

Abstract
ASP2151 (amenamevir) is a helicase-primase inhibitor against herpes simplex virus 1 (HSV-1), HSV-2, and varicella zoster virus. Here, to determine and analyze the correlation between the pharmacodynamic (PD) and pharmacokinetic (PK) parameters of ASP2151, we examined the PD profile of ASP2151 using in vitro plaque reduction assay and a murine model of HSV-1 infection. ASP2151 inhibited the in vitro replication of HSV-1 with a mean 50% effective concentration (EC50) of 14 ng/ml. In the cutaneously HSV-1-infected mouse model, ASP2151 dose dependently suppressed intradermal HSV-1 growth, with the effect reaching a plateau at a dose of 30 mg/kg of body weight/day. The dose fractionation study showed that intradermal HSV-1 titers were below the detection limit in mice treated with ASP2151 at 100 mg/kg/day divided into two daily doses and at 30 or 100 mg/kg/day divided into three daily doses. The intradermal HSV-1 titer correlated with the maximum concentration of drug in serum (Cmax), the area under the concentration-time curve over 24 h (AUC24h), and the time during which the concentration of ASP2151 in plasma was above 100 ng/ml (T>100). The continuous infusion of ASP2151 effectively decreased intradermal HSV-1 titers below the limit of detection in mice in which the ASP2151 concentration in plasma reached 79 to 145 ng/ml. Our findings suggest that the antiviral efficacy of ASP2151 is most closely associated with the PK parameter T>100 in HSV-1-infected mice. Based on these results, we propose that a plasma ASP2151 concentration exceeding 100 ng/ml for 21 to 24 h per day provides the maximum efficacy in HSV-1-infected mice.
INTRODUCTION
Herpes simplex virus 1 (HSV-1), HSV-2, and varicella-zoster virus (VZV) are widely prevalent pathogens belonging to the human herpesvirus family (1). Both HSV and VZV establish a lifetime latent infection in sensory ganglia after primary infection and eventually reactivate, leading to recurrent disease episodes. HSV-1 and HSV-2 cause genital herpes, herpes labialis, or herpetic keratitis, and frequent disease recurrence dramatically affects the quality of life of afflicted individuals (2). VZV infection is characterized by two distinct disease episodes: varicella as the primary episode and herpes zoster as the recurrent episode (3). As viral gene products, such as DNA polymerase and the helicase-primase complex, are essential for herpesvirus replication, they represent ideal targets for therapeutic agents.
Since the late 1970s, several synthetic nucleoside analogues, including acyclovir (ACV), penciclovir, valaciclovir (VCV), and famciclovir, targeting viral DNA polymerase have been developed for the safe and effective treatment of HSV and VZV infections (4, 5). Recently, a new class of nonnucleoside antivirals, termed herpesvirus helicase-primase inhibitors (HPIs), has been identified (6–8). A few clinical trials with HPIs have been completed (9; http://clinicaltrials.gov/ct2/show/NCT00487682; http://clinicaltrials.gov/ct2/show/NCT01047540; http://clinicaltrials.gov/ct2/show/NCT00486200), with the results suggesting that these drugs may offer another strategy for treating HSV and VZV infection.
Nonclinical pharmacokinetic/pharmacodynamic (PK/PD) analysis has been used to predict the clinical efficacy of drugs and to develop dosing strategies that optimize treatment outcomes for a number of antibacterial and antiviral drugs (10–13). Once the appropriate PK/PD index of a target drug has been identified, the index can be used to predict clinical efficacy or outcomes (14). For example, by employing MIC as a PD susceptibility parameter for antibiotics, three useful indices have been identified: cumulative time in a 24-h period during which the drug concentration exceeds the MIC (T>MIC), maximum concentration of drug in serum divided by the MIC (Cmax/MIC ratio), and area under the concentration-time curve over 24 h in the steady state divided by the MIC (AUC/MIC ratio) (14, 15). For antivirals, however, no standard PD parameter exists for testing antiviral susceptibility. The identification of an appropriate PK/PD marker for antivirals is complicated by the lack of a standardized measurement endpoint for the determination of antiviral efficacy.
Target concentrations of antiretroviral drugs are often based on concentrations required for inhibition of viral replication in vitro (16–18) or by the retrospective review of drug concentrations in plasma in human immunodeficiency virus (HIV)-infected patients and the clinical response of patients (19–21). Although in vitro 50% effective concentration (EC50) values are often used as a PD parameter for antiretroviral drugs, EC95 may represent a superior parameter, because the ultimate goal of treatment is complete viral suppression (22). However, the extrapolation from in vitro effective concentrations to in vivo effective concentrations is complicated by numerous factors, including protein binding rates, tissue penetration, active metabolite formation, and resistance development. Therefore, it is necessary to conduct in vivo animal studies to identify an appropriate PD correlation parameter. Such studies are possible for drugs targeting HSV, as a few animal infection models are available, including a well-characterized mouse zosteriform spread model of HSV infection (23, 24). However, a PD parameter that correlates with the efficacy of anti-HSV drugs has not been adequately defined.
Recently, we reported the discovery of ASP2151, a novel HPI, which had potent antiviral activity against HSVs both in vitro and in vivo (6, 25–27). Here, we conducted PK, dose fractionation, and continuous-infusion studies in an HSV-1-infected murine model to define the PK and PD of ASP2151 and to analyze the correlation between PK parameters and the in vivo anti-HSV-1 activity of ASP2151. Using this approach, we attempted to identify an appropriate PK/PD parameter to predict the efficacy of ASP2151 against HSV infection.
MATERIALS AND METHODS
Antiviral compounds.
ASP2151 (molecular weight, 482.55; international nonproprietary name, amenamevir) was synthesized at Astellas Pharma Inc. (Tokyo, Japan). ACV was purchased from Sigma-Aldrich (St. Louis, MO).
Viruses and cell lines.
