

Abstract
The increasing resistance of bacteria to conventional antibiotics and the challenges posed by intracellular bacteria, which may be responsible for chronic and recurrent infections, have driven the need for advanced antimicrobial drugs for effective elimination of both extra- and intracellular pathogens. The purpose of this study was to determine the killing efficacy of cationic antimicrobial peptide LL-37 compared to conventional antibiotics against extra- and intracellular Staphylococcus aureus. Bacterial killing assays and an infection model of osteoblasts and S. aureus were studied to determine the bacterial killing efficacy of LL-37 and conventional antibiotics against extra- and intracellular S. aureus. We found that LL-37 was effective in killing extracellular S. aureus at nanomolar concentrations, while lactoferricin B was effective at micromolar concentrations and doxycycline and cefazolin at millimolar concentrations. LL-37 was surprisingly more effective in killing the clinical strain than in killing an ATCC strain of S. aureus. Moreover, LL-37 was superior to conventional antibiotics in eliminating intracellular S. aureus. The kinetic studies further revealed that LL-37 was fast in eliminating both extra- and intracellular S. aureus. Therefore, LL-37 was shown to be very potent and prompt in eliminating both extra- and intracellular S. aureus and was more effective in killing extra- and intracellular S. aureus than commonly used conventional antibiotics. LL-37 could potentially be used to treat chronic and recurrent infections due to its effectiveness in eliminating not only extracellular but also intracellular pathogens.
INTRODUCTION
Conventional antibiotics are becoming increasingly ineffective due to rapidly evolving multidrug-resistant bacterial strains. The heavy use of antibiotics is causing bacteria to mutate and emerge as multidrug-resistant “superbugs” such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant S. aureus, and vancomycin-intermediate S. aureus (1–4). Recent studies reported that MRSA is posing a serious health care issue due to treatment failure, higher mortality rates, and increased health care costs (5–7). MRSA is now killing more people in the United States than AIDS (8). In 2009, the U.S. Centers for Disease Control and Prevention reported that bacterial infections, especially those caused by multidrug-resistant S. aureus, are on the rise globally (9). Each year, approximately 19,000 people die in the United States alone due to recalcitrant and recurrent bacterial infections (8). Moreover, treating recurrent bacterial infections (10, 11) has become a daunting challenge due to the possible presence of intracellular bacteria (12–14); historically, a high infection recurrence (∼17%) was found in combat-related injuries (15). Therefore, the increasing resistance of bacteria to conventional antibiotics and the challenges posed by intracellular bacteria have driven the need for advanced or alternative antimicrobial drugs.
Cationic antimicrobial peptides (CAMPs) have recently emerged as an alternative to conventional antibiotic therapies (16, 17). They are produced by the innate immune system in both vertebrates and invertebrates as a first line of defense against microbial infections (18–20). They have broad-spectrum killing ability against pathogens (21, 22). In addition to their antibacterial and antifungal properties, CAMPs have also been described recently for their role in neutralization of endotoxins, chemokine-like activities, immunomodulating properties, induction of angiogenesis, and wound repair (23–27). Currently, companies like HelixBiomedix are developing arrays of CAMPs in several pharmaceutical programs ranging from topical anti-infective to wound healing and cystic fibrosis (28), and several CAMPs and their derivatives are being investigated in preclinical and clinical trials (28–33).
Conventional antibiotics are relatively large molecules compared to CAMPs and have different types of mechanisms in killing bacteria. Cefazolin, a beta-lactam and frequently used in orthopedic infection treatment (34), has a very low permeability through cell membranes due to its hydrophilic nature and does not accumulate in the cytoplasm because of its rapid efflux (35). However, it binds to bacterial penicillin-binding proteins, thereby disrupting the synthesis of peptidoglycan, the integral part of the bacterial cell wall (36). Doxycycline (tetracycline) and clindamycin (lincosamide) traverse bacterial membranes using the membrane transport system, but they have to cross the threshold limit to interact with the ribosomes (36). Clindamycin was proven effective against intracellular bacteria (36–38).
The mode of action of CAMPs is different from that of conventional antibiotics and is often more effective in destroying bacteria; they interact with bacteria through electrostatic forces (39, 40). CAMPs, including cathelicidin LL-37, are amphiphilic in nature and are comprised of hydrophobic and hydrophilic residues aligned on opposite sides of the peptides, facilitating their easy penetration through cell membranes (19, 41–44). Their positively charged domain allows CAMPs to bind to bacterial membranes like magnets, and the hydrophobic domain facilitates their penetration through phospholipid bilayers (45, 46). This mode of action results in bacterial death (47, 48).
Bacteria could develop resistance to conventional antibiotics by altering their antibiotic binding cell membrane receptors through mutations, thereby making the antibiotics ineffective; however, CAMPs target the lipid matrix of cell membranes whose lipid composition is highly unlikely to change due to bacterial mutation (49). Development of resistance against CAMPs by modifying membrane compositions of bacteria would compromise the bacteria's viability (50) and thereby would not likely occur (16, 19, 41, 42). However, CAMPs may suffer proteolytic digestion (43), which could be minimized via a small alteration of the peptide structure to make them not be recognized or degraded by proteolytic enzymes (51).
