

Abstract
Biofilms are associated with a wide variety of bacterial infections and pose a serious problem in clinical medicine due to their inherent resilience to antibiotic treatment. Within biofilms, persister cells comprise a small bacterial subpopulation that exhibits multidrug tolerance to antibiotics without undergoing genetic change. The low frequency of persister cell formation makes it difficult to isolate and study persisters, and bacterial persistence is often attributed to a quiescent metabolic state induced by toxins that are regulated through toxin-antitoxin systems. Here we mimic toxins via chemical pretreatments to induce high levels of persistence (10 to 100%) from an initial population of 0.01%. Pretreatment of Escherichia coli with (i) rifampin, which halts transcription, (ii) tetracycline, which halts translation, and (iii) carbonyl cyanide m-chlorophenylhydrazone, which halts ATP synthesis, all increased persistence dramatically. Using these compounds, we demonstrate that bacterial persistence results from halted protein synthesis and from environmental cues.
INTRODUCTION
Within every bacterial population there is a small subpopulation that survives when challenged with a lethal antibiotic treatment (1). This subpopulation of cells, known as persisters, exhibits a nonhereditary (1), multidrug tolerance (2). Regrowth of these persisters, by inoculation into fresh media, produces a population both genetically identical to the original culture and equally susceptible to antibiotic treatment (2). Persisters are found in all phases of cell growth, with a frequency of 0.0001 to 0.001% of the population in exponential-phase cultures and as high as 1% in bacterial biofilms and stationary-phase cultures (3, 4). Biofilms consist of an extracellular polymeric matrix, which, in some cases, acts as a physical barrier to protect bacteria from antimicrobials and/or host defense mechanisms and also contains cells with a wide range of metabolic activities (5). Biofilms are associated with numerous infections (6), including recalcitrant infections of cystic fibrosis and gingivitis or infections on indwelling devices (3, 7). Persister cells are the key to biofilm resilience because survival of just a few persisters allows the biofilm to repopulate (8). Persisters also cause recurrence of tuberculosis infections (7), which are particularly difficult to treat in the lungs. Therefore, persister cell research is necessary in order to devise more effective medical treatments to combat infectious diseases.
Treatment of bacterial infections is complicated by both bacterial persistence and resistance, two distinct phenomena occurring through unrelated mechanisms (3). Resistance is caused by genetic variations, which result in the alteration of antibiotic targets to reduce antibiotic binding and efficacy (3). As a result, resistant bacteria are able to survive and even grow in the presence of antibiotics. Persistence is believed to arise from stochastic, physiological differences (9) that induce a state of dormancy or low metabolic activity (3). This is supported by the naturally higher frequency of persister cells in the stationary phase as opposed to the exponential phase. It is thought that persisters neither grow nor die in the presence of antibiotics because antibiotics are unable to corrupt cellular processes in a cell with globally reduced metabolism (3).
Toxin-antitoxin (TA) systems are prevalent in the bacterial genome (at least 37 in Escherichia coli) (10) and are perceived as a redundant, genetic basis for the formation of persisters from normal cells (11, 12). TA systems typically consist of two genes encoding a stable toxin, which disrupts an essential cellular process, and an unstable antitoxin, which mediates the effect of the toxin (7). MqsR/MqsA is one such well-characterized TA system (13–19) that is important to persistence (20–22). MqsR, the toxin, is an RNase (18) which cleaves nearly all mRNA (23), and overproduction of MqsR was shown to significantly increase persistence (20–22); deletion of mqsR resulted in decreased persistence, which was the first time the absence of a toxin was shown to affect persistence (21). TisB/IstR-1 is another TA system in which the toxin TisB decreases ATP levels and induction of tisB transcription through the SOS response increases persistence (24).
Persister cells are typically described as dormant, nondividing cells with globally reduced metabolism (12, 20, 25); however, it is not clear what exactly a persister cell is or what the extent of dormancy is (26). A significant step in understanding the persistence phenotype was the demonstration that cells exhibiting low levels of translation are more likely to be persisters (20). An unstable green fluorescent protein variant was inserted into the chromosome under the control of the ribosomal rrnBP1 promoter, which is highly expressed under conditions of rapid growth (20). Cells from an exponential-phase culture were individually sorted based on the level of fluorescence to isolate the “dimmer” cells with low levels of translation (20). These dimmer cells were 20-fold enriched in persisters, suggesting a correlation between persisters and reduced protein synthesis (20).