HSV-1 strains CI-25, CI-114, and CI-116 and HSV-2 strains CI-27 and CI-5243, clinically isolated in the United States, were kindly provided by Nancy Sawtell (Children's Hospital Medical Center, Cincinnati, OH). HSV-1 strains KOS and WT51, HSV-2 strains G, Lyon, and Kondo, and human embryonic fibroblast (HEF) and African green monkey kidney Vero cells were provided by Rational Drug Design Laboratories (Fukushima, Japan). HEF and Vero cells were grown in Eagle's minimum essential medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin G, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). HSVs were propagated using HEF cells cultured in maintenance medium containing 2% FBS.
Plaque reduction assay.
The antiviral activities of ASP2151 and ACV against HSVs were tested using a plaque reduction assay, as described previously (6). Briefly, HEF cells were seeded into multiwell plates and incubated until they formed a monolayer. After the medium was removed, the cells were infected with HSV-1 or HSV-2, and the plates were further incubated for 1 h at 37°C. The cells were washed twice with maintenance medium and then treated with the test compound until clear plaques appeared. The cells were then fixed with 10% formalin in phosphate-buffered saline, stained with a 0.02% crystal violet solution, and the number of plaques was determined under a light microscope. The EC50, which represents the concentration of test compound needed to reduce the plaque number by 50%, was calculated using nonlinear regression analysis with a sigmoid-maximum effect (Emax) model.
Mouse HSV infection model.
All animal experimental procedures were approved by the Animal Ethical Committee of Astellas Pharma, Inc. Female hairless mice (HOS:HR-1, 7 to 8 weeks old) were infected with a suspension of HSV-1 strain WT51 (15 μl/mouse; titer, 2 × 108 PFU/ml) or CI-116 (15 μl/mouse; titer, 4 × 107 PFU/ml) in the dorsolateral skin stripped as a small square using a needle, under anesthesia. The day of HSV-1 infection was designated day zero postinfection.
Dose fractionation study.
Total daily doses of 1, 3, 10, 30, or 100 mg/kg/day ASP2151 were orally administered to HSV-1-infected mice (n = 5) for 5 days. ASP2151 treatments were started 2 to 3 h after HSV infection either as a single daily dose (every 24 h, q24h) or as two (every 12 h, q12h) or three (every 8 h, q8h) divided doses. Lesion scores and intradermal HSV-1 titers were measured on day 5 postinfection, as described below.
Continuous-infusion study.
Mice in each group (n = 10) were implanted subcutaneously with an Alzet miniosmotic pump (model 2001; ALZA Corp., Palo Alto, CA) filled with ASP2151 solution (1, 3, 10, and 20 mg/g in polyethylene glycol 400 [PEG 400]) or vehicle at 1 to 2 h before infection. It had been preliminarily confirmed that the way of infusion kept ASP2151 concentration in plasma constant at levels corresponding to ASP2151 solutions used over the study period. Based on the pumping rate (23.28 μl/day) and mean animal body weight (20.4 g), the daily dosage of ASP2151 for the groups infused with 1-, 3-, 10-, and 20-mg/g ASP2151 solution corresponded to approximately 1.1, 3.3, 11, and 22 mg/kg of body weight/day, respectively. Infusion was continued for 5 days. Lesion scores, intradermal HSV-1 titers, and ASP2151 concentrations in plasma were measured on day 5 postinfection, as described below.
Measurement of lesion scores, intradermal HSV-1 titers, and concentration of ASP2151 in plasma.
Disease severity for each animal was scored on a composite scale from 0 to 7 based on the severity of zosteriform lesions and general symptoms according to previously published criteria (6). After scoring disease symptoms, skin and/or blood samples from each animal were obtained. Skin samples were homogenized in 10 ml ice-cold saline, and debris was then removed by centrifugation. The HSV-1 titer in each sample was determined using a plaque assay. The concentration of ASP2151 in plasma obtained from mouse blood was determined using the liquid chromatography-tandem mass spectrometry (LC/MS/MS) method.
Simulation of PK after repeated oral administration of ASP2151.
To estimate PK parameters after repeated oral dosing of ASP2151, concentrations of ASP2151 in plasma were measured after a single oral administration to mice. ASP2151 was administered to mice (n = 3) at doses of 0.33, 3, 30, and 100 mg/kg, and blood samples were then collected from the heart at 0.25, 0.5, 1, 2, 4, 8, and 12 h postdosing. The concentrations in plasma after repeated administration of ASP2151 were simulated by the nonparametric superposition method using WinNonlin software (Pharsight, Mountain View, CA), and data were obtained from the single administration of ASP2151 at a dose of 30 mg/kg.
PK/PD modeling.
The relationship between the steady-state PK parameters of peak concentration (Cmax), area under the concentration-time curve (AUC24 h), and time above the threshold concentration and the antiviral efficacy of ASP2151 was examined using the sigmoid-Emax model (28, 29). The threshold concentration was the concentration of ASP2151 required to completely inhibit the growth of HSV-1 in the dose fractionation experiment.
The PK parameters were fitted to the mean intradermal HSV-1 titer (log10 PFU/skin) using the sigmoid dose-response (variable slope) program of GraphPad Prism (GraphPad Software, San Diego, CA). The following equation was used: y = bottom + (top − bottom)/(1 + 10[(log EC50− x) × hill slope]), where x is the logarithm of each PK variable and y is the response intradermal HSV-1 titer (log10 PFU/skin). The “top” value was fixed as the mean value for the vehicle groups, and the “bottom” value was fixed as the log10 250 value, which represents the detection limit value of the HSV-1 titer.
Statistical analyses.
In the plaque reduction assay, results were analyzed using the Student t test for comparisons between ASP2151 and ACV. In the continuous-infusion study, Dunnett's multiple-comparison test was applied to compare mean values between the vehicle and ASP2151-infused groups. P values of <0.05 were considered statistically significant. All statistical analyses were performed using SAS software (SAS Institute, Carey, NC).
RESULTS
In vitro antiviral activity of ASP2151.
The antiviral activities of ASP2151 and ACV against 10 HSV-1 and HSV-2 strains in HEF cells were examined using a plaque reduction assay. Table 1 summarizes the EC50s of ASP2151 and ACV for each of the examined HSV strains. ASP2151 inhibited the replication of the clinical strains isolated in Japan and the United States as well as the laboratory-stocked strains. The mean EC50s of ASP2151 against HSV-1 and HSV-2 were 14 (range, 7.7 to 20) and 30 ng/ml (range, 15 to 58), respectively, whereas those of ACV were 29 (range, 18 to 38) and 71 ng/ml (range, 45 to 95), respectively. The EC50s of ASP2151 against HSV strains were significantly lower than those of ACV.