Cathelicidin LL-37 is a CAMP that has recently attracted great interest (16, 17, 52). The objective of this study was to determine the antimicrobial properties of cathelicidin LL-37 compared to those of conventional antibiotics against extra- and intracellular S. aureus. We hypothesized that LL-37 can be effective in eliminating not only extracellular bacteria but also intracellular bacteria.
(This work was presented [orally] at the 57th Orthopaedic Research Society [ORS] Annual Meeting, Long Beach, CA, January 2011.)
MATERIALS AND METHODS
A clinical strain of S. aureus obtained from a patient's chronic wound at Ruby Memorial Hospital, Morgantown, WV, and an American Type Culture Collection (ATCC; Manassas, VA) strain (ATCC 25923) of S. aureus were investigated in this study. Susceptibility tests showed that the clinical S. aureus strain was susceptible to cefazolin, ciprofloxacin, clindamycin, erythromycin, gentamicin, levofloxacin, linezolid, moxifloxacin, oxacillin, rifampin, tetracycline, tigecycline, and vancomycin and was resistant to ampicillin, cefoxitin, and penicillin. The ATCC S. aureus strain was susceptible to cefazolin, cefoxitin, ciprofloxacin, clindamycin, erythromycin, gentamicin, levofloxacin, linezolid, moxifloxacin, oxacillin, penicillin, rifampin, tetracycline, tigecycline, and vancomycin and was resistant to ampicillin. S. aureus was chosen because it is one of the major pathogens responsible for most bacterial infections, including orthopedic infections (53–57), and is a potential cause of chronic and recurrent infections (15, 58). CAMPs (cathelicidin LL-37 and lactoferricin B) and conventional antibiotics (cefazolin, doxycycline, and clindamycin) were purchased from Sigma-Aldrich (St. Louis, MO). The purity of LL-37 (product no. 94261; molecular weight [MW], 4492) was 98.5% (as determined by high-performance liquid chromatography [HPLC]). Tryptic soy broth was from Becton, Dickinson and Company (Sparks, MD).
Extracellular antimicrobial activities of LL-37 and conventional antibiotics.
The killing efficacies of LL-37, lactoferricin B, doxycycline, and cefazolin were determined against extracellular S. aureus under the same experimental conditions. Sterile tryptic soy broth, prepared based on the manufacturer's instructions, was used for bacterial cultures. Three colonies of S. aureus were inoculated into a sterile tube containing 5 ml of tryptic soy broth and incubated for 16 h at 37°C. The next day, 100 μl of a 16-h-old S. aureus culture (stationary phase) was inoculated into a sterile tube containing 20 ml of fresh tryptic soy broth and was subjected to shaking (80 rpm) at 37°C for 2.5 h to acquire log-phase bacteria (exponential bacterial growth) using a reciprocal shaking bath, made by Precision (El Cajon, CA). The log-phase S. aureus inocula were diluted to 1.0 × 105 CFU/ml with sterile phosphate-buffered saline (PBS, pH 7.0). The assays were run with a total volume of 1 ml comprising S. aureus (1.0 × 105 CFU/ml) and different molar concentrations (ranging from 10 nM to 100 mM) of cathelicidin LL-37, lactoferricin B, doxycycline, and cefazolin, individually. The controls and the treated samples were incubated at 37°C for 30 min in a reciprocal shaking bath. The samples were then diluted and plated on 5% sheep blood agar plates. Dilutions of 10−1, 10−2, and 10−3 were made for control and treated samples with sterile PBS. The drop plate method (59, 60) was used for viable bacterial enumeration and was performed as follows. A sheep blood agar plate was divided into six sectors. A 20-μl bacterial suspension was pipetted and placed as a drop in each sector. After the drops dried, the plates were inverted and incubated at 37°C for 24 h. The procedure was repeated for each dilution. CFU were determined using an Acolyte colony counter made by Synbiosis (Frederick, MD). The killing efficacy of each drug was presented in terms of percent killing at different molar concentrations. Percent killing was calculated by dividing the difference between control and treated samples with a control value and then multiplying by 100. Data were averages of four samples.
LL-37 was next tested from 50 nM to 100 μM (0.05, 0.25, 0.5, 1.0, 2.0, 3.0, 10.0, and 100 μM) concentrations for strain (ATCC strain versus clinical strain; log phase was studied) and phase (log phase versus stationary phase; the ATCC strain was used) comparisons. The inocula were diluted to 1.0 × 105 CFU/ml with sterile PBS. The experiments were carried out with a total volume of 1 ml comprising S. aureus (1.0 × 105 CFU/ml) and different molar concentrations (0.05, 0.25, 0.5, 1.0, 2.0, 3.0, 10.0, and 100 μM) of LL-37. The controls and the treated samples were incubated at 37°C for 30 min in a reciprocal shaking bath. The samples were then diluted and plated on 5% sheep blood agar plates using the drop plate method; the CFU were determined and the percent killing of LL-37 was calculated.