Induction of the SOS response by a low-concentration fluoroquinolone pretreatment increased the persister population that tolerated lethal antibiotic exposure (27). This suggests that induced persistence is a side effect of antibiotic treatment and can be triggered as a response to the environment. Based on the significant increase in persistence observed with overproduction of the toxin MqsR (20–22) and with ciprofloxacin as an environmental trigger for the SOS response (27), we hypothesized that perhaps compounds which reduce protein synthesis could be used to mimic the effects of toxins, like MqsR and TisB, to produce persister cells. Hence, in this study, we disrupted protein synthesis via rifampin (28) and tetracycline (29) and disrupted energy production via carbonyl cyanide m-chlorophenylhydrazone (CCCP) (30). Corroborating our hypothesis, use of these compounds to inhibit transcription, inhibit translation, and inhibit ATP synthesis induced persistence in a large percentage of the population (10 to 100%) in comparison to a maximum of ∼1% observed in stationary-phase cultures (3). Based on our results, we conclude that the level of bacterial persistence is proportional to the level of halted protein synthesis and, additionally, have designed a novel method for inducing a high level of persistence within any bacterial population by utilizing a chemical pretreatment. The ability to work with a culture consisting of almost entirely persister cells is a useful tool for future persister research.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used are listed in Table 1. All experiments were conducted at 37°C in Luria-Bertani (LB) medium (31), with shaking at 250 rpm (liquid cultures).
Table 1.
Bacteria used in this study
| E. coli K-12 strain | Description | Source or reference |
|---|---|---|
| BW25113 | lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | 48 |
| MB411 | hsdR514 glnV44 tryT58 lacY1 galK2 galT22 metB1 trpR55 | This study |
| MB4814 | MB411 F′110 metB+ rpoB+ | This study |
| MB4815 | MB411 F′110 metB+ rpoB+ | This study |
| MB4816 | MB411 F′110–1 metB+ rpoB3 (Rifr) | This study |
| MB4817 | MB411 F′110–1 metB+ rpoB3 (Rifr) | This study |
MIC assay.
The MIC of E. coli K-12 BW25113 to rifampin, tetracycline, ampicillin, and ciprofloxacin was determined by incubating freshly inoculated cultures in LB broth for 16 h with various concentrations of each antibiotic and observing inhibition of growth based on lack of turbidity. Experiments were performed with at least three independent cultures.
Persister cell viability assay.
The number of persister cells was determined based on an assessment of cell viability after antibiotic treatment by serially diluting cultures in 0.85% NaCl solution, plating 10 μl drops on LB agar, and counting colonies (32). Both ciprofloxacin (5 μg/ml) (33) and ampicillin (100 μg/ml) (34) treatments were used for 3 h, except for the prolonged treatment with ciprofloxacin where 8 h was used. Concentrations were chosen to be at least 10× the MIC (ciprofloxacin, 0.01 to 0.05 μg/ml; ampicillin, 5 to 10 μg/ml) to minimize survival of potential spontaneous resistant mutants, and treatments lasted 3 h to ensure eradication of nonpersisters. Cultures of E. coli K-12 BW25113 were diluted 1:1,000 in fresh LB broth from a 16-h overnight inoculum and grown to mid-exponential phase (turbidity of 0.8 at 600 nm) before either adding antibiotics (ciprofloxacin or ampicillin) and quantifying the number of persisters or pretreating with compounds that reduce protein synthesis and then adding antibiotics (ciprofloxacin or ampicillin) and quantifying the number of persisters. Pretreated cultures were exposed to rifampin (100 μg/ml for 30 min), tetracycline (50 μg/ml for 30 min), or CCCP (50 μg/ml for 3 h). Cultures were centrifuged and resuspended in fresh LB broth to remove the pretreatment compounds prior to quantifying persister cells after exposure to ciprofloxacin or ampicillin. The pretreatment concentrations and durations were optimized to produce the highest survivability for this study. Concentrations of rifampin (100 μg/ml) and tetracycline (50 μg/ml) were both significantly higher than the MICs, 10 to 20 μg/ml and 0.5 to 1 μg/ml, respectively. Experiments were performed with at least four independent cultures.
Persister revival assay.
The time required for the revival of induced persister cells was determined based on growth curves for cultures after pretreatment. Cultures of E. coli K-12 BW25113 were diluted 1:1,000 in fresh LB broth from a 16-h overnight inoculum and grown to mid-exponential phase (turbidity of 0.8 at 600 nm) before being pretreated with rifampin (100 μg/ml for 30 min), tetracycline (50 μg/ml for 30 min), or CCCP (50 μg/ml for 3 h). Following pretreatment, cultures were centrifuged and resuspended in fresh LB broth to remove the pretreatment compounds and then diluted to a turbidity of 0.1 at 600 nm. Experiments were performed with at least four independent cultures.
