

Abstract
Blue light has attracted increasing attention due to its intrinsic antimicrobial effect without the addition of exogenous photosensitizers. However, the use of blue light for wound infections has not been established yet. In this study, we demonstrated the efficacy of blue light at 415 nm for the treatment of acute, potentially lethal Pseudomonas aeruginosa burn infections in mice. Our in vitro studies demonstrated that the inactivation rate of P. aeruginosa cells by blue light was approximately 35-fold higher than that of keratinocytes (P = 0.0014). Transmission electron microscopy revealed blue light-mediated intracellular damage to P. aeruginosa cells. Fluorescence spectroscopy suggested that coproporphyrin III and/or uroporphyrin III are possibly the intracellular photosensitive chromophores associated with the blue light inactivation of P. aeruginosa. In vivo studies using an in vivo bioluminescence imaging technique and an area-under-the-bioluminescence-time-curve (AUBC) analysis showed that a single exposure of blue light at 55.8 J/cm2, applied 30 min after bacterial inoculation to the infected mouse burns, reduced the AUBC by approximately 100-fold in comparison with untreated and infected mouse burns (P < 0.0001). Histological analyses and terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays indicated no significant damage in the mouse skin exposed to blue light at the effective antimicrobial dose. Survival analyses revealed that blue light increased the survival rate of the infected mice from 18.2% to 100% (P < 0.0001). In conclusion, blue light therapy might offer an effective and safe alternative to conventional antimicrobial therapy for P. aeruginosa burn infections.
INTRODUCTION
Burns are one of the most common and devastating forms of trauma (1–4). Data from the National Center for Injury Prevention and Control in the United States show that approximately 2 million fires are reported each year, resulting in 1.2 million people with burn injuries. Significant thermal injuries induce a state of immunosuppression that predisposes burn patients to infectious complications (1). Burn infection is one of the most important and potentially serious complications that occur in the acute period following injury (2, 3, 5). Over the last several decades, Gram-negative organisms have emerged as the most common etiologic agents of invasive infection by virtue of their large repertoire of virulence factors and antimicrobial resistance traits (1, 6). Among Gram-negative organisms, Pseudomonas aeruginosa is one of the more feared bacterial pathogens because it is often resistant to current therapeutic modalities (5). Emerging antimicrobial resistance trends in burn wound bacterial pathogens represent a serious therapeutic challenge for clinicians caring for burn patients (1). As a result, a major research effort has been led to find alternative antimicrobial approaches to which, it is hypothesized, bacteria will not be easily able to develop resistance. In a recent paper published in Nature Reviews Microbiology, Karen Bush and a group of 30 scientists from academia and industry pointed out that, “The investigation of novel non-antibiotic approaches for the prevention of and protection against infectious diseases should be encouraged, and such approaches must be high-priority research and development projects” (7).
As a nonantibiotic approach, the development of light-based antimicrobial therapy, including photodynamic therapy (PDT) (8–13) and UVC irradiation therapy (14–19), has been extensively investigated as an alternative to conventional antibiotics. An advantage of light-based antimicrobial therapies includes their equal killing effectiveness regardless of antibiotic resistance. However, two major disadvantages of PDT as a two-part (photosensitizer plus light) combination approach are the challenge of introducing photosensitizers into certain bacteria (20) and into infected tissues and the less-than-perfect selectivity of many photosensitizers for microbial cells over host tissue. The use of UVC irradiation, on the other hand, has different limitations due to its detrimental effects on mammalian cells and possible damage to host tissue, including carcinogenesis (17).
Another novel light-based approach, blue light therapy, is attracting increasing attention due to its intrinsic antimicrobial effect without the addition of exogenous photosensitizers (21–25). In addition, it is accepted that blue light is much less detrimental to mammalian cells than UV irradiation (26, 27). Blue light has already been used clinically for the treatment of inflammatory acne (28–31). However, the efficacy of blue light for wound infections has not been established. The majority of the publications on the antimicrobial effect of blue light have been confined to in vitro studies (22–24, 32, 33). There have been (rather surprisingly) no published preclinical or clinical reports to demonstrate blue light therapy for wound infections.
In this study, we investigated the use of blue light for mouse burns infected with Pseudomonas aeruginosa. To the best of our knowledge, this study is the first in vivo study on the use of blue light for wound infections.
MATERIALS AND METHODS
Light source.
The light source we used was an Omnilux Clear-U light-emitting diode (LED) array (Photo Therapeutics, Inc., Carlsbad, CA) that emitted blue light at a center wavelength of 415 nm with a full width at half-maximum (FWHM) of 20 nm (Fig. 1). The irradiance of blue light on the target surface was adjusted by manipulating the distance between the LED array aperture and the target (cell culture or mouse burns), and it was measured using a PM100D power/energy meter (Thorlabs, Inc., Newton, NJ).
Fig 1.
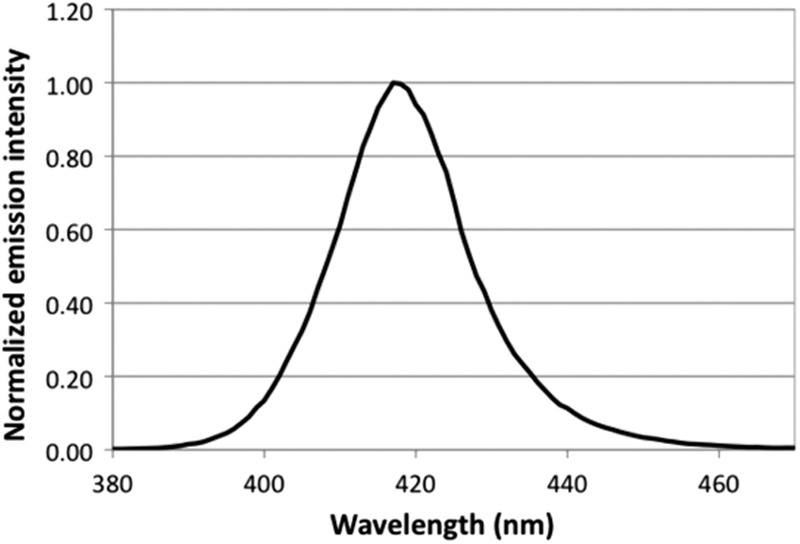
Emission spectrum of Omnilux blue LED.