In addition, kinetic studies were conducted individually for LL-37 (250 nM), lactoferricin B (25.0 μM), and cefazolin (1.0 mM) at given time intervals (5, 10, 15, and 30 min). LL-37 at 250 nM, lactoferricin B at 25.0 μM, and cefazolin at 1.0 mM had approximately the same percent killing from the aforementioned experiments. The kinetic experiments were run with a total volume of 1 ml comprising log-phase bacteria (1.0 × 105 CFU/ml) and LL-37 (250 nM), lactoferricin B (25.0 μM), or cefazolin (1.0 mM) and incubated separately for 5, 10, 15, and 30 min at 37°C in a reciprocal shaking bath. At the predetermined time, the control and treated samples were diluted and plated on 5% sheep blood agar plates using the drop plate method and the CFU were determined. The percent killing data were calculated and normalized by assuming that LL-37 (250 nM), lactoferricin B (25.0 μM), and cefazolin (1.0 mM) had 100% killing at 30 min. Data were averages of four samples.
Intracellular antimicrobial activities of LL-37 and conventional antibiotics.
An infection model of osteoblasts and S. aureus (61–65) was used to obtain intracellular S. aureus; S. aureus can internalize into osteoblasts and survive within them (61–65). The clinical strain of S. aureus in the log phase was studied, and a 500:1 ratio of S. aureus to osteoblasts was used. Experiments were conducted using a 12-well plate in a laminar-flow hood under aseptic conditions. Dulbecco's modified Eagle's medium–F-12 (DMEM–F-12) and PBS were used for osteoblast culture. One milliliter of osteoblasts (UMR-106, passage 2) with a cell density of 4 × 105 cells/ml was seeded in each well and incubated at 37°C with 5% CO2 for 36 h to form a confluent monolayer. After 36 h, the wells were washed twice with 1 ml of PBS to remove growth medium. One milliliter of log-phase S. aureus (2 × 108 CFU/ml) was then added to each well, and the 12-well plate was incubated at 37°C. After culture for 2 h, the wells were washed twice with 1 ml of PBS; 50 μg of lysostaphin was added to each well, and the plate was incubated for 2 h to eliminate extracellular S. aureus. Lysostaphin is an antimicrobial agent that does not penetrate eukaryotic cells, and 50 μg/ml of lysostaphin (Sigma-Aldrich) was found to be effective at eradicating any extracellular S. aureus organisms (63, 66). The wells were washed twice with 1 ml of PBS. Osteoblasts in three wells were immediately lysed with 0.1% Triton X-100 in PBS for 10 min at 37°C; the cell lysates were diluted in PBS and plated on blood agar plates overnight, and the count of intracellular S. aureus was 4 × 104 CFU. Different molar concentrations (10, 30, 50, and 100 μM) of LL-37 or plain DMEM were added to the remaining wells. After incubation at 37°C for 2 h, osteoblasts were rinsed twice with 1 ml of PBS and then lysed with 0.1% Triton X-100, and the intracellular S. aureus was plated on 5% sheep blood agar plates using the aforementioned drop plate method. Dilutions of 10−1, 10−2, and 10−3 were made for control and treated samples with sterile PBS. The colony numbers of viable intracellular S. aureus were determined. The same experiments were also carried out with conventional antibiotics, including cefazolin and clindamycin at 100 μM, for comparison; clindamycin was chosen due to its effectiveness against intracellular bacteria (36–38) and cefazolin due to its wide applications in orthopedic infection treatment (34). Data were averages of four samples.
Kinetic studies of LL-37 (100 μM) were also conducted against intracellular S. aureus at different time intervals (i.e., 0.5, 2, 12, and 24 h). Log-phase S. aureus was internalized within the osteoblasts in a 12-well plate as described above in the osteoblast-S. aureus infection model. The extracellular S. aureus was eliminated using lysostaphin, and the wells were washed twice with 1 ml of PBS; 100 μM LL-37 was added to each well, and the plate was incubated at 37°C. Controls were run separately for each time point. After 0.5, 2, 12, and 24 h, osteoblasts were rinsed twice with 1 ml of PBS and then lysed with 0.1% Triton X-100; the intracellular S. aureus was plated on 5% sheep blood agar plates. Percent killing was calculated; data were averages of four samples.
Statistical analysis.
Values of percent killing were expressed as means ± standard deviations. Differences in percent killing of extracellular S. aureus between the ATCC and clinical strains and between log phase and stationary phase and differences in percent killing of intracellular S. aureus among cefazolin, clindamycin, and LL-37 were analyzed using JMP-V9 statistical visualization software (SAS Institute Inc., Cary, NC). The data were transformed as the arcsin of the square root of percent killing, and a t test was run to compare the two groups; in the case where there were three groups, an analysis of variance (ANOVA) followed by Tukey's honestly significant difference (HSD) test was used to determine significance. A P value of <0.05 was considered statistically significant.
RESULTS
Extracellular bacterial killing efficacy versus concentration of LL-37, lactoferricin B, and conventional antibiotics.
