

Abstract
The protozoan parasite Leishmania donovani undergoes several developmental transitions in its insect and vertebrate hosts that are induced by environmental changes. The roles of protein kinases in these adaptive differentiation steps and their potential as targets for antiparasitic intervention are only poorly characterized. Here, we used the generic protein kinase inhibitor staurosporine to gain insight into how interference with phosphotransferase activities affects the viability, growth, and motility of L. donovani promastigotes in vitro. Unlike the nonkinase drugs miltefosine and amphotericin B, staurosporine strongly reduced parasite biosynthetic activity and had a cytostatic rather than a cytotoxic effect. Despite the induction of a number of classical apoptotic markers, including caspase-like activity and surface binding of annexin V, we determined that, on the basis of cellular integrity, staurosporine did not cause cell death but caused cell cycle arrest and abrogated parasite motility. In contrast, targeted inhibition of the parasite casein kinase 1 (CK1) protein family by use of the CK1-specific inhibitor D4476 resulted in cell death. Thus, pleiotropic inhibition of L. donovani protein kinases and possibly other ATP-binding proteins by staurosporine dissociates apoptotic marker expression from cell death, which underscores the relevance of specific rather than broad kinase inhibitors for antiparasitic drug development.
INTRODUCTION
Leishmaniasis is an important parasitic disease affecting over 12 million people worldwide and causing a variety of pathologies, ranging from self-healing cutaneous lesions to fatal visceral infection causing hepatosplenomegaly (http://apps.who.int/tdr/svc/diseases/leishmaniasis). Treatments available for visceral leishmaniasis include pentavalent antimony (SbV) compounds as first-line drugs and pentamidine and amphotericin B (AmpB) as second-line drugs, the uses of which are limited by toxicity and availability. In addition, the clinical value of antimony therapy is threatened by the emergence of drug resistance. Recently, miltefosine (hexadecylphosphocholine [HePC]), an alkylphosphocholine originally developed as an anticancer drug, was proven to be effective and safe for use against visceral leishmaniasis in India (1) and was successfully applied to treat patients infected with antimony-resistant parasites. However, the therapeutic window of this drug might be very short, given the appearance of drug resistance in vitro (2). Thus, in the absence of vaccination and given the limitations of current therapies in cost, efficacy, and safety, there is an urgent need for the identification of novel targets and new chemical entities with antileishmanial activity.
Parasite-specific signaling pathways have recently attracted increasing attention as potential drug targets (3). Biochemical and genetic studies revealed important roles for trypanosomatid protein kinases in parasite growth and infectivity (4, 5), and as a result this class of proteins is the subject of several ongoing drug development efforts (6) (www.leishdrug.org). In vitro studies have been performed to investigate the activities of kinase inhibitors toward specific recombinant Leishmania kinases, such as CRK3 (7), casein kinase 1 (CK1) (8), and protein kinase A (PKA) (9), whose inhibition reduced parasite cell growth. Surprisingly, despite the presence of stage-specific phosphotransferase activities and their relevance in parasite differentiation and the establishment of intracellular infection (10, 11), little information is available on how generic protein kinase inhibitors affect Leishmania.
Staurosporine, like many competitive kinase inhibitors, is an ATP analogue that binds to the ATP-binding pocket of protein kinases and likely other ATP-binding proteins, thus abrogating their respective enzymatic activities. As such, staurosporine is an excellent tool to enable us to gain insight into kinase- and ATP-dependent biological activities in Leishmania and to identify novel drug targets. Staurosporine and 1-(5-isoquinolinesulfonyl)-2-methylpiperazine) dihydrochloride (H7) were the first generic kinase inhibitors used to investigate the role of protein kinases in Leishmania cell growth, morphology, and infectivity. Treatment of L. major and Leishmania amazonensis promastigotes with 10 μM staurosporine resulted in parasites with morphological differences in the size and appearance of the flagellar pocket (12). Because of its ability to induce apoptosis in various eukaryotic systems, staurosporine was used in investigations of programmed cell death (PCD) in Leishmania, which revealed the expression of classical apoptotic markers, including annexin V (AV) surface binding, loss of mitochondrial transmembrane potential, and DNA fragmentation (13). As a result, staurosporine has been used in Leishmania as an apoptosis-inducing compound to define the modes of action of other kinase inhibitors, such as withaferin (14), and antimicrobial peptides (15). However, because of the lack of the classical annexin V ligand phosphatidylserine in Leishmania promastigotes (16) and the link between annexin V binding and apoptotic mimicry rather than PCD, the question of whether the expression of apoptotic markers alone is a reliable readout for parasite cell death was raised (17). Here, we present a comprehensive and temporal analysis of morphological, molecular, and biochemical events in staurosporine-treated L. donovani, which reveal a highly complex response with regard to parasite motility, cell shape, growth, and viability. We show that the inhibitor abrogates flagellar activity and induces cell cycle arrest, annexin V binding, and caspase 3/7-like protease activities without significant loss of parasite viability and integrity. Our data thus demonstrate the dissociation of cell death from apoptotic marker expression and call into question the use of generic kinase inhibitors for assessing the druggability potential of the Leishmania kinome.
MATERIALS AND METHODS
Cell and culture conditions.
The L. donovani strain 1S2D (MHOM/SD/62/1S-CL2D), clone LdB, was cultured in vitro as described previously (18). Briefly, promastigotes were grown at 26°C in M199 supplemented with 10% fetal calf serum (FCS), 25 mM HEPES (pH 6.9), 12 mM NaHCO3, 1 mM glutamine, 1× RPMI 1640 vitamin mix, 10 μM folic acid, 100 μM adenosine, 7.6 mM hemin, 50 U/ml penicillin, and 50 μg/ml streptomycin. Axenic amastigote conversion was performed as described previously (19).
Growth inhibition assay.
The cell cytotoxicity and antileishmanial activity levels of selected drugs (miltefosine, amphotericin B, and staurosporine) were determined by using the alamarBlue assay. Briefly, L. donovani promastigotes or axenic amastigotes at a cell density of either 1 × 106 or 5 × 106 cells/ml were incubated in the presence of various concentrations of each drug at 26°C for 24 h before the addition of the resazurin dye (0.01%). After another 24 h of incubation, the fluorescence of the reduced resazurin was measured (excitation wavelength [λex], 530 nm; emission wavelength [λem], 585 nm). Values obtained from control wells with cells grown either in the presence of vehicle (0.5% dimethyl sulfoxide [DMSO]) or in the absence of drugs were used as maximum values (100%). All assays were performed in triplicate in 96-well microtiter plates.
Scanning electron microscopy.
Parasites were washed twice in ice-cold phosphate-buffered saline (PBS) and then fixed with 2% (wt/vol) glutaraldehyde (Sigma) in PBS with 0.1 M sodium cacodylate (pH 7.2). Briefly, the fixed cells were treated with 1% (wt/vol) OsO4 and dehydrated, followed by critical-point drying (CPD7501 critical-point dryer; Polaron) and coating with gold powder (ion beam coater 681; Gatan). Samples were visualized with an SEM 500 (Philips) scanning electron microscope as described previously (20).
Flow cytometry analysis.