Table 1.
Anti-herpes simplex virus 1 and 2 activities of ASP2151 and acyclovir in HEF cells
| Virus straina | Mean EC50 ± SE (ng/ml)b |
|
|---|---|---|
| ASP2151 | Acyclovir | |
| HSV-1 | ||
| KOS | 18 ± 1.9 | 36 ± 0.0 |
| WT51 | 20 ± 1.0 | 34 ± 2.3 |
| CI-25 | 14 ± 2.4 | 38 ± 4.5 |
| CI-114 | 8.7 ± 1.9 | 21 ± 3.4 |
| CI-116 | 7.7 ± 0.48 | 18 ± 2.3 |
| HSV-2 | ||
| G | 27 ± 2.9 | 92 ± 20 |
| Lyon | 58 ± 0.0 | 79 ± 6.8 |
| Kondo | 31 ± 2.9 | 45 ± 2.3 |
| CI-27 | 17 ± 2.4 | 95 ± 18 |
| CI-5243 | 15 ± 0.97 | 45 ± 9.0 |
The virus strains used in this study consisted of three laboratory stock strains (KOS, G, and Lyon), two clinical isolates from Japan (WT51 and Kondo), and five clinical isolates from the United States (CI-25, CI-114, CI-116, CI-27, and CI-5243).
The antiviral activity (EC50) was determined by a plaque reduction assay in infected HEF cells. The data are from four independent experiments using each strain. All results for ASP2151 were significantly different from results for acyclovir (P < 0.05, Student's t test).
Effect of ASP2151 on intradermal HSV-1 titer.
Following the cutaneous infection of mice with HSV-1 strain WT51, cutaneous lesions developed beginning on day 4 postinfection, and intradermal HSV-1 titers rapidly increased, peaking on days 4 to 5 postinfection (Fig. 1). In this model, almost all mice infected with HSV-1 strain WT51 in the vehicle group are dead from day 6 postinfection and show neurological signs, while ASP2151 and VCV reduce the mortality of HSV-1-infected mice (6). Lesion scores and intradermal HSV-1 titers in the mice infected with another HSV-1 strain, CI-116, showed almost similar time courses (data not shown). Using the cutaneously HSV-1-infected mouse model, a repeated oral dose of ASP2151 was administered three times daily at total doses of 3, 10, 30, 100, and 300 mg/kg/day for 3 days from day 4 postinfection. The skin of treated mice was collected, and the intradermal HSV-1 titer was then determined using the plaque method (Fig. 2).
Fig 1.
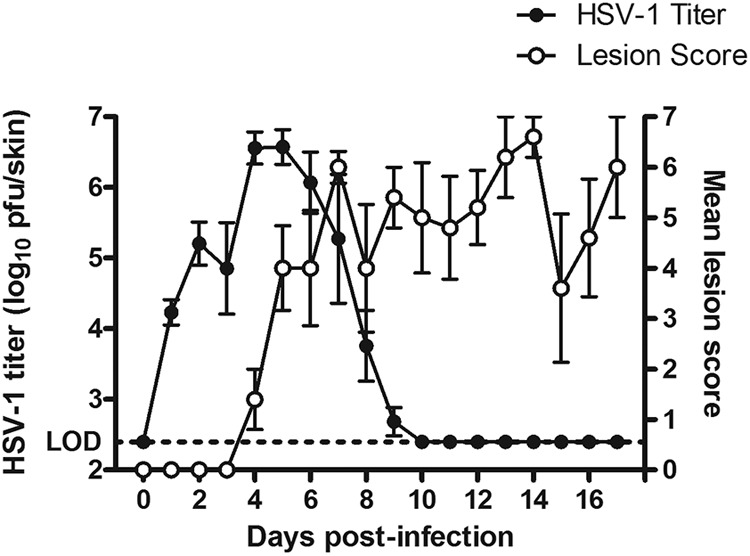
Time course (days postinfection) of changes in lesion score and intradermal virus titer in HSV-1-infected mice. Mice were infected with HSV-1 strain WT51 in the dorsolateral skin. Lesion scores (0 to 7) were determined for each animal on day 5 postinfection. Intradermal HSV-1 titers (expressed as log10 PFU/skin) were determined by plaque assay on day 5 postinfection. Data represent the means ± standard errors from 1 to 5 animals for lesion scores and HSV-1 titers. The dotted line indicates the lower limit of detection (LOD, 2.40 log10 PFU/skin) for the titration assay. The lesion scores and intradermal HSV-1 titers were plotted versus days postinfection.
Fig 2.
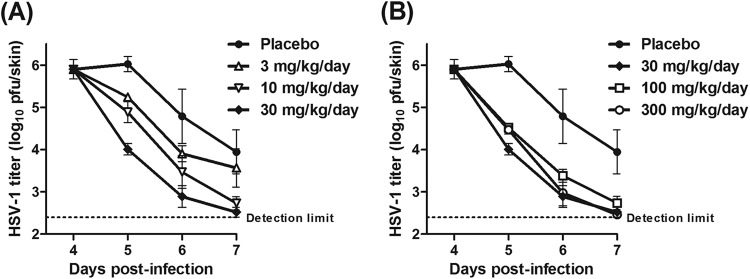
Reducing effect of ASP2151 on intradermal HSV-1 titers. HSV-1-infected mice were orally treated for 3 days from day 4 postinfection with vehicle or ASP2151 at 3, 10, and 30 mg/kg/day (A) and at 30, 100, and 300 mg/kg/day (B) administered as three daily doses. Intradermal HSV-1 titers (log10 PFU/skin) were determined by plaque assay on days 4, 5, 6, and 7 postinfection. Data are expressed as means ± standard errors from four or five animals. The dotted line indicates the lower detection limit (2.40 log10 PFU/skin) for the titration assay.