S. aureus was treated with two CAMPs (i.e., cathelicidin LL-37 and lactoferricin B), and their killing efficacies were compared with those of cefazolin and doxycycline, two commonly used antibiotics, under the same experimental conditions. Overall, LL-37 was effective in killing S. aureus at nanomolar concentrations, while lactoferricin B was effective at micromolar concentrations and doxycycline and cefazolin were effective at millimolar concentrations (Fig. 1). LL-37 was found to exhibit over 90% killing efficacy at as low as 250 nM, over 99% at 500 nM, and 100% at 3.0 μM (Fig. 1). Lactoferricin B had approximately 2% killing potency at 250 nM, 15% at 500 nM, 67% at 3.0 μM, and over 90% at 25 μM. On the other hand, doxycycline and cefazolin were found to have significant killing abilities only at much higher concentrations; they had no killing efficacy at 3.0 μM, more than 90% killing efficacy at 1.0 mM, and 100% killing potency at 10 mM or higher (Fig. 1).
Fig 1.

Killing potencies of LL-37, lactoferricin B, and conventional antibiotics (i.e., cefazolin and doxycycline) against extracellular S. aureus (clinical strain) in log phase.
Extracellular bacterial killing efficacy of LL-37 against S. aureus strains.
LL-37 was tested on both the clinical and ATCC S. aureus strains with different molar concentrations, ranging from 0.05 μM to 100 μM for strain comparison. LL-37 exhibited 100% killing against both strains at higher concentrations (10 and 100 μM). However, at concentrations lower than 3 μM, LL-37 was surprisingly more effective in killing the clinical strain than the ATCC strain (Fig. 2). There was a 24% increase in the ability to kill the clinical strain compared to the ATCC strain at 1.0 μM; the difference was more prominent (over 40%) at lower concentrations, e.g., 0.5, 0.25, and 0.05 μM (Fig. 2).
Fig 2.
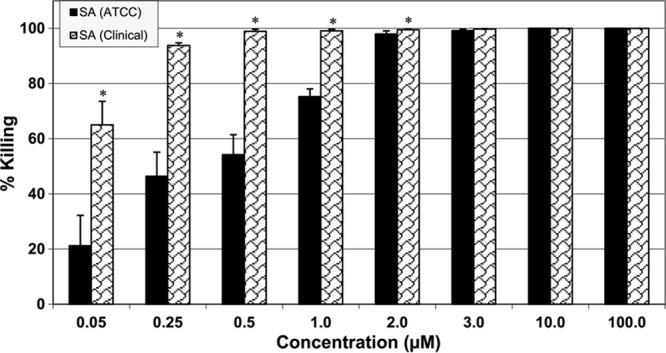
Strain-specific killing efficacy of LL-37 against S. aureus (ATCC and clinical strains) in log phase. Incubation time was 30 min. *, P < 0.05 compared to ATCC strain at the same concentration.
Extracellular bacterial killing efficacy of LL-37 against S. aureus phases.
LL-37 seemed to kill significantly more S. aureus organisms in the stationary phase than S. aureus organisms in the log phase at concentrations at or lower than 1.0 μM; no differences in percent killing were observed at concentrations higher than 2.0 μM (Fig. 3).
Fig 3.
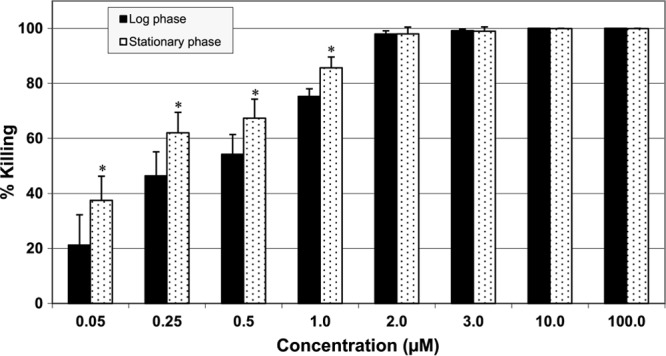
Killing efficacy of LL-37 against S. aureus in log and stationary phases. Incubation time was 30 min. *, P < 0.05 compared to log phase at the same concentration.
Extracellular bacterial killing kinetics of LL-37.
The extracellular bacterial killing kinetics of LL-37 were compared with those of lactoferricin B and cefazolin. Incredibly, LL-37 was able to eliminate more than 70% of S. aureus organisms within just 5 min and more than 90% within 15 min (Fig. 4). In contrast, lactoferricin B and cefazolin had much slower kinetics and showed almost no bacterial killing within the first 5 min and less than 40% killing within 15 min (Fig. 4).
Fig 4.
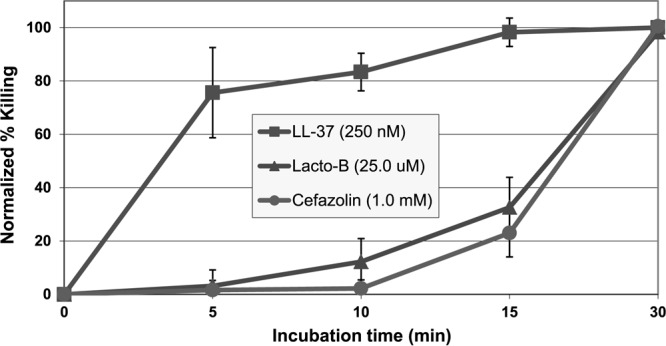
Kinetics of LL-37 killing against extracellular S. aureus (clinical strain) in log phase.
Intracellular antimicrobial activities of LL-37.