To determine changes in the mitochondrial membrane potential (ΔΨm), we used the fluorescent dye tetramethylrhodamine ethyl ester (TMRE), which is a cell-permeable cationic dye that accumulates in the mitochondria of healthy cells. We tested the sensitivity of the TMRE fluorescent dye to changes in mitochondrial membrane potential by treating L. donovani promastigotes with carbonyl cyanide m-chlorophenylhydrazone (CCCP), a mitochondrial uncoupling agent. Total depolarization of the L. donovani promastigote was obtained by using 1 mM CCCP for 10 min (data not shown). Cells were grown to 5 × 106 cells/ml and then treated with amphotericin B (0.055 and 0.111 μM), miltefosine (hexadecylphosphocholine [HePC]) (26 and 46 μM), or staurosporine (0.261 and 0.428 μM) and incubated at 26°C. At various time points, 250 μl of the culture was pelleted and resuspended in 400 μl of HEPES buffer (10 mM HEPES-NaOH [pH 7.4], 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2), and 20.8 μg/ml of TMRE was added. Cells were incubated for 7 min in the dark prior to analysis by flow cytometry on a Becton Dickinson FACSCalibur system using the fluorescence 2 (FL2) channel.
Double staining with annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) was performed to measure the effects of the drugs on the plasma membrane of Leishmania promastigote cells. The expression of phosphatidylserine in the outer membrane of treated and untreated L. donovani promastigote cells was monitored by labeling with annexin V-FITC (Photeomix, Paris, France), and staining with propidium iodide was used to measure the permeability of the plasma membrane. Cells were grown to 5 × 106 cells/ml and then treated with amphotericin B (0.055 and 0.111 μM corresponding to one-half and 1× the 50% inhibitory concentration [IC50], respectively), miltefosine (hexadecylphosphocholine [HePC]) (26 and 46 μM), or staurosporine (0.261 and 0.428 μM) and incubated at 26°C. At various time points, 250 μl of culture was centrifuged (2,000 rpm, 2 min) and resuspended in 400 μl of HEPES buffer (10 mM HEPES-NaOH [pH 7.4], 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2) containing 4 μg/ml of annexin V-FITC and 10 μg/ml of propidium iodide. A FACSCalibur flow cytometer (Beckton Dickinson, San Jose, CA) was used to detect annexin V-FITC and propidium iodide fluorescence. Untreated controls corresponding to L. donovani promastigotes from a logarithmic culture (5 × 106 cells/ml) were negative for annexin V and propidium iodide (classified as healthy) over the 24 h of the experiment, demonstrating that growth up to at least 2 × 107 cells/ml does not induce cell death (data not shown).
Cell division of L. donovani promastigotes was evaluated using carboxyfluorescein diacetate succinimidyl ester (CFSE) as described previously (21). Log-phase parasites were washed and resuspended in 1 ml of M199 without serum to obtain a final concentration of 4 × 107 cells/ml, and CFSE (Molecular Probes) was added to obtain a final concentration of 5 μM. Cells were incubated at 26°C for 15 min with gentle mixing, and 5 volumes of complete M199 medium was added to quench the CFSE. Stained cells were washed to remove free CFSE. Cells were then resuspended in fresh complete M199 medium (containing 10% heat-inactivated fetal calf serum), and miltefosine (26 μM) or staurosporine (0.261 and 0.428 μM) was added. Cells were cultured in 25-cm2 tissue culture flasks at 26°C. At 2 h and 24 h after staining, CFSE fluorescence was determined by flow cytometry using the Becton, Dickinson FACSCalibur system.
Caspase 3/7 activation assay.
The caspase 3/7 substrate rhodamine 110, bis-(N-CBZ-l-aspartyl-l-glutamyl-l-valyl-l-aspartic acid amine) (Z-DEVD-R110), is a profluorescent substrate for which cleavage of the Asp-Glu-Val-Asp (DEVD) peptides produces an intensely fluorescent rhodamine 110. The amount of fluorescent product generated is proportional to the amount of caspase 3/7-like cleavage activity present in the sample. Promastigote cells of L. donovani (5 × 106 cells/ml) were incubated in the presence of miltefosine (26 μM and 46 μM), amphotericin B (0.111 μM), or staurosporine (0.261 μM and 0.428 μM) at 26°C. Cells were collected after 3 h, 6 h, and 24 h of incubation, washed in PBS, and then incubated in the presence of the caspase 3/7 reagents as detailed in the manufacturer's instructions. Fluorescence emanating from the cleaved DEVD substrate was measured at various time points by using a Tecan Infinite F500 fluorometer (λex, 480 nm; λem, 520 nm). Promastigotes grown in the absence of drugs were used as controls. In addition, degradation of Z-DEVD-R110 was measured in the presence of M199 medium and PBS to evaluate the effect of these buffers on the fluorescent substrate.
Activity assay for endogenous Leishmania kinases.
L. donovani log-phase promastigotes were harvested by centrifugation, washed three times in PBS, and resuspended in 1 ml extraction buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2% Triton X-100, 10% glycerol, and a protease inhibitor cocktail (2 mM 4-[2-aminoethyl]-benzenesulfonyl fluoride [AEBSF], 1 μM phosphoramidon, 130 μM bestatin, 14 μM E64, 1 μM leupeptin, 0.2 μM aprotinin, 10 μM papstatin). Kinase activity levels were measured in the promastigote lysates (3 μg of proteins) using 250 μM ATP in the reaction buffer. Peptide substrates specific for casein kinases 1 and 2 (RRKDLHDDEEDEAMSITA and RRASADDSDDEDL), PKA (PRRASLIFGDI and LRRASLG), and PKB (RPRAASF) were used in the kinase assays. Substrate specificity was further determined by using recombinant PKA and a combination of inhibitors (not shown). In addition, a random peptide library (Photeomix, France) was screened for kinase activity levels (30 peptide substrates), from which three peptides were selected because phosphotransferase activities were detectable on them (PSHSNSWIIR, RARHRSDSSR, QKGIASRRNS). Kinase activity was determined using the proprietary biophysical assay Activomics (Photeomix, France) and is reported as a percentage of substrate phosphorylation.
Macrophage culture and infection.
Murine bone marrow-derived macrophages, obtained and differentiated from C57BL/6 bone marrow exudate cells, were incubated at a ratio of 10 parasites per macrophage with bona fide L. donovani amastigotes purified from the livers of infected hamsters. After 24 h, cells were treated with either 0.5% DMSO (control), 0.5 μM amphotericin B, 7 μM miltefosine, or 0.2 μM staurosporine. Intracellular parasites were detected by nuclear staining with Hoechst 33342 in paraformaldehyde (PFA)-fixed cells at 24 h, 48 h, and 72 h after contact with the drugs. One hundred macrophages were analyzed per coverslip, and the experiment was performed in triplicate. For IC50 determination, 105 macrophages were distributed in a 96-well plate and incubated for 24 h, and then the medium was removed and replaced by medium containing either amphotericin B (AmpB) (0.1 to 100 μM), miltefosine (HePC) (0.1 to 100 μM), or staurosporine (Stauro) (0.05 to 50 μM) for 48 h. Macrophage viability was assessed using the alamarBlue viability test to measure mitochondrial metabolic activities. The experiment was performed in triplicate.
RESULTS
Staurosporine-treated L. donovani promastigotes show a reduction in biosynthetic activity and cell proliferation.