ASP2151 administration accelerated the reduction in virus titer in a dose-dependent manner in the range of 3 to 30 mg/kg/day (Fig. 2A). However, the efficacy of ASP2151 was comparable at doses of 30 mg/kg/day and higher (Fig. 2B), suggesting that ASP2151 completely suppressed HSV-1 growth at the dose of 30 mg/kg/day.
The goal in treating herpetic disease is to prevent disease progress and accelerate the cure of the virus infection-induced lesions by suppressing viral growth at an early stage. Accordingly, the clinical efficacy of ASP2151 can be considered to have maximal effect at the dose that completely suppresses viral growth. However, it is difficult to estimate the dosing regimen of antiviral agents needed for viral suppression in clinical studies based solely on experiments in murine models due to possible differences in PK patterns between mice and humans. For this reason, we conducted a series of nonclinical PK/PD analyses to estimate the plasma ASP2151 concentration (PK/PD parameters) at which HSV-1 growth would be completely suppressed.
Dose fractionation study in HSV-1-infected mice.
Dose fractionation efficacy studies in animal models are a useful approach to elucidate PK/PD relationships of a target drug, as different patterns of plasma drug concentrations can result from dose fractionation, even on administration of identical daily doses (14, 30). As part of the PK/PD analyses of ASP2151, we examined the effect of daily dose fractionation on the antiviral efficacy of ASP2151 in the HSV-1-infected mouse model. ASP2151 doses of 1, 3, 10, 30, and 100 mg/kg/day were administered to mice for 5 days from the day of HSV-1 infection either as a single dose or as two or three divided doses. Mean HSV-1 titers and lesion scores were then assessed on day 5 postinfection (Fig. 3). ASP2151 treatment decreased both lesion scores and HSV-1 titers in a dose-dependent manner, irrespective of the dosing interval. Lesion development was not observed in mice treated with the following ASP2151 dosages and administration intervals: 30 and 100 mg/kg/day every 24 hours (q24h); 10, 30, and 100 mg/kg/day q12h; or 30 and 100 mg/kg/day q8h (Fig. 3A). HSV-1 titers were close to the lower detection limit in infected mice treated with ASP2151 at dosages of 100 mg/kg/day q12h and 30 or 100 mg/kg/day q8h (Fig. 3B).
Fig 3.

Effect of ASP2151 on lesion development and intradermal virus titer in HSV-1-infected mice treated with various doses of ASP2151 administered in a single or two or three divided doses. HSV-1-infected mice were orally administered placebo (vehicle) or ASP2151 (1, 3, 10, 30, or 100 mg/kg/day) either as a single dose (q24h) or as two (q12h) or three (q8h) divided doses for 5 days starting 2 to 3 h after HSV-1 infection. (A) A lesion score (0 to 7) was determined for each animal on day 5 postinfection. (B) Intradermal HSV-1 titer (log10 PFU/skin) was determined by plaque assay on day 5 postinfection. Data are expressed as the means ± standard errors from four or five animals. (C) Intradermal HSV-1 titers were plotted versus lesion scores for the corresponding animals. The dotted line indicates the lower detection limit (2.40 log10 PFU/skin) for the titration assay.
The relationship between lesion score and HSV-1 titer in each animal on day 5 postinfection is shown in Fig. 3C. Animals with an HSV-1 titer below 5 log10 PFU/skin did not develop cutaneous lesions (score, 0), indicating that HSV-1 titer was more sensitive than lesion score as a direct PD marker of HSV-1 infection. These results suggested that dividing the daily dose of ASP2151 had greater efficacy in reducing HSV-1 titer than did the same daily dose as a single dose.
PK of ASP2151 in mice.
Concentrations of ASP2151 in plasma after a single oral administration to mice at doses of 0.33, 3, 30, and 100 mg/kg increased in proportion to the dose and reached Cmax 0.25 to 1.0 h after dosing (Fig. 4A). Doses of 0.33, 3, 30, and 100 mg/kg resulted in Cmax of 50, 646, 5,199, and 14,903 ng/ml, and AUC0–∞ values of 0.22, 2.37, 21.97, and 56.12 μg · h/ml, respectively. The measured concentration in plasma after a single oral administration of ASP2151 at 30 mg/kg was analyzed using a one-compartment model. The simulated concentrations of ASP2151 in plasma at dosages of 100 mg/kg/day q24h, 100 mg/kg/day q12h, and 30 mg/kg/day q8h are shown in Fig. 4B. The trough concentration of ASP2151 in plasma with the dosage regimens (30 mg/kg/day q8h and 100 mg/kg/day q12h) that reduced the intradermal HSV-1 titers to below the lower limit of detection was estimated to be approximately 100 ng/ml.
Fig 4.
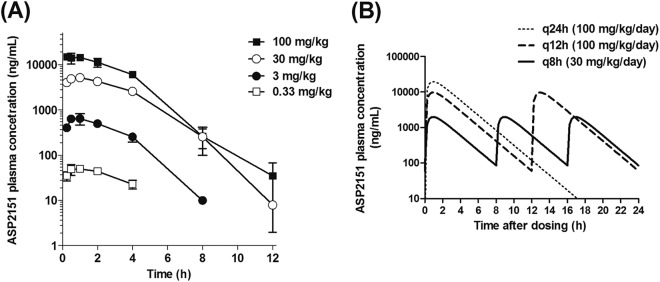
Concentrations of ASP2151 in plasma after single or repeated oral administration to mice. (A) Concentrations of ASP2151 in plasma were determined at 0.25, 0.5, 1, 2, 4, 8, and 12 h after a single oral administration to mice at doses of 0.33, 3, 30, and 100 mg/kg. Data are expressed as the means ± standard deviations for two or three animals. (B) Simulation of concentrations in plasma after repeated oral administration. The concentration of ASP2151 in plasma measured after a single oral administration at a dose of 30 mg/kg was analyzed using the one-compartment model. Based on the result, the ASP2151 concentration in plasma after repeated administration was simulated at 15-min intervals. Simulated concentrations of ASP2151 in plasma at dosages of 100 mg/kg/day q24h, 100 mg/kg/day q12h, and 30 mg/kg/day q8h are plotted.