Pseudomonas aeruginosa strain and culture conditions.
The P. aeruginosa strain that we used was ATCC 19660 (strain 180), which causes septicemia after intraperitoneal injection (34) and has been shown to be invasive in mice with skin burns (35). The stable bioluminescent variant (strain Xen 05) carried the entire bacterial lux operon integrated into its chromosomes for stable luciferase expression, which allowed it to be used for bioluminescent imaging (strain Xen 05 was a kind donation from Xenogen Inc., Alameda, CA) (36).
The bacteria were grown in brain heart infusion (BHI) medium supplemented with 50 μg/ml kanamycin in an orbital incubator (37°C, 100 rpm) to an optical density of 0.6 to 0.8 at 600 nm, which corresponds to approximately 108 cells/ml. The suspension was centrifuged, washed with phosphate-buffered saline (PBS), and resuspended in PBS at the same density for experimental use.
Keratinocytes and culture conditions.
Human keratinocytes (HaCaT) (37) were cultured in 75-cm3 tissue culture flasks in 20 ml Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) (Sigma, St. Louis, MO). Cells were incubated at 37°C in 95% air-5% CO2 in a humidified incubator for 2 to 3 days until the cell monolayer became confluent. The growth medium was replaced every 3 days. Upon reaching at least 70% confluence, the cells were washed with PBS and trypsinized for 10 min at 37°C with 0.25% trypsin-0.02% EDTA (Sigma). The cell suspension was centrifuged, washed with PBS, and resuspended in HEPES buffer (catalog no. A14291 DJ; Life Technologies Corp., Grand Island, NY) to a defined cell density (measured by hemocytometer) for experimental use.
Blue light inactivation of P. aeruginosa in vitro.
Three milliliters P. aeruginosa suspension at 108 CFU/ml in PBS was placed into 35-mm petri dishes. The suspension was irradiated with a blue light LED array at an irradiance of 19.5 mW/cm2 with the lid of the petri dish removed. During the irradiation, the P. aeruginosa suspension was stirred by a mini-magnetic bar (Fisher Scientific Co., Norcross, GA). Aliquots of 40 μl of the suspension were withdrawn at 0, 12, 24, 48, 72, and 96 min, respectively, when 0, 14.0, 28.0, 56.1, 84.2, and 109.9 J/cm2 blue light had been delivered. The numbers of CFU were then determined by serial dilutions on BHI agar plates according to the method of Jett et al. (38). Colonies were allowed to grow for 18 to 24 h at 37°C. The experiments were performed in triplicate.
Blue light irradiation of keratinocytes in vitro.
Three milliliters keratinocyte suspension at 106 cell/ml in HEPES buffer was placed into 35-mm petri dishes at room temperature (21°C). The suspension was irradiated with the blue light LED array at an irradiance of 19.5 mW/cm2 with the lid of the petri dish removed. During the irradiation, the keratinocyte suspension was stirred by a mini-magnetic bar. Aliquots of 40 μl of the suspension were withdrawn at 0, 12, 24, 48, 72, and 96 min, respectively, when 0, 14.0, 28.0, 56.1, 84.2, and 109.9 J/cm2 blue light had been delivered. Viable counts were determined immediately by mixing each sample with an equal volume of 0.4% (wt/vol) trypan blue and transferring the mixture to a hemocytometer. The cell survival percentage was calculated as the ratio of the number of viable cells (unstained cells) to the total number of cells. The experiments were performed in triplicate.
Transmission electron microscopy.
Untreated and blue light-treated P. aeruginosa cells were fixed in 2.5% glutaraldehyde plus 2% paraformaldehyde immediately after blue light illumination and stored overnight at 4°C. After spinning down (1,200 rpm, 10 min) and decanting the fixative, 0.1 M sodium cacodylate buffer (pH 7.2) was added to the pellets. After fixation, hot agar (2% in distilled water, heated to boiling) was immediately added to each pellet. As soon as the agar had solidified, the cell pellets were then processed routinely, as any other tissue, for transmission electron microscopy (TEM). The cell pellets were postfixed in 2% OsO4 in sodium cacodylate buffer, dehydrated in a graded alcohol series, and embedded in Epon t812 (Tousimis, Rockville, MD). Ultrathin sections were cut on a Reichert-Jung Ultracut E microtome (Vienna, Austria), collected on uncoated 200-mesh copper grids, stained with uranyl acetate and lead citrate, and examined on a Philips CM-10 transmission electron microscope (Eindhoven, The Netherlands).
Fluorescence spectroscopy.
To identify the porphyrins within P. aeruginosa cells, an overnight P. aeruginosa culture was centrifuged, washed with PBS, and centrifuged again, and then the supernatant was removed. The P. aeruginosa pellets were added to 1 ml of a mixture of 0.1 M NaOH-1% sodium dodecyl sulfate (SDS) and allowed to stand in the dark for 1 day. Fluorescence of the dissolved P. aeruginosa pellets in NaOH-SDS (in a cuvette 1 cm thick) was measured on a fluorimeter (FluoroMax 3; SPEX Industries, Edison, NJ), with excitation set at 405 nm and emission scanned from 580 to 700 nm.
