmacol. Ther. 79:362–370 [DOI] [PubMed] [Google Scholar]
- 36. Romero AJ, Le Pogamp P, Nilsson LG, Wood N. 2002. Effect of voriconazole on the pharmacokinetics of cyclosporine in renal transplant patients. Clin. Pharmacol. Ther. 71:226–234 [DOI] [PubMed] [Google Scholar]
- 37. Madan A, Graham RA, Carroll KM, Mudra DR, Burton LA, Krueger LA, Downey AD, Czerwinski M, Forster J, Ribadeneira MD, Gan LS, LeCluyse EL, Zech K, Robertson P, Jr, Koch P, Antonian L, Wagner G, Yu L, Parkinson A. 2003. Effects of prototypical microsomal enzyme inducers on cytochrome P450 expression in cultured human hepatocytes. Drug Metab. Dispos. 31:421–431 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.