The killing potency of LL-37 against intracellular S. aureus was determined at different molar concentrations (10, 30, 50, and 100 μM). LL-37 was found to be very effective in eliminating intracellular S. aureus. The intracellular bacterial percent killing increased with increasing LL-37 concentration, and 100 μM LL-37 completely killed the intracellular S. aureus organisms (Fig. 5). In contrast, at the same concentration (i.e., 100 μM), cefazolin and clindamycin eliminated only 2% and 23% of the intracellular S. aureus organisms, respectively (Fig. 6). Kinetic studies further showed that LL-37 killed approximately 50% of the intracellular S. aureus organisms within 30 min and all bacteria within 2 h (Fig. 7).
Fig 5.
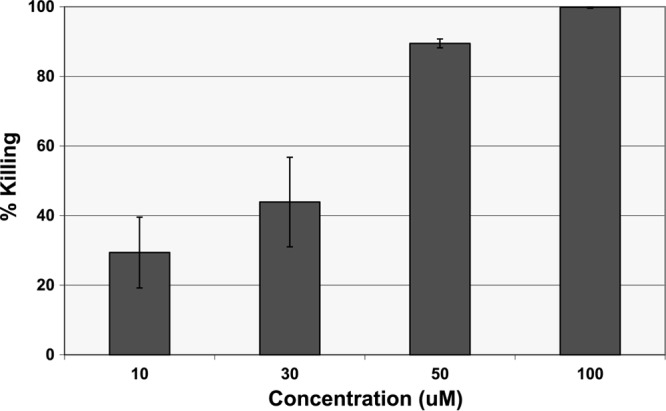
Intracellular killing efficacy of LL-37 against S. aureus (clinical strain) within osteoblasts. Incubation time was 2 h.
Fig 6.

Intracellular killing efficacies of cefazolin, clindamycin, and LL-37 against S. aureus (clinical strain) within osteoblasts. The concentration of cefazolin, clindamycin, and LL-37 was 100 μM; incubation time was 2 h. (A) Percent killing; (B) images at 10−1 dilution: control (a), cefazolin (b), clindamycin (c), and LL-37 (d). *, P < 0.05 compared to cefazolin and clindamycin; **, P < 0.05 compared to cefazolin.
Fig 7.
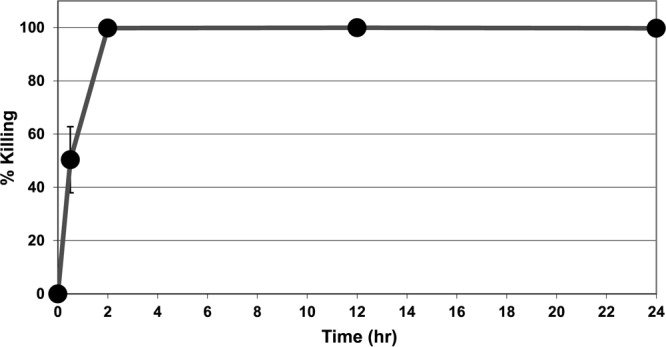
Kinetics of LL-37 killing against intracellular S. aureus (clinical strain) within osteoblasts. The concentration of LL-37 was 100 μM.
DISCUSSION
It is well known that a wide variety of pathogens, including bacteria and viruses, are capable of internalizing into human cells, thereby causing intracellular diseases like human immunodeficiency virus/AIDS (HIV/AIDS), hepatitis, and tuberculosis (TB) (reviewed in reference 67). One of the critical challenges in treating these types of infections is the intracellular nature of the pathogens, which may protect the pathogens from a variety of antibiotic therapies and host immune responses. Antibiotics such as aminoglycosides and beta-lactams have limited cellular penetration, whereas antibiotics like fluoroquinolones or macrolides have poor retention within cells and therefore are inefficient at killing intracellular pathogens (68). Moreover, some bacteria such as S. aureus, which has long been considered an extracellular pathogen, have now been found to be able to internalize and survive within host cells, e.g., osteoblasts (13, 64, 69–72), and may contribute to chronic and recurrent infections (54). Therefore, advanced drugs for effectively destroying both extra- and intracellular pathogens are needed in order to reduce or prevent chronic and recurrent infections. In this study, the potential bacterial killing activities of LL-37 against intracellular S. aureus were examined and compared with those of conventional antibiotics. The bacterial killing activities of LL-37 against extracellular bacteria were also investigated and compared with those of conventional antibiotics.
Our studies indicated that LL-37 is very potent and fast (Fig. 1 and 4) at eliminating extracellular S. aureus, the common culprit of many bacterial infections. Among LL-37, lactoferricin B, doxycycline, and cefazolin, LL-37 was apparently foremost in eliminating extracellular S. aureus. LL-37 was remarkably potent in killing more than 90% of S. aureus organisms even at 250 nM (Fig. 1). Our experiments showed that a substantially smaller quantity of LL-37 (100 times less than lactoferricin B and 4,000 times less than doxycycline and cefazolin) was needed to eliminate extracellular S. aureus (Fig. 1). Moreover, LL-37 was not only potent but also expeditious in eliminating extracellular S. aureus. LL-37 was found to be much faster in killing extracellular S. aureus than were lactoferricin B and cefazolin (Fig. 4).