P. aeruginosa burn infection in mice.
Adult 7- to 8-week-old female BALB/c mice weighing 17 to 21 g were obtained from Charles River Laboratories (Wilmington, MA). The animals were housed one per cage with access to food and water ad libitum and were maintained on a 12-hour light-dark cycle at a room temperature of around 21°C and a relative humidity range of 30 to 70%. All animal procedures were approved by the Subcommittee on Research Animal Care (IACUC) of the Massachusetts General Hospital and were in accordance with the guidelines of the National Institutes of Health (NIH).
Before the creation of burns, the mice were anesthetized by intraperitoneal (i.p.) injection of a ketamine-xylazine cocktail and shaved on the dorsal surfaces. Burns were incurred by applying a preheated (≈95°C) brass block to the dorsal surface of each mouse for 3 s, resulting in nonlethal full-thickness third-degree burns measuring approximately 1.2 cm by 1.2 cm. Five min after incurrence of the burn, a 60-μl bacterial suspension containing 3 × 106 CFU was topically applied to the eschar of each burn.
Bioluminescence imaging.
The setup consisted of an intensified charge-coupled-device (ICCD) camera (model C2400-30H; Hamamatsu Photonics, Bridgewater, NJ), a camera controller, an imaging box, an image processor (C5510-50; Hamamatsu), and a color monitor (PVM 1454Q; Hamamatsu). Light-emitting diodes are mounted inside the imaging box and supply the light required for obtaining dimensional imaging of the sample. Under the photo-counting mode, a clear image can be obtained even at extremely low-light levels by detecting and integrating individual photons one by one.
Prior to imaging, the mice were anesthetized by i.p. injections of a ketamine-xylazine cocktail. The mice were then placed on an adjustable stage in the specimen chamber, and the infected burns were positioned directly under the camera. A grayscale background image of each wound was made, and this was followed by a photon count of the same region. This entire burn photon count was quantified as relative luminescence units (RLUs) and was displayed in a false-color scale ranging from pink (most intense) to blue (least intense).
Blue light therapy of mouse burns infected with P. aeruginosa.
Blue light was initiated at 30 min after bacterial inoculation with the irradiance of 14.6 mW/cm2. The mice were given a total light exposure of up to 55.8 J/cm2 in aliquots with bioluminescence imaging taking place after each aliquot of light. To record the time course of the extent of bacterial infection, the bacterial luminescence from mouse burns was measured daily after blue light therapy until the infections were cured (characterized by the disappearance of bacterial luminescence) or the burns were healed.
TUNEL assays.
Mouse skin was exposed to blue light at an antimicrobial dose of 55.8 J/cm2. Skin biopsy specimens were taken before and at 0, 24, and 48 h after blue light exposure. The biopsy samples were preserved in 10% phosphate-buffered formalin (Fisher Scientific Co.) for 18 to 24 h, processed, and then embedded in paraffin. Serial 4-μm-thick tissue sections were subjected to a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay using the FragEL DNA fragmentation detection kit (EMD Millipore, MA) according to the manufacturer's instructions. Briefly, following deparaffinization and rehydration, sections were permeabilized with proteinase K for 20 min, incubated with a reaction mixture containing terminal deoxynucleotidyl transferase (TdT) and fluorescein-labeled and unlabeled deoxynucleotides for 2 h at room temperature, washed with Tris-buffered saline (TBS), and coverslipped with mounting medium including 4′,6-diamidino-2-phenylindole (DAPI) (SlowFade Gold anti-fade reagent; Invitrogen, CA). Negative controls were treated by substituting distilled water (dH2O) for the TdT enzyme in the reaction mixture. Stained samples were observed by confocal microscopy (FluoView FV1000-MPE; Olympus Corporation, Tokyo, Japan) by using fluorescein isothiocyanate (FITC) as the fluor and DAPI as the nuclear counterstain. Images were acquired using Olympus FluoView FV10-ASW software (version 3.0a, Olympus Corporation).
Since penetration depth is less than 1 mm for blue light and is therefore confined to the epidermis (37), we only examined the epidermal cell DNA fragmentation by TUNEL staining.
Statistical analyses.
The cell inactivation rates (slopes of the survival curves) were compared for statistical significance using a Student t test. In a two-dimensional coordinate system, the area-under-the-bioluminescence-curve (AUBC) data, which represent the time courses of bacterial luminescence of the mouse burns and also a common approach for the analysis of antimicrobial effects of drugs (39), were calculated using numerical integration (40). The difference in the AUBC between the untreated control and the blue light-treated groups was also compared for statistical significance using a Student t test. Kaplan-Meier survival curves were compared by the use of a log rank test. P values of <0.05 were considered significant for all statistical analyses.
RESULTS
Blue light selectively inactivated P. aeruginosa in vitro over keratinocytes.
The results for the blue light inactivation of P. aeruginosa and keratinocytes in vitro are shown in Fig. 2. The inactivation curves approximately followed first-order kinetics (41), a linear relation between the log-transformed cell survival fraction log10(N/N0) and blue light exposure H, i.e., log10(N/N0) = −kHH, where N is the CFU count at the blue light exposure H, N0 is the initial CFU count, and kH is the cell inactivation rate coefficient (or the slope of the inactivation curve) (42).
Fig 2.