The effect of staurosporine, miltefosine, and amphotericin B on the biosynthetic activity of L. donovani promastigotes was assessed by using the alamarBlue assay. This assay measures the reduction of the cell-permeable dye resazurin into fluorescent resorufin by intracellular enzymes. Treatment of cultured L. donovani promastigotes with the three drugs staurosporine, miltefosine, and amphotericin B for 24 h resulted in concentration- and density-dependent inhibition of parasite biosynthetic activity with IC50 values of 0.261 ±0.04 μM, 26.72 ±0.36 μM, and 0.111 ±0.005 μM, respectively, at a density of 5 × 106, which was decreased by 1.67-fold for staurosporine, 3.2-fold for amphotericin B, and 1.62-fold for miltefosine at a density of 1 × 106 (Fig. 1A; Table 1). Significantly, the IC50 for staurosporine was substantially increased in axenic amastigotes at both cell densities (Table 1). This might have been the result of either enhanced resistance to the drug or reduced uptake. Unfortunately, the toxicity of staurosporine toward bone marrow-derived macrophages precluded us from establishing an IC50 for intracellular amastigotes. However, a substantial reduction in the intracellular parasite burden was observed at a staurosporine concentration similar to the IC50 established for the host cells (Table 2). Whether the reduced parasite proliferation was due to a direct toxic effect on intracellular amastigotes or caused by effects on the host cells remains to be elucidated.
Fig 1.

Staurosporine causes a dose-dependent effect on growth of L. donovani promastigotes. (A) alamarBlue viability test. Promastigotes of L. donovani at initial cell densities of 1 × 106 (●) and 5 × 106 (○) cells/ml were treated for 48 h with amphotericin B (AmpB, 0.0012 to 10 μM), miltefosine (hexadecylphosphocholine [HePC], 0.024 to 75 μM), and staurosporine (Stauro, 0.0012 to 10 μM), and parasite viability was assessed using the alamarBlue viability test to measure mitochondrial metabolic activities. IC50 values were calculated from the dose response inhibition (n = 3). The standard deviation of one triplicate experiment is denoted by the bars. (B) Cell growth analysis. Cell growth was determined by the optical density (OD) at 600 nm. The experiment was performed twice in triplicate. Each bar denotes the standard deviation of one experiment performed in triplicate.
Table 1.
IC50 for amphotericin B, HePC, and staurosporine obtained in promastigotes, axenic amastigotes, and noninfected bone marrow-derived macrophages at the indicated cell densities
| Treatment | Density (cells/ml) | IC50 (μM) (mean ± SD) | Fold diff.a |
|---|---|---|---|
| Promastigotes | |||
| Amphotericin B | 1 × 106 | 0.035 ± 0.0006 | 3.20 |
| 5 × 106 | 0.111 ± 0.005 | ||
| HePC | 1 × 106 | 16.53 ± 1.12 | 1.62 |
| 5 × 106 | 26.72 ± 0.36 | ||
| Staurosporine | 1 × 106 | 0.16 ± 0.009 | 1.67 |
| 5 × 106 | 0.26 ± 0.04 | ||
| Axenic amastigotes | |||
| Amphotericin B | 1 × 106 | 0.22 ± 0.02 | 1.36 |
| 5 × 106 | 0.3 ± 0 | ||
| HePC | 1 × 106 | 3.3 ± 0.6 | 2.88 |
| 5 × 106 | 9.5 ± 0 | ||
| Staurosporine | 1 × 106 | 12.3 ± 2.3 | 2.44 |
| 5 × 106 | 30 ± 0 | ||
| Macrophages | |||
| Amphotericin B | 1 × 105 | 10.67 ± 3.75 | NAb |
| HePC | 1 × 105 | 92.5 ± 10.61 | NA |
| Staurosporine | 1 × 105 | 0.15 ± 0 | NA |
The fold difference corresponds to the ratio of the IC50 values obtained by using the two different cell densities.
NA, not applicable.
Table 2.
Percentage of infected macrophages and relative intracellular amastigote survival with treatmenta
| Treatment | Time after treatment (h) | % Mpb (mean ± SD) | % Pc (mean ± SD) |
|---|---|---|---|
| Amphotericin B (0.5 μM) | 24 | 19 ± 11.50 | 5.1 ± 4.6 |
| 48 | 5 ± 5.34 | 1 ± 1 | |
| 72 | 0 ± 0.58 | 0 | |
| HePC (7 μM) | 24 | 94 ± 5.13 | 66.9 ± 32.2 |
| 48 | 84 ± 6.11 | 45.1 ± 14.2 | |
| 72 | 85 ± 2.08 | 60.0 ± 18.3 | |
| Staurosporine (0.2 μM) | 24 | 99 ± 0.58 | 97.2 ± 8.7 |
| 48 | 99 ± 1.15 | 51.5 ± 12.9 | |
| 72 | 96 ± 7.51 | 47.4 ± 20.9 |
Data are normalized to the values obtained for untreated (HePC) or 0.5% DMSO-treated controls at the respective time point (set to 100%).
% Mp, percentage of infected macrophages.
% P, relative intracellular amastigote survival (number of parasites/100 macrophages).
To gain further insight into the mechanism of action of these drugs in L. donovani, we measured the culture densities over 24 h for untreated and inhibitor-treated parasites. While untreated parasites showed a linear increase in culture densities, parasites treated with staurosporine at 0.261 μM showed 30% growth inhibition compared to that of the controls. Increasing the staurosporine concentration by 2-fold did not abrogate growth but reduced it by 70% (Fig. 1B, left). Similarly, miltefosine (HePC) at 26 μM partially inhibited cell growth by 42% but efficiently killed the parasites at 46 μM, as indicated by the decrease in culture densities compared to the density at the 0-h time point (Fig. 1B, middle). In contrast, amphotericin B treatment rapidly decreased the parasite culture density at 0.111 μM (IC50) and even at 0.055 μM, suggesting efficient parasite killing (Fig. 1B, right). Our data enabled us to define different modes of action of the tested inhibitors. Staurosporine causes a cytostatic rather than a cytotoxic effect, which limits the usefulness of enzyme-based assays (e.g., alamarBlue, 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide [MTT], etc.) in determining the IC50 for this inhibitor.
Phenotypic characterization of staurosporine-treated L. donovani promastigotes.
The absence of cell death in staurosporine-treated cells suggests an inhibition of cell proliferation as the cause of the observed growth reduction shown in Fig. 1. To confirm this possibility, we analyzed cell division using a quantitative fluorometric assay based on the cytoplasmic dye CFSE, which is passed to daughter cells during division (21). Cells were stained with CFSE and then placed in culture in the presence of either miltefosine (HePC, 26 μM) or staurosporine (0.261 and 0.428 μM). Amphotericin B was not analyzed, as it has a fast necrotic effect on the cells even at 0.055 μM (one-half its IC50). The fluorescence level of the cells was evaluated by fluorescence-activated cell sorting (FACS) after 2 h and 24 h of treatment. At 2 h, all samples showed narrow peaks of high fluorescence intensity, demonstrating homogenous staining of the cells (Fig. 2A, top). The control population not subjected to drug pressure showed a significant shift of the fluorescence peak, from 1,200 to 110 of FL2 intensity at 585 nm, indicating robust cell proliferation (Fig. 2A, bottom). The presence of miltefosine at 26 μM in the culture medium had little or no effect on the cell division of surviving L. donovani promastigotes. In contrast, the quantification of cell division following treatment with 0.261 and 0.428 μM staurosporine showed cell cycle arrest for 22% and 58% of the cells, respectively, confirming a dose-dependent cytostatic effect of staurosporine on L. donovani promastigotes.