Estimation of the PK/PD parameters of ASP2151 at which HSV-1 growth is completely suppressed.
To determine the plasma ASP2151 concentration at which HSV-1 growth is completely suppressed in mice, Cmax, AUC24h, and time during which the plasma concentration is 100 ng/ml or higher (T>100) per day were estimated for each dosage regimen used in the dose fractionation study. The correlation between these values and the intradermal HSV-1 titer was then analyzed with an equation of the sigmoid Emax model. The intradermal HSV-1 titer correlated with the Cmax, AUC24h, and T>100 with coefficient of determination (R2) values of 0.7665, 0.9005, and 0.8227, respectively (Fig. 5). Based on the correlation curves, the PK parameters at which HSV-1 growth was completely suppressed by oral administration of ASP2151 were estimated to be 10,000 ng/ml or higher for Cmax, 60 μg · h/ml or higher for AUC24h, and 21 to 24 h for T>100 (Fig. 5).
Fig 5.
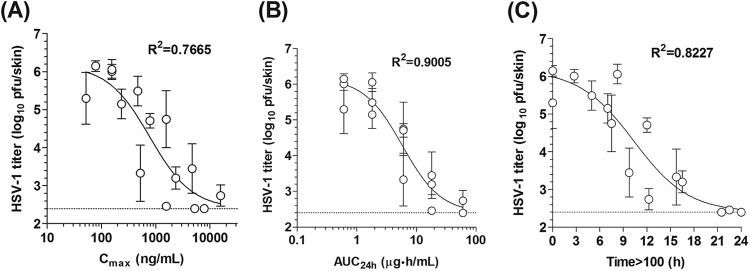
Relationship between intradermal HSV-1 titer and three pharmacokinetic (PK) parameters. The PK parameters Cmax, AUC24h, and time above 100 ng/ml (Time>100) were calculated under conditions of repeated ASP2151 administration either as a single dose or as two or three divided doses (1, 3, 10, 30, and 100 mg/kg/day) in mice. The PK parameters were plotted versus intradermal HSV-1 titers for animals treated with each regimen. Correlations were analyzed using a sigmoid-Emax model. Data are expressed as the means ± standard errors for five animals. The dotted line and R2 indicate the lower limit of detection (2.40 log10 PFU/skin) for the titration assay and the coefficient of determination, respectively.
Although AUC24h had the highest R2, complete inhibition of HSV-1 replication appeared to be achieved with less variation in T>100 than that for AUC24h (Fig. 5). In addition, ASP2151 had the best efficacy on three-times-daily dosing in the dose fractionation study, although the AUC24h did not vary between the three dosing regimens, which used the same total daily dose (Fig. 3A and B). These findings require additional research to confirm which PK parameter, AUC24h or T>100, is a more reliable predictor of drug efficacy; however, such a determination is difficult, as a strong interrelationship exists between them.
PK/PD parameters at which HSV-1 growth is completely suppressed by continuous ASP2151 infusion.
The validity of the three PK parameters (Cmax, AUC24h, and T>100) was further investigated by changing the dosing method from oral to continuous subcutaneous infusion. For the experiment, an osmotic pump filled with ASP2151 solution (1, 3, 10, and 20 mg/g in PEG 400) was subcutaneously implanted in HSV-1-infected mice from 1 to 2 h before virus infection and was used to continuously infuse ASP2151 at a constant rate for 5 days. Intradermal HSV-1 titer and concentration of ASP2151 in plasma were then determined for each animal (Table 2 and Fig. 6). The mean concentration of ASP2151in plasma at 5 days postinfection increased in a dose-dependent manner, with doses of 3 mg ASP2151/g or higher significantly reducing the intradermal HSV-1 titer (Table 2). When the HSV-1 titer was plotted versus the ASP2151 concentration in plasma, the intradermal HSV-1 titers were clearly below the limit of detection in all mice whose plasma ASP2151 concentrations were 145 ng/ml or higher (Fig. 6). The results of the continuous-infusion study suggested that a constant concentration of 79 to 145 ng/ml ASP2151 in plasma was necessary to completely suppress HSV-1 growth (Fig. 6).
Table 2.
Concentration of ASP2151 in plasma and intradermal HSV-1 titers 5 days after starting subcutaneous infusion to HSV-1-infected micea
| Dosing solution amt of ASP2151 (mg/g) | Doseb (mg/kg/day) | Mean ASP2151 concn in plasma (ng/ml) | Minimum–maximum concn (ng/ml) | Mean intradermal HSV-1 titerc (log10 PFU/skin) |
|---|---|---|---|---|
| None (vehicle) | 5.5 | |||
| 1 | 1.1 | 17 | 9–24 | 5.3 |
| 3 | 3.3 | 60 | 36–79 | 4.8* |
| 10 | 11 | 235 | 145–307 | 2.4* |
| 20 | 22 | 316 | 28–627 | 2.6* |
Each group consisted of 10 animals.
Calculated from the mean body weight and daily output from the pump.
The limit of detection of the intradermal HSV-1 titer was 2.4 log10 PFU/skin; *, significantly different from the vehicle group at P values of <0.05 (Dunnett's multiple comparison test).
Fig 6.