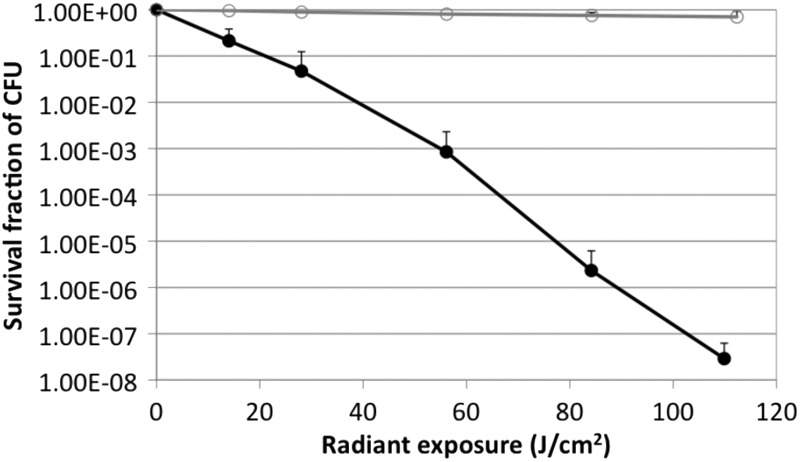
Dose response of blue light inactivation of P. aeruginosa (●) and keratinocytes (○) in vitro.
When 109.9 J/cm2 blue light had been delivered (96 min of illumination at an irradiance of 19.5 mW/cm2), an approximately 7.64-log10-cycle CFU inactivation of P. aeruginosa was achieved. In contrast, the inactivation rate for HaCaT was much lower than that for P. aeruginosa under the same blue light irradiation condition. When 109.9 J/cm2 blue light had been delivered, only a 0.16-log10-cycle loss of viability of HaCaT was observed (Fig. 1), resulting in a 7.48-log10 inactivation selectivity of P. aeruginosa cells over HaCaT. The mean inactivation rate coefficients (kH) of P. aeruginosa and HaCaT were 0.067 and 0.002 cm2/J, respectively, indicating an approximately 34-fold higher inactivation rate of P. aeruginosa by blue light than HaCaT (P = 0.0014).
Transmission electron microscopy (Fig. 3) revealed apparent steps in blue light-mediated inactivation of P. aeruginosa, beginning with the development of vacuoles within the cytoplasm (Fig. 3B), the release of cytoplasmic material to the surrounding environment (Fig. 3C), and, finally, significant cytoplasmic disruption (Fig. 3D).
Fig 3.

Transmission electron microscopy of P. aeruginosa cells. (A) Untreated P. aeruginosa cells. Bar = 100 nm. (B to D) P. aeruginosa cells after being exposed to 109.9 J/cm2 blue light: development of vacuoles within the cytoplasm (B) (bar = 100 nm), release of cytoplasmic material to the surrounding environment (C) (bar = 500 nm), and complete disappearance of cytoplasm (D) (bar = 100 nm).
Intracellular coproporphyrin III and/or uroporphyrin III was associated with the blue light inactivation of P. aeruginosa.
The fluorescence spectrum (excitation at 405 nm) of the P. aeruginosa cells dissolved in NaOH-SDS is shown in Fig. 4. The spectra peaked at 613 and 667 nm, which are very close to the typical fluorescence emissions of coproporphyrin III and uroporphyrin III at the same excitation of 405 nm (43), suggesting that coproporphyrin III and/or uroporphyrin III within the P. aeruginosa cells was the photosensitizing chromophore associated with the antimicrobial effect of blue light.
Fig 4.
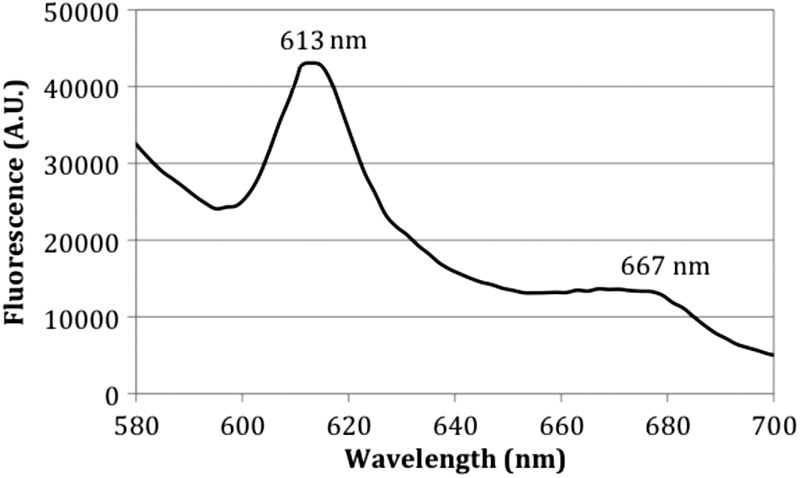
Fluorescence spectra of P. aeruginosa cell pellets from overnight culture dissolved in NaOH-SDS. Excitation wavelength, 405 nm.
Blue light rescued mice from otherwise lethal P. aeruginosa burn infection.
Figure 5A and B show the successive bioluminescence images of representative full-thickness mouse burns (1.2 cm by 1.2 cm) infected with 3 × 106 CFU of luminescent P. aeruginosa, with and without blue light therapy, respectively. Blue light (415 nm) was delivered at 30 min after bacterial inoculation. Bacterial luminescence was completely eliminated after 55.8 J/cm2 blue light had been delivered (62 min of illumination at an irradiance of 14.6 mW/cm2) (Fig. 5A), while in the untreated mouse burn, infection steadily developed with time (Fig. 5B), and the mouse died at 72 h (day 3) after bacterial inoculation. Luminescent P. aeruginosa was detected in the blood culture of the dead mouse, indicating that the mouse died of sepsis.
Fig 5.