Fig 2.

Staurosporine treatment affects promastigote proliferation, morphology, and motility. (A) Proliferation assay of L. donovani in the presence of 26 μM miltefosine (hexadecylphosphocholine [HePC]) or 0.261 μM and 0.428 μM staurosporine (Stauro). L. donovani promastigotes from a logarithmic culture were stained with CFSE, and fluorescence levels were determined by FACS analysis using the fluorescence channel FL2-H at 2 h (top) and 24 h (bottom) after inoculation into new culture medium. A representative experiment of two independent tests is shown. (B) Scanning electron microscopy (SEM). L. donovani promastigotes from the logarithmic growth phase were treated for 4 h with miltefosine (HePC, 46 μM), staurosporine (Stauro, 0.428 μM), and amphotericin B (AmpB, 0.055 μM) and processed for SEM as described in Materials and Methods. The bar corresponds to 1 μm. Control corresponds to untreated promastigotes from a logarithmic culture. (C) Motility assay. The movement of L. donovani promastigotes in untreated and staurosporine-treated cultures was assessed by taking 200 pictures in 20 s using a phase-contrast microscope. Images were analyzed using the image analysis medeaLAB tracking software (version 5.5). Each line represents the path taken by a single L. donovani promastigote over the 20 s of the analysis.
We next assessed the effects of the inhibitors on the morphology of logarithmic-phase promastigotes 4 h after treatment. Untreated parasites showed the characteristic oval-shaped cell body, which became more elongated and slender after exposure to 0.428 μM staurosporine (Fig. 2B). In contrast, cells treated with miltefosine (HePC, 46 μM) exhibited membrane damage and showed the characteristic morphology of stressed parasites undergoing cell death, i.e., reduction in cell size and rounding up and shortening of the flagellum (Fig. 2B). Finally, cells treated with amphotericin B (0.055 μM) retained the shape and length of untreated cells but were severely damaged, as indicated by the loss of surface integrity. Based on morphological criteria, our data revealed different morphological responses associated with apoptotic-like and necrotic cell death induced by antileishmanial miltefosine and amphotericin B, respectively, and confirmed the absence of parasite death in the presence of staurosporine. However, despite the maintenance of cellular integrity, we noticed in the presence of staurosporine a variation in flagellar movement showing a wave-like rather than the propeller-like motion characteristic of untreated controls. Live imaging analysis coupled with video microscopy revealed that 100% of the staurosporine-treated cells gyrated and lost their capacity for forward movement (Fig. 2C). Thus, pleiotropic inhibition of Leishmania protein kinases and other ATP-dependent enzymes by staurosporine causes a cytostatic rather than cytotoxic effect under the conditions and at the concentrations used in our study.
Staurosporine induces the expression of classical eukaryotic apoptotic markers without causing L. donovani cell death.
Staurosporine has been shown to activate certain markers of apoptosis in Leishmania. To better define the mode of action of staurosporine on L. donovani promastigotes, we evaluated the extent of caspase 3/7-like protease activity in the presence of this inhibitor. Despite the absence of conserved caspases in Leishmania, corresponding proteolytic activity has been recently linked to a cathepsin B-like protein involved in parasite cell death (22). Promastigotes from a logarithmic culture (5 × 106 cells/ml) were incubated in the presence of 0.261 and 0.428 μM staurosporine, 26 and 46 μM miltefosine, and 0.111 μM amphotericin B at 26°C. After 3 h, 6 h, and 24 h of treatment, cells were lysed and the caspase 3/7-like proteolytic activity was measured using the fluorescent peptide substrate Z-DEVD-R110 (Fig. 3). Caspase 3/7-like activity was induced in a dose-dependent fashion by miltefosine, with 46 μM of this drug inducing stronger activity at 3 h and 6 h than did 26 μM miltefosine, as indicated by the kinetics of proteolysis. However, no activity was detected at 24 h of treatment with 46 μM miltefosine, probably due to the large amount of cell death. Cell lysates obtained from staurosporine-treated cultures were able to proteolyze the Z-DEVD-R110 substrate in a time- and dose-dependent manner, suggesting the activation of caspase 3/7-like proteases despite the absence of detectable cell death.
Fig 3.
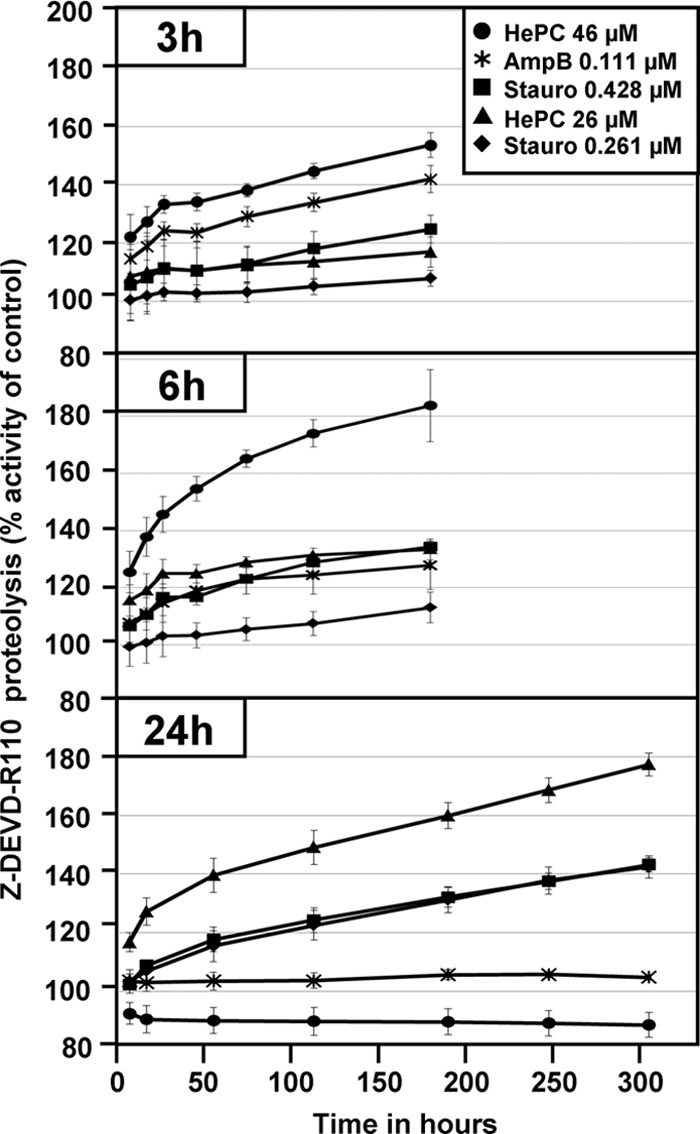
Caspase 3/7-like activity is induced by miltefosine, amphotericin B, and staurosporine. L. donovani promastigote cells were incubated in the presence of miltefosine (hexadecylphosphocholine [HePC]) at 26 μM (▲) and 46 μM (●), staurosporine at 0.261 μM (⧫) and 0.428 μM (■), and amphotericin B at 0.111 μM (⁎) for 3 h, 6 h, and 24 h. Following treatment, cells were lysed and incubated in the presence of Z-DEVD-R110 substrate. Fluorescence intensities corresponding to proteolysis of the DEVD substrate (λex, 480 nm; λem, 520 nm) are reported as percentages of the fluorescence intensity obtained in untreated culture. The bars represent the standard deviation of a representative experiment performed in triplicate.