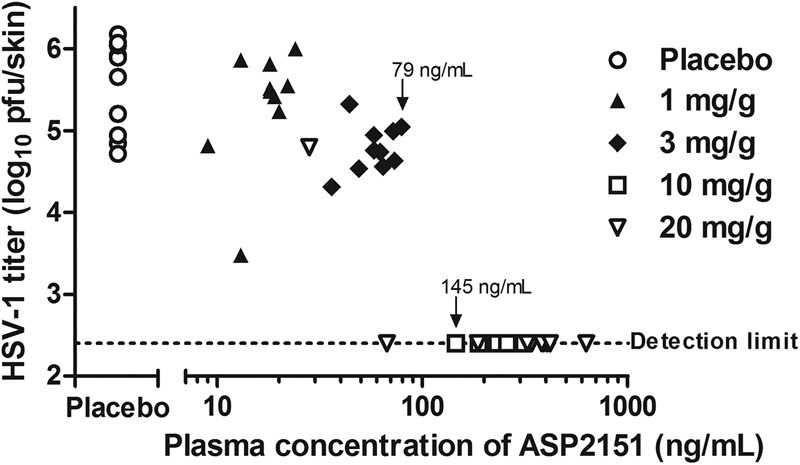
Relationship between intradermal HSV-1 titer and ASP2151 concentration in plasma in mice continuously infused with ASP2151. HSV-1-infected mice (10 animals per group) were infused for 5 days with placebo (vehicle) or ASP2151 solution (1, 3, 10, or 20 mg/g dissolved in PEG 400) starting 1 to 2 h before HSV-1 infection. Intradermal HSV-1 titer (log10 PFU/skin) was determined by plaque assay at day 5 postinfection and was then plotted for each animal. The dotted line indicates the lower limit of detection (2.40 log10 PFU/skin) for the assay. The highest maintained concentration in plasma at which the intradermal HSV-1 titer could be detected and the lowest maintained concentration in plasma to reduce the intradermal HSV-1 titer below the detection limit are indicated (79 and 145 ng/ml, respectively).
Most appropriate PK parameter to predict complete inhibition of viral replication by ASP2151.
To determine the most appropriate PK parameter to predict the complete inhibition of viral replication in mice by ASP2151, the Cmax, AUC24h, and T>100 values at which HSV-1 growth was completely suppressed in mice were compared between the oral and continuous-infusion routes of administration. The predicted values for oral administration and continuous infusion of ASP2151 were 10,000 and 145 ng/ml for Cmax, 60 and 3.48 μg · h/ml for AUC24h, and 21 to 24 h and 24 h for T>100, respectively (Table 3). As T>100 showed consistent values in the two different dosing methods, the T>100 of 21 to 24 h was suggested to indicate the concentration of ASP2151 in plasma needed to completely suppress intradermal virus growth in the cutaneous HSV-1 infection model.
Table 3.
Minimum values of Cmax, AUC24h, and T>100 to reduce the intradermal HSV-1 titer to below the detection limit on oral administration and continuous infusion of ASP2151 in HSV-1-infected mice
| Route of ASP2151 treatment | Cmax (ng/ml) | AUC24h (μg · h/ml) | T>100 (h) |
|---|---|---|---|
| Oral administrationa | 10,000 | 60 | 21–24 |
| Continuous infusionb | 145 | 3.48c | 24 |
The values for oral administration were estimated from fitted sigmoid curves.
For continuous infusion, a constant ASP2151 concentration in plasma of 145 ng/ml, which was the lowest concentration to reduce the intradermal HSV-1 titer below the limit of detection, was used to calculate the PK parameter.
AUC24h was calculated by multiplying 145 ng/ml by 24 h.
DISCUSSION
In the present study, we attempted to identify an appropriate PK/PD marker for predicting the efficacy of ASP2151 by investigating the effect of dose-fractionated oral and continuous subcutaneous administration on the antiviral activity of ASP2151 in cutaneously HSV-1-infected mice. Our results revealed that the PK/PD parameter T>100, which is the length of time the ASP2151 concentration in plasma exceeds 100 ng/ml, most accurately predicted the efficacy of ASP2151 with respect to the complete inhibition of HSV-1 replication. The identified PK/PD parameter may be of utility for determining dosing regimens during the clinical development of ASP2151 for treating herpesvirus infections.
This study focuses on infectious virus titer in the skin for analyzing the PK-PD relationship to explore the PK benchmark at which ASP2151 is expected to completely inhibit viral replication in mice. It was suggested that cutaneous infection of hairless mice with HSV-1 caused not only skin lesions but also abnormalities of neurons, in which infectious virus replication was transiently observed with titer-time kinetics similar to those of the skin, peaking on day 4 postinfection (31). In this study, since virus replication in neurons was not evaluated, our estimation might be limited to predict effective PK just in the skin. In our histopathological and immunohistochemical studies, however, ASP2151 at a dosage of 100 mg/kg/day q12h, one of the dosing regimens to completely inhibit HSV replication in the skin, exhibited complete protection of neuronal tissue from pathological destruction caused by HSV-1 infection (our unpublished data). The PK-PD relation in the nervous system could not be significantly different from that in skin, although further evaluations are warranted to obtain an accurate estimation.
A number of physiological factors, including protein binding and tissue distribution, affect drug efficacy in vivo and should be considered when attempting to predict the clinical efficacy of a target drug. Understanding PK/PD parameters may allow for the correction of differences in PK and intrinsic antimicrobial activity, as the PK goal of antimicrobial therapy is to achieve adequate drug concentrations in target tissues (14). Here, we determined that the protein-binding ratio of ASP2151 in plasma is nearly the same for mice and for humans (approximately 77% and 75%, respectively) and that the ASP2151 concentration in the plasma and skin of mice given radiolabeled ASP2151 is also similar (data not shown). As the PK/PD parameter values required for efficacy are expected to be similar among animal species (14), these physiological factors likely had minimal impact on the present PK/PD analysis of ASP2151.
To predict the clinical efficacy of ASP2151, comparison with VCV, which is the standard therapeutic agent for HSV and VZV infections, is highly informative. The PK/PD parameter to best estimate the efficacy of VCV was also determined using the HSV-1-infected mouse model (see Fig. S1 in the supplemental material). We detected a clear correlation (R2, 0.8559) between HSV-1 titer and the PK parameter time above 0.01 μg/ml of ACV, which is the reported 50% inhibitory concentration (IC50) of ACV against HSV isolated from patients with genital herpes (32), in the plasma of mice treated with VCV. Our findings are consistent with the results of a PK/PD study that found the time above the in vitro ACV IC50 for HSV-2 replication to be the most relevant PK parameter for predicting the efficacy of VCV in terms of the prevention of recurrent HSV eruptions in patients with genital herpes (33). In addition, a few reports regarding the use of VCV and ACV in humans have shown that the duration that the ACV concentration in plasma remains above a given threshold is an important criterion for antiviral efficacy (34–36). Although differences existed in the PD parameters and analysis conditions between these previous reports and our present nonclinical PK/PD analysis, the results obtained using a murine infection model are consistent with those of clinical PK/PD analysis.