(A and B) Successive bacterial luminescence images of representative mouse burns infected with 3 × 106 CFU of luminescent P. aeruginosa with and without blue light (415 nm) exposure, respectively. Blue light was delivered at 30 min after bacterial inoculation. (C) Dose response of mean bacterial luminescence of mouse burns infected with 3 × 106 CFU of P. aeruginosa and exposed to blue light (415 nm) at 30 min after bacterial inoculation (n = 11). Bars, standard deviations. (D) Time courses of mean bacterial luminescence of the infected skin abrasions with (n = 11) and without (n = 11 at days 1 and 2, n = 4 at day 3) blue light exposure, respectively. Bars, standard deviations. RLU values in the blue light group versus RLU values in the untreated groups: day 1, P = 0.0008; day 2, P = 6.11 × 10−5; day 3, P = 0.049. (E) Mean areas under the bioluminescence versus time curves (from day 1 to day 2 in the two-dimensional coordinate system in panel D), representing the overall bacterial burden of mouse wounds. Bars, standard deviations. (F) Kaplan-Meier survival curves of blue light-treated (n = 11) and untreated (n = 11) mouse burns (P < 0.0001).
Figure 5C shows the average reduction in bacterial luminescence from 11 mice, each of which was exposed to blue light. The in vivo inactivation curve also approximately followed first-order kinetics. After 55.8 J/cm2 blue light had been delivered, an average 3.5-log10-cycle reduction of bacterial luminescence was achieved in a light dose-dependent manner, with the bacterial inactivation rate coefficient kH at approximately 0.064 J/cm2.
Figure 5D shows the time courses of the mean bacterial luminescence (in RLUs) from day 1 to day 3 of the blue light-treated mice (n = 11) and untreated mice (n = 11). The RLU values of the blue light-treated group were significantly lower than those of the untreated group from day 1 to day 3 (P = 0.0008 on day 1; P = 6.11 × 10−5 on day 2; P = 0.049 on day 3). The AUBC of the bioluminescence time course (from day 1 to day 2, i.e., the time period before most of the mortalities occurred) were (1.19 ± 2.57) × 105 and (8.91 ± 4.53) × 106 for blue light-treated mice and untreated mice, respectively (P < 0.0001) (Fig. 5E), indicating an approximately 100-fold reduction of the AUBC resulting from acute blue light treatment.
All the treated mice (n = 11) survived after acute blue light treatment, while only 18.2% (2 out of 11) of the mice survived without acute blue light treatment (P < 0.0001) (Fig. 5F). Most of the mortalities (6 out of 9) occurred on day 3 (72 h) after bacterial inoculation.
No significant or irreversible damage was observed in the mouse skin exposed to blue light at the effective antimicrobial dose.
Figure 6A shows hematoxylin and eosin-stained histological sections of a representative mouse skin exposed to blue light at a dose of 55.8 J/cm2. Immediately after the blue light exposure, swelling of the nuclei of basal cells and slight edema in the upper dermis were observed. However, at 48 h after blue light exposure, the epithelium returned to its normal composition, and the collagenous fibers were lined up in order in the dermis.
Fig 6.
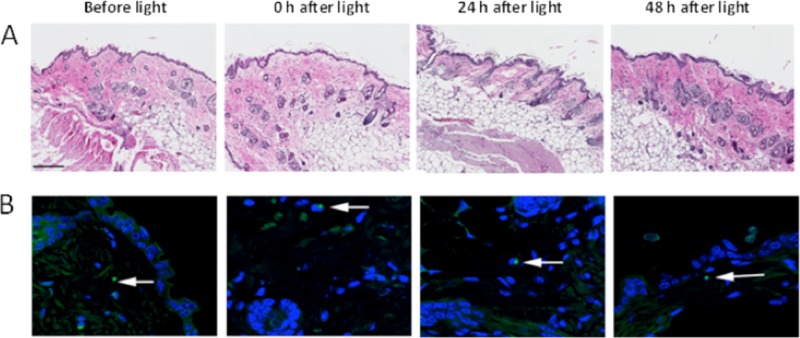
(A) Hematoxylin-and-eosin-stained histological sections of skin samples from a representative mouse exposed to blue light at a dose of 55.8 J/cm2. Skin samples were taken before blue light exposure and 0 h, 24 h, and 48 h after blue light exposure. Bar, 200 μm. (B) TUNEL analyses of DNA damage in the same mouse skin shown in panel A (100×). Skin samples were taken before blue light exposure and 0 h, 24 h, and 48 h after blue light exposure. Fluorescein and DAPI are represented by green and blue, respectively. DAPI was used for nuclear counterstaining. Arrows, TUNEL-positive cells.
Figure 6B shows the representative results of TUNEL assays of mouse skin exposed to blue light. A blue light exposure of 55.8 J/cm2 led to almost no apoptotic cells in the epidermis immediately after blue light exposure (only one TUNEL-positive cell was observed in the confocal image). Similarly, a lack of TUNEL-positive epidermal cells after 24 or 48 h was observed (only one TUNEL-positive cell was observed in each confocal image). These results demonstrate that blue light irradiation at the therapeutic antimicrobial dose is safe, and no adverse effects in terms of DNA damage were observed up to 48 h after blue light treatment.
DISCUSSION
We report here for the first time an in vivo study demonstrating the efficacy of blue light for P. aeruginosa burn infection in mice. The study might serve as an initial effort in the pursuit of a novel therapeutic option, blue light therapy, for wound infections, especially those caused by multidrug-resistant bacteria. Thus, the first and most important impact is through opening a new area of study on a different therapeutic regimen for wound infections.