The dissociation of caspase 3/7-like protease activity from cell death in staurosporine-treated promastigotes primed us to investigate the expression levels of three other classical markers of programmed cell death: (i) changes in the mitochondrial membrane potential (ΔΨm) that occur with the utilization of the fluorescent dye tetramethylrhodamine ethyl ester (TMRE), which accumulates in the mitochondria of healthy cells; (ii) surface binding of annexin V-FITC, which enables monitoring of the reorganization of the plasma membrane (i.e., translocation of phosphatidylserine phospholipid from the inside to the outside the membrane); and (iii) staining of the DNA with propidium iodide, which reveals permeabilization of the plasma membrane. The cells were classified into four categories on the basis of their annexin V (AV) and propidium iodide (PI) signals: AV− and PI− healthy cells, AV+ and PI− early apoptotic-like cells that undergo changes in transbilayer phospholipid arrangement, AV+ and PI+ late apoptotic-like cells that lose membrane integrity, and finally AV− and PI+ necrotic cells.
Based on the three apoptotic markers analyzed, we determined that the cells reacted very slowly to the presence of 0.428 μM staurosporine during the first 5 h of treatment, with the first effect observed on depolarization of the mitochondrial membrane after 4 h in 20% of the cells (Fig. 4, top). Depolarization was followed 20 h later by a hyperpolarization of the membrane in 70% of the cells. After 24 h of incubation, 43% of the cells were decorated with annexin V without evidence of propidium iodide incorporation. Only after 48 h of staurosporine-induced cytostasis did a minor fraction (8%) of the cells undergo cell death, as indicated by AV+ and PI+ staining, which might have been a consequence of prolonged cell cycle arrest rather than a direct toxic effect of the inhibitor.
Fig 4.
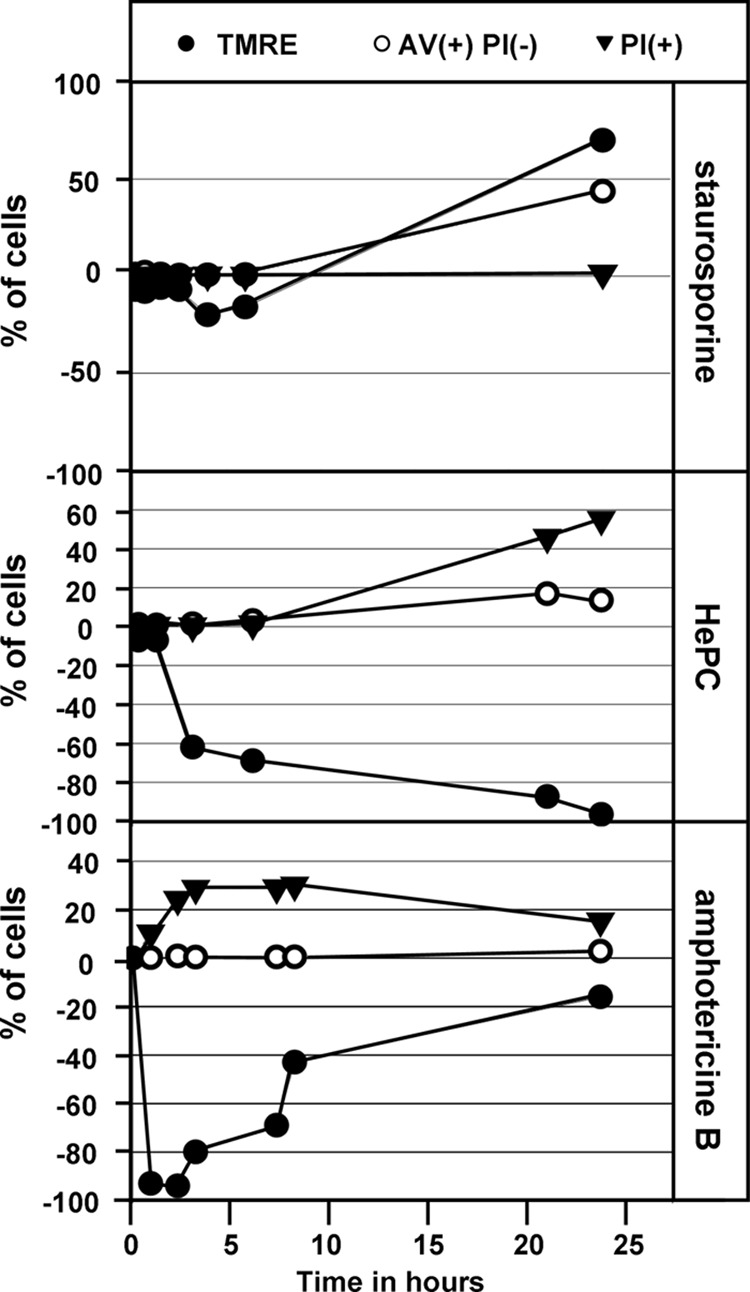
FACS analysis of three markers used to define the mode of cell death induced in L. donovani in the presence of staurosporine, miltefosine, and amphotericin B at 0.428 μM, 46 μM, and 0.055 μM, respectively. The graphs represent the percentages of cells, out of 10,000 cells counted, that showed mitochondrial membrane depolarization (negative %) or hyperpolarization (positive %) (●), annexin V binding (○), and propidium iodide incorporation (▼). The experiments were carried out three times, and a representative result is shown. TMRE, tetramethylrhodamine ethyl; AV, annexin V; PI, propidium iodide.
Treatment of L. donovani promastigotes with miltefosine at 46 μM caused a time-dependent depolarization of the mitochondrial membrane in 40% and 100% of the cells after 3 h and 24 h of treatment, respectively (Fig. 4, middle). Based on AV staining and PI incorporation, we determined that miltefosine-treated parasites followed the classical apoptotic stages, with 3% of cells staining AV+ PI− after 7 h of exposure, which increased to 16% at 24 h and was accompanied by 25% of cells staining PI+.
In contrast to incubation with staurosporine and miltefosine, incubation of parasites with 0.055 μM amphotericin B for 10 min caused a strong depolarization of the mitochondrial membrane in almost 100% of the cells (Fig. 4, bottom), which was accompanied by the appearance of AV+ PI+ cells. The rapid action of amphotericin B correlated with a fast decrease in culture density (Fig. 1B) and damage of the plasma membrane (Fig. 2B). In the course of our analysis, the number of propidium iodide-positive cells fell from 30% at 5 h to 20% at 24 h as a consequence of cell death and subsequent cell disintegration. In addition, at no point during the 24 h of treatment did we detect AV+ PI− cells, suggesting that death occurs through a direct cytotoxic effect distinct from miltefosine-induced cell death. Our data thus reveal three different mechanisms of antileishmanial action, with staurosporine inducing various classical apoptotic markers in viable cells.
The casein kinase 1-specific inhibitor D4476 leads to fast cell death.