Merodiploid construction.
The merodiploid strains were constructed by conjugation between a multiply auxotrophic donor carrying F′110 or F′110-1 and selecting for Met+ prototrophy with MB411. Rifampin resistance was scored for confirmation. Persistence was tested as above, except with 25 μg/ml rifampin pretreatment, a concentration allowing normal growth of the resistant merodiploid, with at least three independent cultures.
RESULTS
For this study, ciprofloxacin and ampicillin were the bactericidal antibiotics chosen for treatments to isolate persister cells. Ciprofloxacin, a fluoroquinolone, binds to DNA and affects the activity of DNA gyrase, inhibiting DNA replication (33). Ciprofloxacin is effective at killing cells in all phases of growth (33), making it an appropriate antibiotic to isolate persister cells regardless of whether pretreatment affects replication. Ampicillin, a β-lactam, binds to transpeptidase and carboxypeptidase, inhibiting cell wall synthesis (34). The disruption of these enzymes leads to lethal defects of the cell wall and also cell lysis during the process of cell replication (34). It should be noted that ampicillin has reduced efficacy in treating nongrowing or slow-growing cells (35). Ampicillin was used in this study, in addition to ciprofloxacin, to isolate persister cells and test for multidrug tolerance of pretreated cultures, indicative of bacterial persistence.
Rifampin pretreatment increases persistence.
Rifampin is a bacteriostatic antibiotic (36) which inhibits mRNA synthesis (37) by binding and inactivating RNA polymerase (28), thereby preventing transcription. We reasoned that rifampin pretreatment could be used to increase persistence by inhibiting transcription, consistent with overproduction of MqsR, which cleaves nearly all mRNA. Hence, we tested whether a pretreatment of E. coli with rifampin would increase persistence. To pretreat the cells, we exposed cells to rifampin (100 μg/ml) during the mid-exponential phase for 30 min. Rifampin pretreatment conditions were optimized for maximum survivability, and altering the concentration and duration led to varied survivability. Our results showed that cultures pretreated with rifampin displayed significantly increased persistence (∼1,000- to 10,000-fold increase over untreated cultures), with 59% of the pretreated population surviving ciprofloxacin treatment and 69% surviving ampicillin treatment (Fig. 1).
Fig 1.
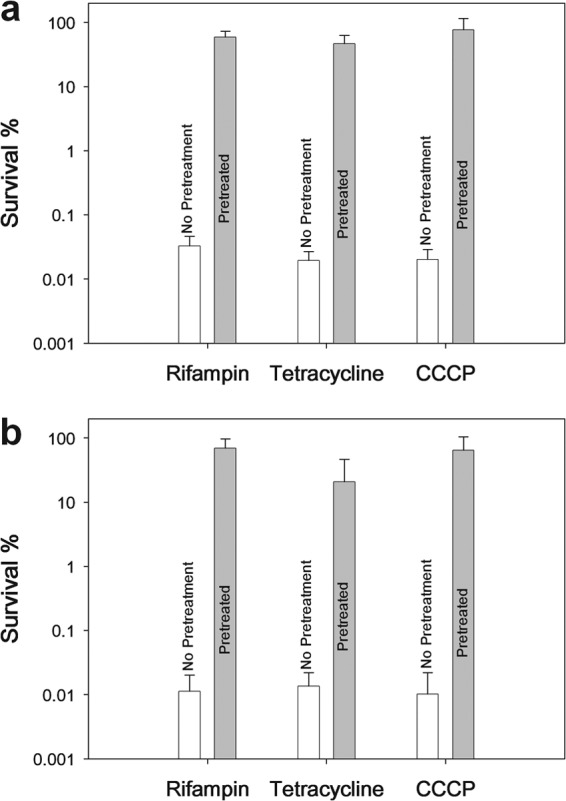
Persistence induced with rifampin, tetracycline, and CCCP pretreatments. Cell survival (%) after treatment with ciprofloxacin (5 μg/ml) (a) and ampicillin (100 μg/ml) (b) for 3 h. Exponential-phase cultures of the E. coli K-12 BW25113 wild type were pretreated with rifampin (100 μg/ml for 30 min), tetracycline (50 μg/ml for 30 min), and CCCP (50 μg/ml for 3 h) prior to antibiotic exposure. Data from four independent cultures are shown along with one standard deviation.