It was found in our in vitro study that P. aeruginosa was much more susceptible to blue light inactivation than were HaCaT. As a result, there exists a therapeutic window where P. aeruginosa can be selectively inactivated by blue light while the host tissue cells can be preserved.
We found from the in vivo study that blue light at a 415-nm wavelength, when applied at 30 min after bacterial inoculation, effectively reduced the P. aeruginosa burden in mouse burns and prevented otherwise lethal bacteremia in mice. The P. aeruginosa strain we used is lethal to mice. Without treatment, the bacteria can invade deep into the mouse tissue and, subsequently, the bloodstream within hours and cause bacteremia and mortality of the animals. Therefore, we initiated blue light therapy very soon (at 30 min) after bacterial inoculation. Histological analyses and TUNEL assays revealed no significant or reversible damage in the mouse skin exposed to blue light at the effective antimicrobial dose.
An interesting finding of the present study is the equal susceptibilities of P. aeruginosa cells to blue light inactivation in vitro and in vivo. The in vitro and in vivo bacterial inactivation rate coefficients were 0.067 and 0.064 J/cm2, respectively. In our previous studies on antimicrobial PDT for wound infections (8, 11, 44), we observed that, to achieve equivalent amounts of inactivation of microorganisms, orders-of-magnitude-higher light exposures (and higher doses of photosensitizers) were required for in vivo than for in vitro treatment. One possible major factor responsible for this phenomenon is that the host tissue and cells compete with the microorganisms for binding to the exogenous photosensitizer, resulting in a reduced efficacy of in vivo microorganism inactivation when the microorganisms are embedded in the tissue. In contrast, it is suggested that the inactivation of bacteria by blue light treatment is caused by the photoexcitation of naturally occurring porphyrins within the bacterial cells, which act as endogenous photosensitizers. Therefore, competition from surrounding host tissue and cells for binding photosensitizers does not exist. One more advantage of antimicrobial blue light therapy is the highly selective inactivation of bacterial cells over mammalian cells, because blue light targets the photosensitizing porphyrins only within the bacterial cells, while mammalian cells (in the absence of porphyria or added 5-aminolevulanic acid) do not contain free porphyrins.
We also investigated the mechanism of blue light inactivation of P. aeruginosa. TEM images showed that blue light-mediated damage to P. aeruginosa cells started from the development of vacuoles within the cytoplasm, implying that the damage was associated with the intracellular chromophores excited by the blue light. By using fluorescence spectroscopy, we observed that the emission maxima of the P. aeruginosa pellets dissolved in NaOH-SDS were 613 nm and 667 nm at an excitation of 405 nm. These emission spectra are very close to those of uroporphyrin III (emission maxima, 618 nm and 670 nm) and coproporphyrin III (emission maxima, 615 nm and 674 nm) (43). Therefore, it is likely that the photosensitizing porphyrin within P. aeruginosa cells is uroporphyrin III or coproporphyrin III or that both uroporphyrin III and coproporphyrin III exist within P. aeruginosa cells. To further identify and quantify the intracellular porphyrins, future studies using techniques such as high-performance liquid chromatography (HPLC) or capillary electrophoresis together with authentic porphyrin standards are warranted (45).
As we have already stated, we understand that there remain many unanswered questions. One question that will have to be addressed in future studies is, “Can bacterial cells develop resistance to blue light inactivation?” To our knowledge, this question has not yet been experimentally addressed. The possible development of bacterial resistance to PDT has been studied. After repeated cycles of partial inactivation followed by regrowth, different bacterial species failed to develop resistance to PDT after 10 (46) or even 20 (47) cycles. It is commonly accepted that PDT acts at multiple sites within bacterial cells (structural proteins, enzymes, nucleic acids, unsaturated lipids, etc.) (48) and therefore would offer less potential for the development of bacterial resistance than conventional antibiotics, which are usually specific for a single target. As the mechanism for the antimicrobial effect of blue light is suggested to be similar to that of PDT, one can expect that the potential of bacterial resistance development to blue light is also less than that of conventional antibiotics. However, at the very least, it will be necessary to repeatedly deliver suberadication doses of blue light to susceptible cultures with regrowth between cycles to investigate whether resistant clones can be selected or whether mutants with lower accumulation of porphyrins or increased blue light damage-repairing enzymes can be produced.
In addition, more studies are warranted to deepen our understanding of the blue light therapy approach. Examples of such studies include comparison of the susceptibilities to blue light inactivation in vitro between pathogenic bacteria and host cells, including not only keratinocytes but also fibroblasts, endothelial cells, macrophages, and muscle cells; evaluation of the effects of blue light on the phagocytosis and reactive oxygen species production of macrophages in vitro; and identification of the maximum safe exposure of mice to blue light in this model.