Given the pleiotropic effect of staurosporine on various cellular kinomes and likely other ATP-dependent enzymes, the failure of this inhibitor to induce Leishmania cell death challenges the notion of parasite kinases as a source for novel drug targets. To investigate if a specific rather than generic kinase inhibitor causes parasite death, we analyzed the effects of the casein kinase 1 (CK1) inhibitor D4476 (CAS 301836-43-1) on parasite growth, apoptotic marker expression, and viability. D4476 is a triaryl-substituted imidazolo compound that acts as a potent, reversible, and relatively specific ATP-competitive inhibitor of CK1 in various cellular systems. First, we used a novel activity-based procedure to investigate the effects of staurosporine and D4476 on endogenous protein kinase activities, including that of CK1, present in L. donovani lysates. In this procedure, we used synthetic peptides as the substrates to monitor the phosphotransferase activity levels of various protein kinase families. In the absence of inhibitor, phosphorylation occurred on canonical substrates for PKA, PKB, CK1, and CK2, as well as three peptides from a random peptide library (Fig. 5A). As expected from its pleiotropic action, staurosporine treatment of parasite lysates abrogated phosphorylation of these peptides, with the exception of the CK1- and CK2-specific substrates. In contrast, D4476 abrogated only phosphorylation of the CK1-specific substrate, confirming its specificity to Leishmania CK1 kinases.
Fig 5.

The casein kinase 1 inhibitor D4476 causes death in L. donovani promastigotes. (A) Modulation of endogenous Leishmania kinase activities by staurosporine and D4476. L. donovani cell extracts were treated with vehicle alone (0.5% DMSO, control [co]) or treated with either 10 μM staurosporine (S) or D4476 (D) for 5 min before adding the peptide substrate and ATP. The percentage of phosphorylation of the substrate compared to that of untreated cell extracts (determined as described in Materials and Methods) is reported in the form of a heat map, with gray corresponding to phosphorylation and black to the absence or inhibition of phosphorylation. Abbreviations: cyclic AMP-dependent protein kinase (PKA), protein kinase B (PKB), protein kinase CΔ (PKC), casein kinases (CK1 and -2), cyclin-dependent kinases (CDK2 and -5), and AMP-activated protein kinase (AMPK). (B) Inhibition of CK1 activity by D4476 kills Leishmania through necrosis. Cell growth analysis (left). Cell growth was determined following measurement of optical density (OD) at 600 nm in the presence of 30 μM (●) and 60 μM (○) D4476. Right, FACS analysis. The three markers TMRE (tetramethylrhodamine ethyl), AV (annexin V), and PI (propidium iodide) were followed to define the mode of cell death induced in L. donovani in the presence of D4476 at 60 μM. The percentages of cells showing mitochondrial membrane depolarization (negative %) or hyperpolarization (positive %) (●), annexin V binding (○), and propidium iodide incorporation (▼) are represented. The experiment was carried out twice, and one representative result is shown.
We next tested if specific inhibition of CK1 kinases by D4476 causes parasite death. The IC50 of this inhibitor, determined with L. donovani promastigotes by using the alamarBlue assay, was 30 μM (N. Rachidi, unpublished data). At 30 μM an inhibition of cell growth by 88% was observed, while 60 μM completely abrogated cell growth as shown by a decrease in culture density (Fig. 5B, left). At this concentration, D4476 treatment resulted in 12% PI+ cells after only 4 h of incubation, which increased steadily to 75% at 24 h (Fig. 5B, right), demonstrating that the inhibition of a single protein kinase family, such as CK1, has dramatic effects on cell survival. It must be noted that at no time point in the analysis did we see Leishmania cells that were exclusively annexin V positive, not even with the use of 30 μM D4476 (data not shown), suggesting that D4476 induces an annexin V-independent mode of cell death. These data showed that specific rather than pleiotropic kinase inhibition can be exploited for drug development. The antileishmanial effect of D4476 was confirmed using bone marrow-derived primary macrophages infected with bona fide amastigotes obtained from infected hamster spleen, thus validating L. donovani casein kinase family members as interesting drug targets (Rachidi, unpublished).
DISCUSSION
Staurosporine treatment of L. donovani promastigotes caused a pleiotropic response and revealed important functions of respective cellular targets in regulating parasite morphology, motility, and cell cycle progression. As indicated by its effect on protein phosphorylation, staurosporine largely targets protein kinases, although effects of this inhibitor on other ATP-dependent and autophosphorylation activities cannot be excluded (10). The first phenotypic manifestation in response to staurosporine was represented by a drastic change in L. donovani flagellar activity from a propeller-like to a wave-like motion, thus preventing parasite propulsion. This defect might arise from inhibition of the ATP-dependent flagellar motor proteins such as dyneins (23); flagellar protein kinases such as the mitogen-activated protein (MAP) kinase homologs mitogen-activated protein kinase 3 (MPK3) and MPK9, which have been genetically linked in Leishmania mexicana to flagellar biogenesis (24, 25); or phosphoinositide 3-kinase (PI3K) and adenylate kinase homologs, which were identified in the Trypanosoma brucei flagellar proteome (26). Given the potential role of L. donovani motility in establishing host cell infection (27), our data lead us to propose ATP-driven flagellar activities as potential new drug targets.
A second major phenotypic manifestation in staurosporine-treated L. donovani promastigotes corresponds to a dose-dependent inhibition of cell proliferation, a phenomenon previously observed in other eukaryotic systems (28). Similar to its effect on the human cyclin-dependent kinases (CDKs) CDK2, CDK3, CDK5, and CDK7 (29), staurosporine might inhibit 1 or more of the 11 conserved L. donovani CDK homologs (30). Significantly, this cytostatic effect was accompanied by an inhibition of the mitochondrial enzymatic activity that is used as a readout for cell survival in the alamarBlue viability assay. Using staurosporine at the IC50 determined by this assay did not affect parasite survival as indicated by the absence of propidium iodide incorporation. Hence, drug development efforts targeting Leishmania protein kinases must include the consideration of these kinase-dependent effects on metabolic activity, and the establishment of IC50 values for potential kinase inhibitors should be performed by direct monitoring of cell death.
Finally, staurosporine treatment induced expression of several classical apoptotic markers in L. donovani promastigotes in the absence of cell death. This observation feeds important new information to the somewhat controversial discussion of the existence of programmed cell death in Leishmania (31, 32). Clearly, the dissociation of marker expression and cell death raises important questions on the applicability of current apoptotic readouts to determine Leishmania cell death. This ambiguity is further fueled by the absence of conserved caspase homologs from the Leishmania genomes (33) and by conflicting results regarding the presence of the annexin V ligand phosphatidylserine in parasite membrane fractions (16, 34).