LL-37 furthermore exhibited a strain-specific, higher ability to kill the clinical strain than the ATCC strain at concentrations lower than 3.0 μM (Fig. 2). These findings indicated that the S. aureus clinical strain was surprisingly more susceptible to LL-37 than the ATCC strain; the reason is unknown. In our previous in vivo studies, we found that the S. aureus clinical strain was much more virulent in inducing infections than the ATCC strain (73). LL-37 also presented a phase-specific response (Fig. 3) at concentrations lower than 1.0 μM, with a higher ability to kill bacteria in the stationary phase than in the log phase. This may suggest that it is relatively easier to eliminate stationary-phase bacteria than log-phase bacteria.
More interestingly, we found that LL-37 was very effective in eliminating intracellular pathogens. LL-37 had remarkable intracellular killing ability against S. aureus compared to conventional antibiotics like cefazolin and clindamycin; clindamycin was reported to have potent antimicrobial properties against intracellular S. aureus due to its good penetration, retention, and distribution properties in eukaryotic cells (36, 37). Our results indicated that a 100 μM concentration of LL-37 completely eliminated intracellular S. aureus within just 2 h, whereas cefazolin and clindamycin eliminated only 2% and 23%, respectively (Fig. 6). However, due to the intracellular nature of the pathogen, a much higher (100 μM versus 3 μM) concentration of LL-37 was needed (Fig. 1 and 5), and relatively slower kinetics were observed (Fig. 4 and 7) in killing intracellular S. aureus than for extracellular S. aureus. Note that 10 mM concentrations of cefazolin and doxycycline were needed to completely eliminate extracellular S. aureus alone (Fig. 1).
The current study therefore demonstrated that LL-37 is very potent and fast at eliminating both extra- and intracellular S. aureus compared to conventional antibiotics. Moreover, LL-37 may exhibit synergistic antibacterial activities with β-defensin and lysozyme in both neutral and acidic environments (74). However, the antibacterial properties of LL-37 may be reduced by serum proteins. It was reported that certain biological fluids containing glycosaminoglycans and serum may hamper the antibacterial properties of LL-37 (75). Serum proteins such as apolipoproteins could bind to LL-37 and reduce its antimicrobial efficacy (75–77). Interestingly, the removal of N-terminal hydrophobic amino acids from LL-37 may reduce the effect of serum without compromising its antimicrobial properties (78).
One limitation of this study is that the potential toxicity of LL-37 was not examined. It was reported that LL-37 could prevent sepsis in neonatal rats (79), and a low dose (100 μg/kg of body weight) of LL-37 did not induce observable toxicity, but a high dose (3,000 μg/kg) resulted in adverse effects and appeared to be toxic to organs affected by sepsis (79). It is noteworthy that studies on human cathelicidin analogs reveal that removal of hydrophobic amino acids from the N-terminal end of native LL-37 could decrease its cytotoxicity without compromising the peptide's antimicrobial efficacy toward both Gram-positive and Gram-negative bacteria (78). Wang et al. (80) recently mapped and unmasked the potential roles of cationic residues of human cathelicidin LL-37 against different bacterial strains. The cationic side chains of the major antimicrobial region of human cathelicidin LL-37 were fragmented, and their functional roles were studied in detail. The GF-17 fragment, comprising residues 17 to 32, was found to be more potent against methicillin-resistant S. aureus in vitro than was intact LL-37. It also indicated that the conversion of amino acids from lysines (K) to arginines (R) increased the ability of the peptide to kill S. aureus. Therefore, the use of the GF-17 fragment of LL-37 may lead to lower dosages and therefore reduced toxicity (80).
In summary, S. aureus and S. aureus internalized within osteoblasts were treated with LL-37 and conventional antibiotics. LL-37 was found to have rapid and robust killing efficacy against both extra- and intracellular S. aureus, one of the most common causes of bacterial infections. In eliminating extracellular S. aureus, LL-37 is 100 times more potent than lactoferricin B and 4,000 times more potent than conventional antibiotics such as doxycycline and cefazolin. LL-37 also eliminates the majority (more than 70%) of S. aureus organisms within just 5 min, compared to almost no killing by lactoferricin B and cefazolin at the same time point. The efficacy of LL-37 was found to be bacterial strain and phase specific. Surprisingly, LL-37 was more effective at killing the clinical strain than the ATCC strain of S. aureus. In eliminating intracellular S. aureus, 100 μM LL-37 killed approximately 50% of intracellular S. aureus organisms within the first 30 min and completely eradicated the bacteria within 2 h. However, at the same concentration, cefazolin and clindamycin only eliminated 2% and 23% of the intracellular S. aureus organisms, respectively, within 2 h. Therefore, we conclude that LL-37 has rapid and remarkable killing abilities toward both extra- and intracellular S. aureus compared to conventional antibiotics. In future studies, we will examine the in vivo antimicrobial activities of LL-37 in our animal model (81–83) and may evaluate in vitro whether LL-37 will induce resistance.
ACKNOWLEDGMENTS
This work was supported by NIH grant 5P20RR016477 to the West Virginia IDeA Network for Biomedical Research Excellence, the National Science Foundation (no. 1003907), and NASA WV Space Grant Consortium and WVNano SURE fellowships.