ACKNOWLEDGMENTS
This study was supported in part by Airlift Research Foundation Extremity Trauma Research grant 109421 (to T.D.), COTA/Smith & Nephew grant 2012-16 (to T.D.), CIMIT Innovation grant 13-1033 (to T.D.), and NIH grant RO1AI050875 (to M.R.H.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
T.D., A.G., Y.-Y.H., C.K.M., M.S.V., G.P.T., and M.R.H. conceived and designed the experiments, T.D., A.G., Y.-Y.H., and M.S. performed the experiments, T.D., A.G., Y.-Y.H., R.Y., and M.R.H. analyzed the data, and T.D., A.G., R.Y., and M.R.H. wrote the paper.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Church D, Elsayed S, Reid O, Winston B, Lindsay R. 2006. Burn wound infections. Clin. Microbiol. Rev. 19:403–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rafla K, Tredget EE. 2011. Infection control in the burn unit. Burns 37:5–15 [DOI] [PubMed] [Google Scholar]
- 3. Rowley-Conwy G. 2010. Infection prevention and treatment in patients with major burn injuries. Nurs. Stand. 25:51– 52,, 54,, 56–58 passim [DOI] [PubMed] [Google Scholar]
- 4. Weber J, McManus A. 2004. Infection control in burn patients. Burns 30:A16–A24 [DOI] [PubMed] [Google Scholar]
- 5. Polavarapu N, Ogilvie MP, Panthaki ZJ. 2008. Microbiology of burn wound infections. J. Craniofac. Surg. 19:899–902 [DOI] [PubMed] [Google Scholar]
- 6. Azzopardi EA, Azzopardi SM, Boyce DE, Dickson WA. 2011. Emerging gram-negative infections in burn wounds. J. Burn Care Res. 32:570–576 [DOI] [PubMed] [Google Scholar]
- 7. Bush K, Courvalin P, Dantas G, Davies J, Eisenstein B, Huovinen P, Jacoby GA, Kishony R, Kreiswirth BN, Kutter E, Lerner SA, Levy S, Lewis K, Lomovskaya O, Miller JH, Mobashery S, Piddock LJ, Projan S, Thomas CM, Tomasz A, Tulkens PM, Walsh TR, Watson JD, Witkowski J, Witte W, Wright G, Yeh P, Zgurskaya HI. 2011. Tackling antibiotic resistance. Nat. Rev. Microbiol. 9:894–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dai T, Bil de Arce VJ, Tegos GP, Hamblin MR. 2011. Blue dye and red light, a dynamic combination for prophylaxis and treatment of cutaneous Candida albicans infections in mice. Antimicrob. Agents Chemother. 55:5710–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dai T, Huang YY, Hamblin MR. 2009. Photodynamic therapy for localized infections—state of the art. Photodiagnosis Photodyn. Ther. 6:170–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai T, Tegos GP, Lu Z, Huang L, Zhiyentayev T, Franklin MJ, Baer DG, Hamblin MR. 2009. Photodynamic therapy for Acinetobacter baumannii burn infections in mice. Antimicrob. Agents Chemother. 53:3929–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai T, Tegos GP, Zhiyentayev T, Mylonakis E, Hamblin MR. 2010. Photodynamic therapy for methicillin-resistant Staphylococcus aureus infection in a mouse skin abrasion model. Lasers Surg. Med. 42:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hamblin MR, Hasan T. 2004. Photodynamic therapy: a new antimicrobial approach to infectious disease? Photochem. Photobiol. Sci. 3:436–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kharkwal GB, Sharma SK, Huang YY, Dai T, Hamblin MR. 2011. Photodynamic therapy for infections: clinical applications. Lasers Surg. Med. 43:755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dai T, Garcia B, Murray CK, Vrahas MS, Hamblin MR. 2012. UVC light prophylaxis for cutaneous wound infections in mice. Antimicrob. Agents Chemother. 56:3841–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dai T, Kharkwal GB, Zhao J, St Denis TG, Wu Q, Xia Y, Huang L, Sharma SK, d'Enfert C, Hamblin MR. 2011. Ultraviolet-C light for treatment of Candida albicans burn infection in mice. Photochem. Photobiol. 87:342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dai T, Tegos GP, St Denis TG, Anderson D, Sinofsky E, Hamblin MR. 2011. Ultraviolet-C irradiation for prevention of central venous catheter-related infections: an in vitro study. Photochem. Photobiol. 87:250–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dai T, Vrahas MS, Murray CK, Hamblin MR. 2012. Ultraviolet C irradiation: an alternative antimicrobial approach to localized infections? Expert Rev. Anti Infect. Ther. 10:185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor GJ, Leeming JP, Bannister GC. 1993. Effect of antiseptics, ultraviolet light and lavage on airborne bacteria in a model wound. J. Bone Joint Surg. Br. 75:724–730 [DOI] [PubMed] [Google Scholar]
- 19. Thai TP, Keast DH, Campbell KE, Woodbury MG, Houghton PE. 2005. Effect of ultraviolet light C on bacterial colonization in chronic wounds. Ostomy Wound Manage. 51:32–45 [PubMed] [Google Scholar]
- 20. Wainwright M. 1998. Photodynamic antimicrobial chemotherapy (PACT). J. Antimicrob. Chemother. 42:13–28 [DOI] [PubMed] [Google Scholar]
- 21. Dai T, Gupta A, Murray CK, Vrahas MS, Tegos GP, Hamblin MR. 2012. Blue light for infectious diseases: Propionibacterium acnes, Helicobacter pylori, and beyond? Drug Resist. Updat. 15:223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Enwemeka CS, Williams D, Enwemeka SK, Hollosi S, Yens D. 2009. Blue 470-nm light kills methicillin-resistant Staphylococcus aureus (MRSA) in vitro. Photomed. Laser Surg. 27:221–226 [DOI] [PubMed] [Google Scholar]
- 23. Enwemeka CS, Williams D, Hollosi S, Yens D, Enwemeka SK. 2008. Visible 405 nm SLD light photo-destroys methicillin-resistant Staphylococcus aureus (MRSA) in vitro. Lasers Surg. Med. 40:734–737 [DOI] [PubMed] [Google Scholar]