To verify our results, we observed that pretreatment only caused a modest amount of cell death, with 57% of the initial population surviving rifampin exposure, confirming that persistence is not artificially increased by eradication of nonpersisters via pretreatment. We also tested whether the formation of spontaneous resistant mutants affected our results by checking for growth of the persister cultures plated on LB agar plates containing ampicillin or ciprofloxacin, respective of prior treatment. Our results showed that spontaneous resistant mutants contribute to less than 0.0001% of the induced persister population, with no observable growth up to the limit of detection. An important consideration, regarding the choice of antibiotics, is that pretreatment may lead to a state of diminished growth, similar to stationary phase, which would reduce the efficacy of ampicillin. However, increased survival to ciprofloxacin after rifampin treatment confirms the persistence phenotype because ciprofloxacin eradicates cells with reduced growth. Therefore, we achieved a consistent increase in persistence to antibiotics of two separate classes.
Rifampin-induced persisters show a characteristic lag in revival and slow loss of viability.
To provide further evidence that the rifampin pretreatment induced persistence, we performed a revival assay of cultures pretreated with rifampin since persister cells exhibit a lag before resumption of normal growth (25). As expected for persister cells, we found that rifampin-pretreated cultures displayed a lag of ∼4 h (Fig. 2). The delay before growth following exposure to rifampin has previously been described as a “post-antibiotic effect,” caused by the lack of mRNA (38). The lag before resumption of normal growth, reduced levels of mRNA, and increased survival to antibiotics are all characteristics that are associated with both persister cells and rifampin-treated cells. These correlations suggest that the “post-antibiotic effect” of rifampin is physiologically the same as that in persister cells, so we conclude that rifampin pretreatment induces a persister-like state.
Fig 2.
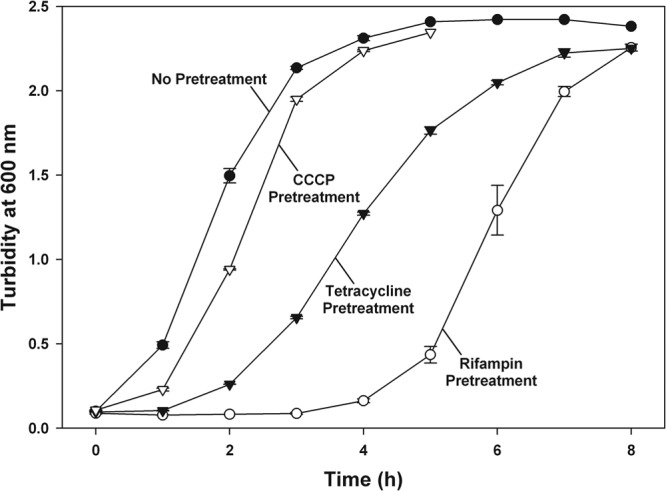
Revival of rifampin-, tetracycline-, and CCCP-induced persisters. Resumption of growth (monitored with turbidity) for persisters subsequent to pretreatments. Exponential-phase cultures of the E. coli K-12 BW25113 wild type were pretreated with rifampin (100 μg/ml for 30 min), tetracycline (50 μg/ml for 30 min), and CCCP (50 μg/ml for 3 h) prior to growth in fresh LB media. Representative data from two independent cultures are shown along with one standard deviation.
Since antibiotic treatment of bacterial cultures produces biphasic cell death, with persister cells surviving extended periods of time, exhibiting slow, steady cell death (39), we tested the rifampin-pretreated cells to see if they demonstrated this slow loss of viability that is characteristic of persister cells. Hence, we exposed pretreated cultures to ciprofloxacin for a prolonged period, quantifying the number of viable cells at several time points. Our results reflect slow, steady cell death for up to 8 h of antibiotic treatment (Fig. 3), which is characteristic of persister cell cultures. Therefore, our rifampin-induced persisters are “true” persisters.
Fig 3.
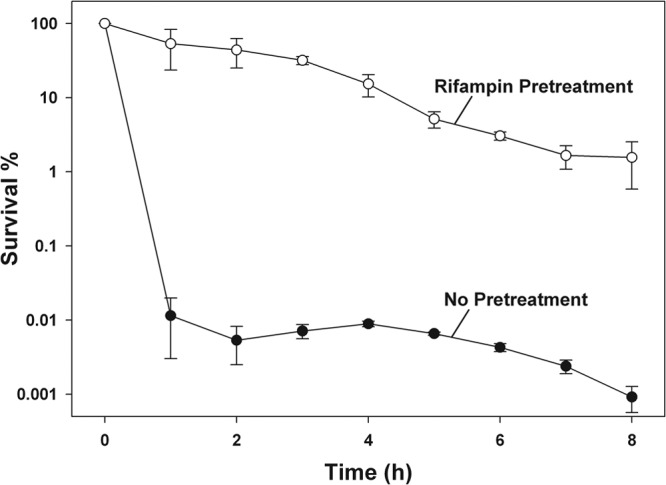
Prolonged antibiotic exposure of rifampin-induced persisters. Cell survival (%) during prolonged treatment with ciprofloxacin (5 μg/ml). Exponential-phase cultures of the E. coli K-12 BW25113 wild type were pretreated with rifampin (100 μg/ml for 30 min) prior to antibiotic exposure. Data from four independent cultures are shown along with one standard deviation.