To describe the association between drug exposure and treatment efficacy, PD parameters based on drug activity (MIC), such as T>MIC, Cmax/MIC, and AUC/MIC, have been used for antibiotics in various organisms (14, 37, 38). As herpes simplex and herpes zoster are caused by HSV-1 or HSV-2 and VZV, respectively, the target concentration of ASP2151 in plasma should be estimated by considering the difference between the degree of anti-HSV-1 activity and that of the anti-HSV-2 and anti-VZV activities. Here, the mean EC50s of ASP2151 against 5 strains of HSV-1 and HSV-2 in HEF cells were 14 and 30 ng/ml, respectively (Table 1). ASP2151 is also reported to display anti-VZV activity in HEF cells, with a mean EC50 of 30 ng/ml (0.063 μM) for 9 ACV-susceptible VZV strains (6). As the activity of ASP2151 against HSV-2 and VZV is approximately 2-fold lower than that against HSV-1, we speculate that HSV-2 and VZV growth can be completely suppressed using a dosage regimen resulting in a T>200 of close to 24 h per day. Consequently, the target concentration of ASP2151 in plasma for treating herpes simplex and herpes zoster is estimated to be 200 ng/ml.
Based on the present nonclinical PK/PD analyses and observed concentrations of ASP2151 in plasma in healthy volunteers who participated in a phase I study, the target center dose of ASP2151 for genital herpes (usually caused by HSV-2) and herpes zoster in phase II clinical studies was set to 200 mg/patient, which was estimated to give approximately 21 h in terms of T>200. The clinical efficacy of ASP2151 was confirmed in the phase II clinical studies of recurrent genital herpes and herpes zoster, in which the efficacy of ASP2151 reached a plateau level at all examined doses (100, 200, and 400 mg) (9; http://clinicaltrials.gov/ct2/show/NCT00487682). Indeed, the concentrations in plasma of most samples were >200 ng/ml in patients dosed with 100 to 400 mg ASP2151. Therefore, our nonclinical PK/PD analyses using the HSV-1-infected mouse model was able to accurately predict the appropriate clinical dose of ASP2151 for treating herpesvirus infections.
In conclusion, our nonclinical PK/PD analyses of the novel helicase-primase inhibitor ASP2151 suggest that in vivo dose fractionation and continuous-infusion studies examining intradermal virus titers in HSV-infected mice might provide useful nonclinical PK/PD information for predicting effective clinical dose regimens for antiherpes drugs.
ACKNOWLEDGMENTS
We thank Nancy Sawtell for providing HSV clinical isolates from the United States. We also thank Naoko Kojima for experimental technical support.
This work was supported by and conducted at Astellas Pharma, Inc.
Footnotes
Published ahead of print 28 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01803-12.
REFERENCES
- 1. Pellet PE, Roizman B. 2007. The family Herpesviridae: a brief introduction, p 2479–2499 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Roizman B, Knipe D, Whitley R. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Levin MJ, Schmader K. 2007. Prevention strategies: herpes zoster, post-herpetic neuralgia and immunogenicity. Herpes 14(Suppl 2):45–47 [PubMed] [Google Scholar]
- 4. De Clercq E. 1993. Antivirals for the treatment of herpesvirus infections. J. Antimicrob. Chemother. 32:121–132 [DOI] [PubMed] [Google Scholar]
- 5. De Clercq E, Field HJ. 2006. Antiviral prodrugs—the development of successful prodrug strategies for antiviral chemotherapy. Br. J. Pharmacol. 147:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chono K, Katsumata K, Kontani T, Kobayashi M, Sudo K, Yokota T, Konno K, Shimizu Y, Suzuki H. 2010. ASP2151, a novel helicase-primase inhibitor, possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J. Antimicrob. Chemother. 65:1733–1741 [DOI] [PubMed] [Google Scholar]
- 7. Crute JJ, Grygon CA, Hargrave KD, Simoneau B, Faucher Bolger A-MG, Kibler P, Liuzzi M, Cordingley MG. 2002. Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease. Nat. Med. 8:386–391 [DOI] [PubMed] [Google Scholar]
- 8. Kleymann G, Fischer R, Betz UA, Hendrix M, Bender W, Schneider U, Handke G, Eckenberg P, Hewlett G, Pevzner V, Baumeister J, Weber O, Henninger K, Keldenich J, Jensen A, Kolb J, Bach U, Popp A, Maben J, Frappa I, Haebich D, Lockhoff O, Rubsamen-Waigmann H. 2002. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 8:392–398 [DOI] [PubMed] [Google Scholar]
- 9. Tyring S, Wald A, Zadeikis N, Dhadda S, Takenouchi K, Rorig R. 2012. ASP2151 for the treatment of genital herpes: a randomized, double-blind, placebo- and valacyclovir-controlled, dose-finding study. J. Infect. Dis. 205:1100–1110 [DOI] [PubMed] [Google Scholar]
- 10. Bilello JA, Bauer G, Dudley MN, Cole GA, Drusano GL. 1994. Effect of 2′,3′-didehydro-3′-deoxythymidine in an in vitro hollow-fiber pharmacodynamic model system correlates with results of dose-ranging clinical studies. Antimicrob. Agents Chemother. 38:1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jumbe N, Louie A, Leary R, Liu W, Deziel MR, Tam VH, Bachhawat R, Freeman C, Kahn JB, Bush K, Dudley MN, Miller MH, Drusano GL. 2003. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Invest. 112:275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Louie A, Kaw P, Liu W, Jumbe N, Miller MH, Drusano GL. 2001. Pharmacodynamics of daptomycin in a murine thigh model of Staphylococcus aureus infection. Antimicrob. Agents Chemother. 45:845–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Preston SL, Piliero PJ, Bilello JA, Stein DS, Symonds WT, Drusano GL. 2003. In vitro-in vivo model for evaluating the antiviral activity of amprenavir in combination with ritonavir administered at 600 and 100 milligrams, respectively, every 12 hours. Antimicrob. Agents Chemother. 47:3393–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26:1–10 [DOI] [PubMed] [Google Scholar]