We acknowledge statistical assistance from Gerald R. Hobbs at West Virginia University. We appreciate access to the West Virginia University Imaging Facility, which is supported in part by the Mary Babb Randolph Cancer Center and NIH grant P20 RR016440. We thank Sara Kurian from Shepherd University, Shepherdstown, WV, for helping with experiments and Suzanne Smith for proofreading the manuscript.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 28 December 2012
REFERENCES
- 1. Arias CA, Murray BE. 2009. Antibiotic-resistant bugs in the 21st century—a clinical super-challenge. N. Engl. J. Med. 360:439–443 [DOI] [PubMed] [Google Scholar]
- 2. Chambers HF. 2005. Community-associated MRSA—resistance and virulence converge. N. Engl. J. Med. 352:1485–1487 [DOI] [PubMed] [Google Scholar]
- 3. Linares J. 2001. The VISA/GISA problem: therapeutic implications. Clin. Microbiol. Infect. 7:8–15 [DOI] [PubMed] [Google Scholar]
- 4. Young LS, Perdreau-Remington F, Winston LG. 2004. Clinical, epidemiologic, and molecular evaluation of a clonal outbreak of methicillin resistant Staphylococcus aureus infection. Clin. Infect. Dis. 38:1075–1083 [DOI] [PubMed] [Google Scholar]
- 5. Boucher HW, Corey GR. 2008. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 46:5344–5349 [DOI] [PubMed] [Google Scholar]
- 6. Jensen AG, Wachmann CH, Espersen F, Scheibel J, Skinhoj P, Frimodt-Moller N. 2002. Treatment and outcome of Staphylococcus aureus bacteremia: a prospective study of 278 cases. Arch. Intern. Med. 162:25–32 [DOI] [PubMed] [Google Scholar]
- 7. Martínez-Aguilar G, Avalos-Mishaan A, Hulten K, Hammerman W, Mason EO, Jr, Kaplan SL. 2004. Community-acquired, methicillin-resistant and methicillin-susceptible Staphylococcus aureus musculoskeletal infections in children. Pediatr. Infect. Dis. J. 23:701–706 [DOI] [PubMed] [Google Scholar]
- 8. Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771 [DOI] [PubMed] [Google Scholar]
- 9. CDC 2009. Rise in life-threatening bacterial infections linked to H1N1 swine flu. http://www.webmd.com/cold-and-flu/news/20091125/worrisome-spike-in-bacterial-infections-with-h1n1-flu Accession date, 16 February 2012
- 10. Chang FY, Peacock JE, Jr, Musher DM, Triplett P, MacDonald BB, Mylotte JM, O'Donnell A, Wagener MM, Yu VL. 2003. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine 82:333–339 [DOI] [PubMed] [Google Scholar]
- 11. Johnson LB, Almoujahed MO, Ilg K, Maolood L, Khatib R. 2003. Staphylococcus aureus bacteremia: compliance with standard treatment, long-term outcome and predictors of relapse. Scand. J. Infect. Dis. 35:782–789 [DOI] [PubMed] [Google Scholar]
- 12. Ciampolini J, Harding KG. 2000. Pathophysiology of chronic bacterial osteomyelitis. Why do antibiotics fail so often? Postgrad. Med. J. 76:479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garzoni C, Kelley WL. 2009. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 17:59–65 [DOI] [PubMed] [Google Scholar]
- 14. Sendi P, Rohrbach M, Graber P, Frei R, Ochsner PE, Zimmerli W. 2006. Staphylococcus aureus small colony variants in prosthetic joint infection. Clin. Infect. Dis. 43:961–966 [DOI] [PubMed] [Google Scholar]
- 15. Murray CK, Obremskey WT, Hsu JR, Andersen RC, Calhoun JH, Clasper JC, Whitman TJ, Curry TK, Fleming ME, Wenke JC, Ficke JR. 2011. Prevention of Combat-Related Infections Guidelines Panel. Prevention of infections associated with combat-related extremity injuries. J. Trauma 71(Suppl 2):235–257 [DOI] [PubMed] [Google Scholar]
- 16. Bucki R, Leszczyńska K, Namiot A, Sokołowski W. 2010. Calthelicidin LL-37: a multitask antimicrobial peptide. Arch. Immunol. Ther. Exp. 85:15–25 [DOI] [PubMed] [Google Scholar]
- 17. Liu L, Xu K, Wang H, Tan PK, Fan W, Venkatraman SS, Li L, Yang YY. 2009. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat. Nanotechnol. 4:457–463 [DOI] [PubMed] [Google Scholar]
- 18. Guaní-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Teran LM. 2010. Antimicrobial peptides: general overview and clinical implications in human health and disease. Clin. Immunol. 135:1–11 [DOI] [PubMed] [Google Scholar]
- 19. Wiesner J, Vilcinskas A. 2010. Antimicrobial peptides: the ancient arm of the human immune system. Virulence 1:440–464 [DOI] [PubMed] [Google Scholar]
- 20. Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395 [DOI] [PubMed] [Google Scholar]
- 21. Yang D, Biragyn A, Kwak LW, Oppenheim JJ. 2002. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol. 23:291–296 [DOI] [PubMed] [Google Scholar]
- 22. Zaiou M, Nizet V, Gallo RL. 2003. Antimicrobial and protease inhibitory functions of the human cathelicidin (hCAP18/LL-37) prosequence. J. Invest. Dermatol. 120:810–816 [DOI] [PubMed] [Google Scholar]