Rifampin induces persistence by arresting transcription.
Our hypothesis is that rifampin increases persistence by halting protein synthesis via inhibition of transcription. This suggests that rifampin-resistant mutants would no longer show a rifampin-induced increase in persistence. Three independent spontaneous rifampin-resistant mutants behaved consistently with this prediction. These strains showed no effect of rifampin pretreatment, producing less than a 4-fold difference in antibiotic persistence with and without pretreatment in comparison to a 13,000-fold increase in persistence for the pretreated wild-type culture. Rifampin-resistant mutants yielded between 0.0001 to 0.002% persistence, identical to the magnitude of survival for the wild-type parent without pretreatment.
Another important consideration was to check whether the induced persistence is actually the result of arrested transcription, rather than a cellular response from the inhibition of RNA polymerase. Merodiploid strains (two independent strains) containing both wild-type and rifampin-resistant (rpoB+/rpoB3) alleles were compared to the wild type (rpoB+/rpoB+) for persister formation upon rifampin pretreatment. If rifampin induces persistence through inhibition of transcription, then carrying a rifampin-resistant allele will allow continued transcription and should reduce the effect of pretreatment. Without pretreatment, both the rpoB+/rpoB3 and rpoB+/rpoB+ merodiploids showed the same magnitude of antibiotic persistence (0.2 to 2%). However, rifampin pretreatment increased persistence ∼100-fold for the rpoB+/rpoB+ strains, with a reduced effect of ∼10-fold for the rpoB+/rpoB3 strains. Therefore, the presence of a rifampin-resistant allele in the merodiploids reduced persistence ∼10-fold, allowing us to conclude that rifampin pretreatment induces persistence by inhibition of transcription and not via a cellular signal from the wild-type polymerase.
Tetracycline induces persistence similarly to rifampin.
Based on our findings that rifampin induces persistence through inhibited transcription, we decided to test whether inhibiting translation would also induce persistence. We found that pretreatment with tetracycline, a translation inhibitor (29), significantly increased persistence. Cultures were pretreated with tetracycline in the same manner as with rifampin (conditions optimized for maximum survivability), resulting in 47% survival to ciprofloxacin (5 μg/ml) and 21% survival to ampicillin (100 μg/ml) (Fig. 1). A majority of cells (60%) survived the tetracycline exposure, confirming that persistence was not artificially increased and that a reduction in protein synthesis increases persistence.
We also performed a revival assay and found that tetracycline-pretreated cultures displayed a lag in revival of ∼2 h (Fig. 2). The “post-antibiotic effects” of tetracycline (40), a combination of the lag before growth, reduced protein levels (from inhibition of protein synthesis), and increased survival to antibiotics, are consistent with rifampin pretreatment, so we conclude that tetracycline pretreatment also produces persister cells. The corroboration of results from rifampin and tetracycline pretreatments suggests that inhibiting protein synthesis is the key to making persister cells. Preliminary tests with streptomycin (100 μg/ml) and trimethoprim (10 μg/ml) had a similar effect, both displaying ∼1% survival to ampicillin (100 μg/ml), but pretreatments with these compounds were not optimized.
CCCP induces persistence.
After our success at inducing persistence through mechanisms acting similarly to the toxin MqsR, we hypothesized that mimicking the toxin TisB would also induce persistence. Since the toxin TisB decreases cellular ATP (24), we tested pretreatment with the metabolic poison CCCP, an uncoupling agent that inhibits ATP synthesis (30). Using a pretreatment of CCCP to reduce cellular ATP (optimized for maximum survivability), we observed 76% persistence to ciprofloxacin and 64% persistence to ampicillin, similar to the results with rifampin and tetracycline (Fig. 1). As with the other pretreatments, 47% of cells survived CCCP exposure, confirming that persistence was not artificially increased and a reduction in metabolism increases persistence.
CCCP-induced persisters show characteristic lag in revival.
As with the rifampin and tetracycline pretreatments, we performed a revival assay following CCCP pretreatment to characterize the induced survivability as persistence. Our results showed a lag in revival of ∼0.5 to 1 h (Fig. 2) following CCCP pretreatment, which is shorter than that induced by antibiotics. We reason that the recovery time is reduced because CCCP diminishes metabolic activity without directly inhibiting mRNA or protein synthesis. Therefore, we conclude that CCCP pretreatment does in fact induce persistence.