Some of our results might support rather than challenge the presence of PCD in Leishmania. We provide evidence that drug-induced cell death is distinct between amphotericin B- and miltefosine-treated parasites, which argues for the presence of defined and regulated cell death mechanisms. Miltefosine-induced parasite death in our experimental system reproduced the sequence of events described for apoptosis in other eukaryotes, including the loss of morphological features and depolarization of the mitochondrial membrane (3 h), induction of caspase 3/7-like protease activities (6 h), annexin V binding (9 h), and finally, propidium iodide uptake and cellular death (24 h). The absence of conserved caspases from the parasite genome might be compensated for by Leishmania metacaspases, as their expression in yeast can functionally replace the yeast metacaspase YCA1 in PCD (35), and their overexpression in Leishmania correlates with enhanced sensitivity to oxidant-induced parasite death (36, 37). Furthermore, a recent study linked a cathepsin B-like protease to Leishmania cell death, which can cleave the canonical caspase substrate and was likely monitored in our study (22). An intriguing observation was the absence of cell death despite pleiotropic inhibition of the parasite kinome and ATP-dependent enzymatic activities in staurosporine-treated parasites. This finding represents a paradox, given that genetic ablation of specific Leishmania protein kinases (5, 38) as well as pharmacological inhibition of confined protein kinase families (7, 39) (our study) results in parasite death. Although we cannot rule out the possibility that staurosporine does not affect any of these essential protein kinases in Leishmania, it is possible that the absence of cell death is caused by inhibition of staurosporine-sensitive ATP-dependent functions required for PCD. Indeed, a regulatory relationship between protein kinases and caspases has been described in other organisms. In this relationship, caspase activities are regulated by phosphorylation (40), and vice versa, cell death-promoting protein kinases, for example protein kinase Cδ (PKCδ) (41), MEKK1 (42), and Rho-associated, coiled-coil containing protein kinase 1 (ROCK1) (43), are activated by caspase-dependent proteolysis. It is therefore possible to hypothesize that inhibition of Leishmania homologous protein kinases that might recognize cathepsin B or metacaspases as substrates can abrogate PCD in staurosporine-treated promastigotes. In support of this hypothesis, our data provide evidence that staurosporine treatment of parasite lysates interferes with PKA protein kinase activity, which has proapoptotic functions in mammalian cells through phosphorylation of its downstream targets (44, 45). Future experiments designed to enrich for staurosporine-binding proteins by combining affinity chromatography and proteomic analyses might reveal the Leishmania regulatory proteins required for PCD.
In conclusion, the comparative inhibitor study described here delivered significant new insight into ATP-dependent biological processes in L. donovani with relevance for parasite infectivity, including motility and proliferation, and allowed us to elucidate molecular mechanisms underlying drug action and parasite death. Our study provides information on important aspects relevant for kinase-based drug development efforts, which should be focused on the identification of cytotoxic, rather than cytostatic, kinase inhibitors with specificity toward a limited number of essential protein kinases not implicated in parasite cell death regulation.
ACKNOWLEDGMENTS
We thank Sophie Veillault for editorial help, Pascale Pescher for the isolation of hamster-derived L. donovani amastigotes, and Eric Prina for providing bone marrow-derived macrophages.
This work was supported by the 7th Framework Programme of the European Commission through a grant to the LEISHDRUG (223414) and the ANR-11-RPIB-0016 TRANSLEISH projects.
We report no conflicts of interest, and we alone are responsible for the content and writing of the paper.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Rahman M, Ahmed BN, Faiz MA, Chowdhury MZ, Islam QT, Sayeedur R, Rahman MR, Hossain M, Bangali AM, Ahmad Z, Islam MN, Mascie-Taylor CG, Berman J, Arana B. 2011. Phase IV trial of miltefosine in adults and children for treatment of visceral leishmaniasis (kala-azar) in Bangladesh. Am. J. Trop. Med. Hyg. 85:66–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perez-Victoria JM, Bavchvarov BI, Torrecillas IR, Martinez-Garcia M, Lopez-Martin C, Campillo M, Castanys S, Gamarro F. 2011. Sitamaquine overcomes ABC-mediated resistance to miltefosine and antimony in Leishmania. Antimicrob. Agents Chemother. 55:3838–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doerig C. 2004. Protein kinases as targets for anti-parasitic chemotherapy. Biochim. Biophys. Acta 1697:155–168 [DOI] [PubMed] [Google Scholar]
- 4. Chow C, Cloutier S, Dumas C, Chou MN, Papadopoulou B. 2011. Promastigote to amastigote differentiation of Leishmania is markedly delayed in the absence of PERK eIF2alpha kinase-dependent eIF2alpha phosphorylation. Cell. Microbiol. 13:1059–1077 [DOI] [PubMed] [Google Scholar]
- 5. Madeira da Silva L, Beverley SM. 2010. Expansion of the target of rapamycin (TOR) kinase family and function in Leishmania shows that TOR3 is required for acidocalcisome biogenesis and animal infectivity. Proc. Natl. Acad. Sci. U. S. A. 107:11965–11970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dujardin JC, Gonzalez-Pacanowska D, Croft SL, Olesen OF, Spath GF. 2010. Collaborative actions in anti-trypanosomatid chemotherapy with partners from disease endemic areas. Trends Parasitol. 26:395–403 [DOI] [PubMed] [Google Scholar]
- 7. Grant KM, Dunion MH, Yardley V, Skaltsounis AL, Marko D, Eisenbrand G, Croft SL, Meijer L, Mottram JC. 2004. Inhibitors of Leishmania mexicana CRK3 cyclin-dependent kinase: chemical library screen and antileishmanial activity. Antimicrob. Agents Chemother. 48:3033–3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allocco JJ, Donald R, Zhong T, Lee A, Tang YS, Hendrickson RC, Liberator P, Nare B. 2006. Inhibitors of casein kinase 1 block the growth of Leishmania major promastigotes in vitro. Int. J. Parasitol. 36:1249–1259 [DOI] [PubMed] [Google Scholar]
- 9. Malki-Feldman L, Jaffe CL. 2009. Leishmania major: effect of protein kinase A and phosphodiesterase activity on infectivity and proliferation of promastigotes. Exp. Parasitol. 123:39–44 [DOI] [PubMed] [Google Scholar]
- 10. Schmidt-Arras D, Leclercq O, Gherardini PF, Helmer-Citterich M, Faigle W, Loew D, Spath GF. 2011. Adaptation of a 2D in-gel kinase assay to trace phosphotransferase activities in the human pathogen Leishmania donovani. J. Proteomics 74:1644–1651 [DOI] [PubMed] [Google Scholar]
- 11. Hem S, Gherardini PF, Osorio y Fortea J, Hourdel V, Morales MA, Watanabe R, Pescher P, Kuzyk MA, Smith D, Borchers CH, Zilberstein D, Helmer-Citterich M, Namane A, Spath GF. 2010. Identification of Leishmania-specific protein phosphorylation sites by LC-ESI-MS/MS and comparative genomics analyses. Proteomics 10:3868–3883 [DOI] [PubMed] [Google Scholar]
- 12. Becker S, Jaffe CL. 1997. Effect of protein kinase inhibitors on the growth, morphology, and infectivity of Leishmania promastigotes. Parasitol. Res. 83:273–280 [DOI] [PubMed] [Google Scholar]
- 13. Arnoult D, Akarid K, Grodet A, Petit PX, Estaquier J, Ameisen JC. 2002. On the evolution of programmed cell death: apoptosis of the unicellular eukaryote Leishmania major involves cysteine proteinase activation and mitochondrion permeabilization. Cell Death Differ. 9:65–81 [DOI] [PubMed] [Google Scholar]