- 15. Mouton JW, Dudley MN, Cars O, Derendorf H, Drusano GL. 2005. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: an update. J. Antimicrob. Chemother. 55:601–607 [DOI] [PubMed] [Google Scholar]
- 16. Burger D, Hugen P, Reiss P, Gyssens I, Schneider M, Kroon F, Schreij G, Brinkman K, Richter C, Prins J, Aarnoutse R, Lange J. 2003. Therapeutic drug monitoring of nelfinavir and indinavir in treatment-naive HIV-1-infected individuals. AIDS 17:1157–1165 [DOI] [PubMed] [Google Scholar]
- 17. Durant J, Clevenbergh P, Garraffo R, Halfon P, Icard S, Del Giudice P, Montagne N, Schapiro JM, Dellamonica P. 2000. Importance of protease inhibitor plasma levels in HIV-infected patients treated with genotypic-guided therapy: pharmacological data from the Viradapt Study. AIDS 14:1333–1339 [DOI] [PubMed] [Google Scholar]
- 18. Fletcher CV, Anderson PL, Kakuda TN, Schacker TW, Henry K, Gross CR, Brundage RC. 2002. Concentration-controlled compared with conventional antiretroviral therapy for HIV infection. AIDS 16:551–560 [DOI] [PubMed] [Google Scholar]
- 19. Gatti G, Pontali E, Boni S, De Pascalis CR, Bassetti M, Bassetti D. 2002. The relationship between ritonavir plasma trough concentration and virological and immunological response in HIV-infected children. HIV Med. 3:125–128 [DOI] [PubMed] [Google Scholar]
- 20. Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, Buclin T. 2001. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 15:71–75 [DOI] [PubMed] [Google Scholar]
- 21. Sadler BM, Gillotin C, Lou Y, Stein DS. 2001. Pharmacokinetic and pharmacodynamic study of the human immunodeficiency virus protease inhibitor amprenavir after multiple oral dosing. Antimicrob. Agents Chemother. 45:30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drusano GL, Bilello JA, Preston SL, O'Mara E, Kaul S, Schnittman S, Echols R. 2001. Hollow-fiber unit evaluation of a new human immunodeficiency virus type 1 protease inhibitor, BMS-232632, for determination of the linked pharmacodynamic variable. J. Infect. Dis. 183:1126–1129 [DOI] [PubMed] [Google Scholar]
- 23. de Clercq E. 1984. Topical treatment of cutaneous herpes simplex virus infection in hairless mice with (E)-5-(2-bromovinyl)-2′-deoxyuridine and related compounds. Antimicrob. Agents Chemother. 26:155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simmons A, Nash A. 1984. Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J. Virol. 52:816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Himaki T, Masui Y, Chono K, Daikoku T, Takemoto M, Haixia B, Okuda T, Suzuki H, Shiraki K. 2012. Efficacy of ASP2151, a helicase-primase inhibitor, against thymidine kinase-deficient herpes simplex virus type 2 infection in vitro and in vivo. Antiviral Res. 93:301–304 [DOI] [PubMed] [Google Scholar]
- 26. Katsumata K, Chono K, Sudo K, Shimizu Y, Kontani T, Suzuki H. 2011. Effect of ASP2151, a herpesvirus helicase-primase inhibitor, in a guinea pig model of genital herpes. Molecules 16:7210–7223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Katsumata K, Weinberg A, Chono K, Takakura S, Kontani T, Suzuki H. 2012. Susceptibility of herpes simplex virus isolated from genital herpes lesions to ASP2151, a novel helicase-primase inhibitor. Antimicrob. Agents Chemother. 56:3587–3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Derendorf H, Meibohm B. 1999. Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives. Pharm. Res. 16:176–185 [DOI] [PubMed] [Google Scholar]
- 29. Holford NH, Sheiner LB. 1981. Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin. Pharmacokinet. 6:429–453 [DOI] [PubMed] [Google Scholar]
- 30. Craig WA. 1995. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn. Microbiol. Infect. Dis. 22:89–96 [DOI] [PubMed] [Google Scholar]
- 31. Wharton SB, Meyers NL, Nash AA. 1995. Experimental herpes simplex virus type 1 (HSV-1) infection of the spinal cord and dorsal root ganglia. Neuropathol. Appl. Neurobiol. 21:228–237 [DOI] [PubMed] [Google Scholar]
- 32. McLaren C, Corey L, Dekket C, Barry DW. 1983. In vitro sensitivity to acyclovir in genital herpes simplex viruses from acyclovir-treated patients. J. Infect. Dis. 148:868–875 [DOI] [PubMed] [Google Scholar]
- 33. Patel R, Bell AR. 1998. Valaciclovir for the suppression of recurrent genital herpes: relationship between efficacy and pharmacokinetic parameters. Antiinfect. Drugs Chemother. 16:19 [Google Scholar]
- 34. Saiag P, Praindhui D, Chastang C. 1999. A double-blind, randomized study assessing the equivalence of valacyclovir 1000 mg once daily versus 500 mg twice daily in the episodic treatment of recurrent genital herpes. J. Antimicrob. Chemother. 44:525–531 [DOI] [PubMed] [Google Scholar]
- 35. Spruance SL, Tyring SK, DeGregorio B, Miller C, Beutner K. 1996. A large-scale, placebo-controlled, dose-ranging trial of peroral valaciclovir for episodic treatment of recurrent herpes genitalis. Arch. Intern. Med. 156:1729–1735 [PubMed] [Google Scholar]
- 36. Tod M, Lokiec F, Bidault R, De Bony F, Petitjean O, Aujard Y, Grp APF. 2001. Pharmacokinetics of oral acyclovir in neonates and in infants: a population analysis. Antimicrob. Agents Chemother. 45:150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andes D, Craig WA. 2002. Animal model pharmacokinetics and pharmacodynamics: a critical review. Int. J. Antimicrob. Agents 19:261–268 [DOI] [PubMed] [Google Scholar]
- 38. Drusano GL. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat. Rev. Microbiol. 2:289–300 [DOI] [PubMed] [Google Scholar]