DISCUSSION
Persister cells have frequently been characterized as dormant cells in a state of low metabolism (12, 20, 25), although few experiments have been performed to verify this. Several TA systems have been suggested as the basis of persister cell formation (12, 21, 22). In this study, we have shown that bacterial persistence can be induced with pretreatments that affect transcription and translation. We first attempted to mimic the effects of the toxin MqsR (18) and found that temporary exposure of cells to rifampin, to inhibit transcription, significantly increased subsequent survival to treatment with the bactericidal antibiotics ciprofloxacin and ampicillin (Fig. 1), two different classes of bactericidal antibiotics. This shows that the rifampin-induced survivor cells exhibit multidrug tolerance, which is indicative of bacterial persistence (2). The order of magnitude of these persister cells was consistently between 10 to 100%, a considerably higher level than the ∼0.01% persistence we found in exponentially growing cells (Fig. 1) and the ∼1% persistence observed in stationary-phase cultures (3, 4). This high increase in percentage of persister cells can have several applications, considering the low frequency of naturally forming persisters.
Similarly to our rifampin pretreatment, we found that tetracycline, which binds the ribosome to inhibit translation, also produced a high level of persistence (Fig. 1). Since both rifampin and tetracycline effectively prevent translation, the corroboration of these results shows that lack of protein synthesis is a key to bacterial persistence. Our conclusion is consistent with previous research that indicates a correlation between persister cells and reduced protein synthesis (20). It was shown that cells exhibiting low levels of translation were 20-fold more persistent than a normal culture (20). With our pretreatments, we have induced persistence in nearly 100% of the cell population by blocking protein synthesis (∼1,000- to 10,000-fold increased persistence). Our results suggest that persisters are cells exhibiting essentially a complete lack of protein synthesis.
Pretreating cells with rifampin or tetracycline to induce persistence was effective at imitating the effect of overexpressing the toxin MqsR. However, it is important to note that rifampin and tetracycline block all protein synthesis, while MqsR allows continued translation of 14 mRNA lacking GCU cleavage sites (23) and transcripts whose GCU sites are blocked by mRNA secondary structure or protein binding. Therefore, it seems that naturally formed persisters arrest protein synthesis, except for the continued synthesis of a few select proteins. These specific proteins may have an important role in maintaining the viability of the persisters, or may be coincidental, so further study is warranted.
After success with mimicking the effect of MqsR, we also tried to mimic another toxin linked to persister cell formation, TisB (24). We did this by testing whether the uncoupling agent CCCP had a similar effect on persistence when used as a pretreatment. We found that this metabolic poison induced persistence along the same order of magnitude as that observed for pretreatments with rifampin and tetracycline (Fig. 1). Based on our supposition that bacterial persistence is the general lack of protein synthesis, we postulate that the inhibition of ATP synthesis from CCCP (30) forces the cells to halt metabolic activity and become persister cells. We conclude that any TA system capable of inhibiting protein synthesis, either directly (e.g., inhibition of transcription) or indirectly (e.g., inhibition of ATP synthesis), provides a means for persister cell formation.
Combination therapy with multiple antibiotics is often used to treat infections as an added measure to prevent development of bacterial resistance and to achieve antibiotic synergism (41). Rifampin (42) and tetracycline (43) have often been used in combination therapy against infections from a wide range of microorganisms. The results of this study suggest that the use of these two compounds may induce persistence and lead to a reduced or failed treatment from additional antibiotics.
Our newfound ability to produce a culture consisting almost entirely of persister cells allows utilization of techniques designed to study population-wide characteristics. Previously, the low frequency of persister cells has made it difficult to isolate enough cells for transcriptome and proteome experiments. Using chemical pretreatments to produce high-persister cultures will be useful in studying additional aspects of persistence; for example, we can better study gene expression of persister cells to determine the extent of dormancy required for persistence. Abundant persisters also allow us to more easily study how persister cells revive and determine methods for altering revival. Gaining a better understanding of the extent of dormancy required for persistence and uncovering the dynamics of revival will allow research to progress in designing methods to eliminate persisters.