- 14. Sen N, Banerjee B, Das BB, Ganguly A, Sen T, Pramanik S, Mukhopadhyay S, Majumder HK. 2007. Apoptosis is induced in leishmanial cells by a novel protein kinase inhibitor withaferin A and is facilitated by apoptotic topoisomerase I-DNA complex. Cell Death Differ. 14:358–367 [DOI] [PubMed] [Google Scholar]
- 15. Kulkarni MM, McMaster WR, Kamysz W, McGwire BS. 2009. Antimicrobial peptide-induced apoptotic death of leishmania results from calcium-dependent, caspase-independent mitochondrial toxicity. J. Biol. Chem. 284:15496–15504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weingartner A, Kemmer G, Muller FD, Zampieri RA, Gonzaga Dos Santos M, Schiller J, Pomorski TG. 2012. Leishmania promastigotes lack phosphatidylserine but bind annexin V upon permeabilization or miltefosine treatment. PLoS One 7:e42070 doi:10.1371/journal.pone.0042070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Freitas Balanco JM, Moreira ME, Bonomo A, Bozza PT, Amarante-Mendes G, Pirmez C, Barcinski MA. 2001. Apoptotic mimicry by an obligate intracellular parasite downregulates macrophage microbicidal activity. Curr. Biol. 11:1870–1873 [DOI] [PubMed] [Google Scholar]
- 18. Saar Y, Ransford A, Waldman E, Mazareb S, Amin-Spector S, Plumblee J, Turco SJ, Zilberstein D. 1998. Characterization of developmentally-regulated activities in axenic amastigotes of Leishmania donovani. Mol. Biochem. Parasitol. 95:9–20 [DOI] [PubMed] [Google Scholar]
- 19. Goyard S, Segawa H, Gordon J, Showalter M, Duncan R, Turco SJ, Beverley SM. 2003. An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol. Biochem. Parasitol. 130:31–42 [DOI] [PubMed] [Google Scholar]
- 20. Absalon S, Blisnick T, Bonhivers M, Kohl L, Cayet N, Toutirais G, Buisson J, Robinson D, Bastin P. 2008. Flagellum elongation is required for correct structure, orientation and function of the flagellar pocket in Trypanosoma brucei. J. Cell Sci. 121:3704–3716 [DOI] [PubMed] [Google Scholar]
- 21. Morales MA, Pescher P, Spath GF. 2010. Leishmania major MPK7 protein kinase activity inhibits intracellular growth of the pathogenic amastigote stage. Eukaryot. Cell 9:22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. El-Fadili AK, Zangger H, Desponds C, Gonzalez IJ, Zalila H, Schaff C, Ives A, Masina S, Mottram JC, Fasel N. 2010. Cathepsin B-like and cell death in the unicellular human pathogen Leishmania. Cell Death Dis. 1:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wemmer KA, Marshall WF. 2004. Flagellar motility: all pull together. Curr. Biol. 14:R992–R993 [DOI] [PubMed] [Google Scholar]
- 24. Bengs F, Scholz A, Kuhn D, Wiese M. 2005. LmxMPK9, a mitogen-activated protein kinase homologue affects flagellar length in Leishmania mexicana. Mol. Microbiol. 55:1606–1615 [DOI] [PubMed] [Google Scholar]
- 25. Erdmann M, Scholz A, Melzer IM, Schmetz C, Wiese M. 2006. Interacting protein kinases involved in the regulation of flagellar length. Mol. Biol. Cell 17:2035–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Portman N, Lacomble S, Thomas B, McKean PG, Gull K. 2009. Combining RNA interference mutants and comparative proteomics to identify protein components and dependences in a eukaryotic flagellum. J. Biol. Chem. 284:5610–5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forestier CL, Machu C, Loussert C, Pescher P, Spath GF. 2011. Imaging host cell-Leishmania interaction dynamics implicates parasite motility, lysosome recruitment, and host cell wounding in the infection process. Cell Host Microbe 9:319–330 [DOI] [PubMed] [Google Scholar]
- 28. Crissman HA, Gadbois DM, Tobey RA, Bradbury EM. 1991. Transformed mammalian cells are deficient in kinase-mediated control of progression through the G1 phase of the cell cycle. Proc. Natl. Acad. Sci. U. S. A. 88:7580–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. 2008. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26:127–132 [DOI] [PubMed] [Google Scholar]
- 30. Parsons M, Worthey EA, Ward PN, Mottram JC. 2005. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 6:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaczanowski S, Sajid M, Reece SE. 2011. Evolution of apoptosis-like programmed cell death in unicellular protozoan parasites. Parasit. Vectors 4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smirlis D, Duszenko M, Ruiz AJ, Scoulica E, Bastien P, Fasel N, Soteriadou K. 2010. Targeting essential pathways in trypanosomatids gives insights into protozoan mechanisms of cell death. Parasit. Vectors 3:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O'Neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. 2005. The genome of the kinetoplastid parasite, Leishmania major. Science 309:436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Imbert L, Ramos RG, Libong D, Abreu S, Loiseau PM, Chaminade P. 2012. Identification of phospholipid species affected by miltefosine action in Leishmania donovani cultures using LC-ELSD, LC-ESI/MS, and multivariate data analysis. Anal. Bioanal Chem. 402:1169–1182 [DOI] [PubMed] [Google Scholar]
- 35. Gonzalez IJ, Desponds C, Schaff C, Mottram JC, Fasel N. 2007. Leishmania major metacaspase can replace yeast metacaspase in programmed cell death and has arginine-specific cysteine peptidase activity. Int. J. Parasitol. 37:161–172 [DOI] [PubMed] [Google Scholar]
- 36. Lee N, Bertholet S, Debrabant A, Muller J, Duncan R, Nakhasi HL. 2002. Programmed cell death in the unicellular protozoan parasite Leishmania. Cell Death Differ. 9:53–64 [DOI] [PubMed] [Google Scholar]
- 37. Zalila H, Gonzalez IJ, El-Fadili AK, Delgado MB, Desponds C, Schaff C, Fasel N. 2011. Processing of metacaspase into a cytoplasmic catalytic domain mediating cell death in Leishmania major. Mol. Microbiol. 79:222–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hassan P, Fergusson D, Grant KM, Mottram JC. 2001. The CRK3 protein kinase is essential for cell cycle progression of Leishmania mexicana. Mol. Biochem. Parasitol. 113:189–198 [DOI] [PubMed] [Google Scholar]
- 39. Diaz-Gonzalez R, Kuhlmann FM, Galan-Rodriguez C, Madeira da Silva L, Saldivia M, Karver CE, Rodriguez A, Beverley SM, Navarro M, Pollastri MP. 2011. The susceptibility of trypanosomatid pathogens to PI3/mTOR kinase inhibitors affords a new opportunity for drug repurposing. PLoS Negl. Trop. Dis. 5:e1297 doi:10.1371/journal.pntd.0001297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kurokawa M, Kornbluth S. 2009. Caspases and kinases in a death grip. Cell 138:838–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, et al. 1995. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 14:6148–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Widmann C, Gibson S, Johnson GL. 1998. Caspase-dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti-apoptotic signals. J. Biol. Chem. 273:7141–7147 [DOI] [PubMed] [Google Scholar]
- 43. Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. 2001. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 3:339–345 [DOI] [PubMed] [Google Scholar]
- 44. Zambon AC, Zhang L, Minovitsky S, Kanter JR, Prabhakar S, Salomonis N, Vranizan K, Dubchak I, Conklin BR, Insel PA. 2005. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc. Natl. Acad. Sci. U. S. A. 102:8561–8566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang L, Zambon AC, Vranizan K, Pothula K, Conklin BR, Insel PA. 2008. Gene expression signatures of cAMP/protein kinase A (PKA)-promoted, mitochondrial-dependent apoptosis. Comparative analysis of wild-type and cAMP-deathless S49 lymphoma cells. J. Biol. Chem. 283:4304–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]