- 24. Maclean M, MacGregor SJ, Anderson JG, Woolsey G. 2009. Inactivation of bacterial pathogens following exposure to light from a 405-nanometer light-emitting diode array. Appl. Environ. Microbiol. 75:1932–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDonald R, Macgregor SJ, Anderson JG, Maclean M, Grant MH. 2011. Effect of 405-nm high-intensity narrow-spectrum light on fibroblast-populated collagen lattices: an in vitro model of wound healing. J. Biomed. Opt. 16:048003. doi:10.1117/1.3561903 [DOI] [PubMed] [Google Scholar]
- 26. Kleinpenning MM, Smits T, Frunt MH, van Erp PE, van de Kerkhof PC, Gerritsen RM. 2010. Clinical and histological effects of blue light on normal skin. Photodermatol. Photoimmunol. Photomed. 26:16–21 [DOI] [PubMed] [Google Scholar]
- 27. Liebmann J, Born M, Kolb-Bachofen V. 2010. Blue-light irradiation regulates proliferation and differentiation in human skin cells. J. Investig. Dermatol. 130:259–269 [DOI] [PubMed] [Google Scholar]
- 28. Elman M, Slatkine M, Harth Y. 2003. The effective treatment of acne vulgaris by a high-intensity, narrow band 405–420 nm light source. J. Cosmet. Laser Ther. 5:111–117 [PubMed] [Google Scholar]
- 29. Kawada A, Aragane Y, Kameyama H, Sangen Y, Tezuka T. 2002. Acne phototherapy with a high-intensity, enhanced, narrow-band, blue light source: an open study and in vitro investigation. J. Dermatol. Sci. 30:129–135 [DOI] [PubMed] [Google Scholar]
- 30. Morton CA, Scholefield RD, Whitehurst C, Birch J. 2005. An open study to determine the efficacy of blue light in the treatment of mild to moderate acne. J. Dermatolog. Treat. 16:219–223 [DOI] [PubMed] [Google Scholar]
- 31. Wheeland RG, Dhawan S. 2011. Evaluation of self-treatment of mild-to-moderate facial acne with a blue light treatment system. J. Drugs Dermatol. 10:596–602 [PubMed] [Google Scholar]
- 32. Guffey JS, Wilborn J. 2006. In vitro bactericidal effects of 405-nm and 470-nm blue light. Photomed. Laser Surg. 24:684–688 [DOI] [PubMed] [Google Scholar]
- 33. Lipovsky A, Nitzan Y, Gedanken A, Lubart R. 2010. Visible light-induced killing of bacteria as a function of wavelength: implication for wound healing. Lasers Surg. Med. 42:467–472 [DOI] [PubMed] [Google Scholar]
- 34. Rosenthal SM. 1967. Local and systemic therapy of pseudomonas septicemia in burned mice. Ann. Surg. 165:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Markley K, Smallman E. 1968. Protection by vaccination against Pseudomonas infection after thermal injury. J. Bacteriol. 96:867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rocchetta HL, Boylan CJ, Foley JW, Iversen PW, LeTourneau DL, McMillian CL, Contag PR, Jenkins DE, Parr TR., Jr 2001. Validation of a noninvasive, real-time imaging technology using bioluminescent Escherichia coli in the neutropenic mouse thigh model of infection. Antimicrob. Agents Chemother. 45:129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jett BD, Hatter KL, Huycke MM, Gilmore MS. 1997. Simplified agar plate method for quantifying viable bacteria. Biotechniques 23:648–650 [DOI] [PubMed] [Google Scholar]
- 39. Firsov AA, Mattie H. 1997. Relationships between antimicrobial effect and area under the concentration-time curve as a basis for comparison of modes of antibiotic administration: meropenem bolus injections versus continuous infusions. Antimicrob. Agents Chemother. 41:352–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davis PJ, Rabinowitz P. 2007. Methods of numerical integration, 2nd ed Dover Publications, Mineola, NY [Google Scholar]
- 41. Xiong R, Xie G, Edmondson AE, Sheard MA. 1999. A mathematical model for bacterial inactivation. Int. J. Food Microbiol. 46:45–55 [DOI] [PubMed] [Google Scholar]
- 42. Sinton LW, Hall CH, Lynch PA, Davies-Colley RJ. 2002. Sunlight inactivation of fecal indicator bacteria and bacteriophages from waste stabilization pond effluent in fresh and saline waters. Appl. Environ. Microbiol. 68:1122–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strauss WS, Sailer R, Schneckenburger H, Akgun N, Gottfried V, Chetwer L, Kimel S. 1997. Photodynamic efficacy of naturally occurring porphyrins in endothelial cells in vitro and microvasculature in vivo. J. Photochem. Photobiol. B 39:176–184 [DOI] [PubMed] [Google Scholar]
- 44. Ragas X, Dai T, Tegos GP, Agut M, Nonell S, Hamblin MR. 2010. Photodynamic inactivation of Acinetobacter baumannii using phenothiazinium dyes: in vitro and in vivo studies. Lasers Surg. Med. 42:384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamblin MR, Viveiros J, Yang C, Ahmadi A, Ganz RA, Tolkoff MJ. 2005. Helicobacter pylori accumulates photoactive porphyrins and is killed by visible light. Antimicrob. Agents Chemother. 49:2822–2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tavares A, Carvalho CM, Faustino MA, Neves MG, Tomé JP, Tomé AC, Cavaleiro JA, Cunha A, Gomes NC, Alves E, Almeida A. 2010. Antimicrobial photodynamic therapy: study of bacterial recovery viability and potential development of resistance after treatment. Mar. Drugs 8:91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Giuliani F, Martinelli M, Cocchi A, Arbia D, Fantetti L, Roncucci G. 2010. In vitro resistance selection studies of RLP068/Cl, a new Zn(II) phthalocyanine suitable for antimicrobial photodynamic therapy. Antimicrob. Agents Chemother. 54:637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wainwright M. 2010. “Safe” photoantimicrobials for skin and soft-tissue infections. Int. J. Antimicrob. Agents 36:14–18 [DOI] [PubMed] [Google Scholar]