As with all biological studies, laboratory conditions need to be evaluated with regard to naturally occurring conditions. In this study, chemical pretreatment may introduce unwanted physiological responses that are not relevant to the natural persister phenotype; the pretreatment compounds mimic the activity of toxins, so the high levels of persistence arise as a direct consequence of the chemical pretreatments. Caution is also advised for studying the revival of these chemically induced persisters because it is not known whether resumption of growth is distinct from that of natural persistence (i.e., regulated by TA systems). Therefore, this induced state of dormancy should be regarded as a persister-like state and distinguished from the dormancy of naturally forming persister cells. Other methods may also be used to generate high frequencies of persister cells (e.g., overexpression of a toxin from an inducible plasmid); for example, persistence has been induced in exponential cell cultures through overexpression of mqsR (21), tisB (24), and relE (44) at the same order of magnitude achieved in this study. However, the method developed in this study can be applied to wild-type cultures in nonsupplemented media and provides another viable method to study persisters.
Persister cell formation was first shown to be a stochastic process (25), thought to result from fluctuations in gene expression and protein levels within individual cells of an isogenic population (45). Recent studies have suggested that persistence can also arise as a result of environmental factors, including the presence of ciprofloxacin (24), a bactericidal antibiotic, or indole (46), an intercellular signaling molecule. Many antibiotic compounds are naturally occurring in the environment, are produced by various microorganisms, and are used to provide a competitive advantage against neighboring species (47). The significant increase in persistence we observe, after pretreatment with rifampin, tetracycline, and CCCP, clearly shows several ways in which bacterial persistence is induced via environmental pressure, rather than solely via a stochastic event. Hence, it is apparent that while persistence can result from stochastic fluctuations, it is also directly affected by the presence of numerous extracellular molecules. The response of persister cell formation, when faced with the presence of these natural inhibitory compounds, likely serves as an evolutionarily developed mechanism for bacteria to subsist despite chemical pressure from competing organisms.
ACKNOWLEDGMENTS
This work was supported by the NIH (grant R01 GM089999).
T.K.W. is the Biotechnology Endowed Professor at the Pennsylvania State University.
Footnotes
Published ahead of print 7 January 2013
REFERENCES
- 1. Bigger JW. 1944. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244:497–500 [Google Scholar]
- 2. Wiuff C, Zappala RM, Regoes RR, Garner KN, Baquero F, Levin BR. 2005. Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial populations. Antimicrob. Agents Chemother. 49:1483–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lewis K. 2007. Persister cells, dormancy, and infectious disease. Nat. Rev. Microbiol. 5:48–56 [DOI] [PubMed] [Google Scholar]
- 4. Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322:107–131 [DOI] [PubMed] [Google Scholar]
- 5. Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2:95–108 [DOI] [PubMed] [Google Scholar]
- 6. Wolcott R, Dowd S. 2011. The role of biofilms: are we hitting the right target? Plast. Reconstr. Surg. 127:28S–35S [DOI] [PubMed] [Google Scholar]
- 7. Jayaraman R. 2008. Bacterial persistence: some new insights into an old phenomenon. J. Biosci. 33:795–805 [DOI] [PubMed] [Google Scholar]
- 8. Spoering AL, Lewis K. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183:6746–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lewis K. 2000. Programmed death in bacteria. Microbiol. Mol. Biol. Rev. 64:503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tan Q, Awano N, Inouye M. 2011. YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol. Microbiol. 79:109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X, Wood TK. 2011. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77:5577–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lewis K. 2010. Persister cells. Annu. Rev. Microbiol. 64:357–372 [DOI] [PubMed] [Google Scholar]
- 13. Ren D, Bedzyk LA, Thomas SM, Ye RW, Wood TK. 2004. Gene expression in Escherichia coli biofilms. Appl. Microbiol. Biotechnol. 64:515–524 [DOI] [PubMed] [Google Scholar]
- 14. González Barrios AF, Zuo R, Hashimoto Y, Yang L, Bentley WE, Wood TK. 2006. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 188:305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Kim Y, Hong SH, Ma Q, Brown BL, Pu M, Tarone AM, Benedik MJ, Peti W, Page R, Wood TK. 2011. Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 7:359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Lord DM, Cheng H-Y, Osbourne DO, Hong SH, Sanchez-Torres V, Quiroga C, Zheng K, Herrmann T, Peti W, Benedik MJ, Page R, Wood TK. 2012. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 8:855–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brown BL, Wood TK, Peti W, Page R. 2011. Structure of the Escherichia coli antitoxin MqsA (YgiT/b3021) bound to its gene promoter reveals extensive domain rearrangements and the specificity of transcriptional regulation. J. Biol. Chem. 286:2285–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown BL, Grigoriu S, Kim Y, Arruda JM, Davenport A, Wood TK, Peti W, Page R. 2009. Three dimensional structure of the MqsR:MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog. 5:e1000706 doi:10.1371/journal.ppat.1000706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim Y, Wang X, Zhang X-S, Grigoriu S, Page R, Peti W, Wood TK. 2010. Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ. Microbiol. 12:1105–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shah D, Zhang Z, Khodursky AB, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim Y, Wood TK. 2010. Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 391:209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hong SH, Wang X, O'Connor HF, Benedik MJ, Wood TK. 2012. Bacterial persistence increases as environmental fitness decreases. Microb. Biotechnol. 5:509–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamaguchi Y, Park J-H, Inouye M. 2009. MqsR, a crucial regulator for quorum sensing and biofilm formation, is a GCU-specific mRNA interferase in Escherichia coli. J. Biol. Chem. 284:28746–28753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dörr T, Vulić M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8:e1000317 doi:10.1371/journal.pbio.1000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625 [DOI] [PubMed] [Google Scholar]
- 26. Allison KR, Brynildsen MP, Collins JJ. 2011. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr. Opin. Microbiol. 14:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dörr T, Lewis K, Vulić M. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5:e1000760 doi:10.1371/journal.pgen.1000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wehrli W, Knüsel F, Schmid K, Staehelin M. 1968. Interaction of rifamycin with bacterial RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 61:667–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chopra I, Roberts M. 2001. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 65:232–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cavari BZ, Avi-Dor Y, Grossowicz N. 1967. Effect of carbonyl cyanide m-chlorophenylhydrazone on respiration and respiration-dependent phosphorylation in Escherichia coli. Biochem. J. 103:601–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 32. Donegan K, Matyac C, Seidler R, Porteous A. 1991. Evaluation of methods for sampling, recovery, and enumeration of bacteria applied to the phylloplane. Appl. Environ. Microbiol. 57:51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanders CC. 1988. Ciprofloxacin: in vitro activity, mechanism of action, and resistance. Rev. Infect. Dis. 10:516–527 [DOI] [PubMed] [Google Scholar]
- 34. Fisher JF, Meroueh SO, Mobashery S. 2005. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105:395–424 [DOI] [PubMed] [Google Scholar]
- 35. Tuomanen E, Cozens R, Tosch W, Zak O, Tomasz A. 1986. The rate of killing of Escherichia coli by β-lactam antibiotics is strictly proportional to the rate of bacterial growth. J. Gen. Microbiol. 132:1297–1304 [DOI] [PubMed] [Google Scholar]
- 36. Wehrli W. 1983. Rifampin: mechanisms of action and resistance. Rev. Infect. Dis. 5:S407–S411 [DOI] [PubMed] [Google Scholar]
- 37. Calvori C, Frontali L, Leoni L, Tecce G. 1965. Effect of rifamycin on protein synthesis. Nature 207:417–418 [DOI] [PubMed] [Google Scholar]
- 38. Stubbings W, Bostock J, Ingham E, Chopra I. 2006. Mechanisms of the post-antibiotic effects induced by rifampicin and gentamicin in Escherichia coli. J. Antimicrob. Chemother. 58:444–448 [DOI] [PubMed] [Google Scholar]
- 39. Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230:13–18 [DOI] [PubMed] [Google Scholar]
- 40. Athamna A, Athamna M, Medlej B, Bast DJ, Rubinstein E. 2004. In vitro post-antibiotic effect of fluoroquinolones, macrolides, β-lactams, tetracyclines, vancomycin, clindamycin, linezolid, chloramphenicol, quinupristin/dalfopristin and rifampicin on Bacillus anthracis. J. Antimicrob. Chemother. 53:609–615 [DOI] [PubMed] [Google Scholar]
- 41. Gal K. 1965. Combined antibiotic therapy. Can. Med. Assoc. J. 93:844–847 [PMC free article] [PubMed] [Google Scholar]
- 42. Forrest GN, Tamura K. 2010. Rifampin combination therapy for nonmycobacterial infections. Clin. Microbiol. Rev. 23:14–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roberts MC. 2003. Tetracycline therapy: update. Clin. Infect. Dis. 36:462–467 [DOI] [PubMed] [Google Scholar]
- 44. Maisonneuve E, Shakespeare LJ, Jørgensen MG, Gerdes K. 2011. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U. S. A. 108:13206–13211 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Kint CI, Verstraeten N, Fauvart M, Michiels J. 2012. New-found fundamentals of bacterial persistence. Trends Microbiol. 20:577–585 [DOI] [PubMed] [Google Scholar]
- 46. Vega NM, Allison KR, Khalil AS, Collins JJ. 2012. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 8:431–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fajardo A, Martínez JL. 2008. Antibiotics as signals that trigger specific bacterial responses. Curr. Opin. Microbiol. 11:161–167 [DOI] [PubMed] [Google Scholar]
- 48. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008 doi:10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]