- 23. Alalwani SM, Sierigk J, Herr C, Pinkenburg O, Gallo R, Vogelmeier C, Bals R. 2010. The antimicrobial peptide LL-37 modulates the inflammatory and host defense response of human neutrophils. Eur. J. Immunol. 40:1118–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Izadpanah A, Gallo R. 2005. Antimicrobial peptides. J. Am. Acad. Dermatol. 52:381–390 [DOI] [PubMed] [Google Scholar]
- 25. Lai Y, Gallo RL. 2009. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 30:131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Turner J, Cho Y, Dinh N, Waring AJ, Lehrer RI. 1998. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 42:2206–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaiou M. 2007. Multifunctional antimicrobial peptides: therapeutic targets in several human diseases. J. Mol. Med. 85:317–329 [DOI] [PubMed] [Google Scholar]
- 28. Giuliani A, Pirri G, Nicoletto S. 2007. Antimicrobial peptides: an overview of a promising class of therapeutics. Cent. Eur. J. Biol. 2:1–33 [Google Scholar]
- 29. Andrès E, Dimarcq JL. 2004. Cationic anti-microbial peptides: from innate immunity study to drug development. J. Rev. Med. 25:629–635 [DOI] [PubMed] [Google Scholar]
- 30. Andrès E, Dimarcq JL. 2004. Cationic antimicrobial peptides: update of clinical development. J. Intern. Med. 255:519–520 [DOI] [PubMed] [Google Scholar]
- 31. Dimarcq JL. 2003. Developing insect-derived drug candidates. Drug Discov. Today 8:107–110 [DOI] [PubMed] [Google Scholar]
- 32. Hancock RE, Patrzykat A. 2002. Clinical development of cationic antimicrobial peptides: from natural to novel antibiotics. Curr. Drug Targets Infect. Disord. 2:79–83 [DOI] [PubMed] [Google Scholar]
- 33. Yeung TY, Gellatly SL, Hancock REW. 2011. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 68:2161–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Welch WD, Jantzen J, Johnson K, Bawdon RE. 1985. Effects of general and local anesthesia on the pharmacokinetics of cefazolin in patients undergoing orthopedic surgery. Antimicrob. Agents Chemother. 27:874–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Darouiche RO, Hamill RJ. 1994. Antibiotic penetration of and bactericidal activity within endothelial cells. Antimicrob. Agents Chemother. 38:1059–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tulkens PM. 1991. Intracellular distribution and activity of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 10:100–106 [DOI] [PubMed] [Google Scholar]
- 37. Bernardo K, Pakulat N, Fleer S, Schnaith A, Utermohlen O, Krut O, Müller S, Kronke M. 2004. Subinhibitory concentrations of linezolid reduce Staphylococcus aureus virulence factor expression. Antimicrob. Agents Chemother. 48:546–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van den Broek PJ. 1991. Activity of antibiotics against microorganisms ingested by mononuclear phagocytes. Eur. J. Clin. Microbiol. Infect. Dis. 10:114–118 [DOI] [PubMed] [Google Scholar]
- 39. Ulvatne H, Haukland HH, Samuelsen O, Krämer M, Vorland LH. 2002. Proteases in Escherichia coli and Staphylococcus aureus confer reduced susceptibility to lactoferricin-B. J. Antimicrob. Chemother. 50:461–467 [DOI] [PubMed] [Google Scholar]
- 40. Yu L, Ding JL, Ho B, Feng S-S, Wohland T. 2008. Investigation of the mechanisms of antimicrobial peptides interacting with membranes by fluorescence correlation spectroscopy. Open Chem. Phys. J. 1:62–79 [Google Scholar]
- 41. Hancock RE, Diamond G. 2000. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 8:402–410 [DOI] [PubMed] [Google Scholar]
- 42. Park SC, Park Y, Hahm KS. 2011. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int. J. Mol. Sci. 12:5971–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peschel A, Sahl HS. 2006. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4:529–536 [DOI] [PubMed] [Google Scholar]
- 44. Tossi A, Scocchi M, Zahariev S, Gennaro R. 2012. Use of unnatural amino acids to probe structure activity relationships and mode of action of antimicrobial peptides. Methods Mol. Biol. 794:169–183 [DOI] [PubMed] [Google Scholar]
- 45. Tossi A, Sandri L, Giangaspero A. 2000. Amphipathic alpha-helical antimicrobial peptides. Biopolymers 55:4–30 [DOI] [PubMed] [Google Scholar]
- 46. Wu M, Maier E, Benz R, Hancock RE. 1999. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 38:7235–7242 [DOI] [PubMed] [Google Scholar]
- 47. Scott MG, Hancock RE. 2000. Cationic antimicrobial peptides and their multifunctional role in the immune system. Crit. Rev. Immunol. 20:407–431 [PubMed] [Google Scholar]
- 48. Shai Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1462:55–70 [DOI] [PubMed] [Google Scholar]
- 49. Neville F, Cahuzac M, Nelson A, Gidalevitz D. 2004. The interaction of antimicrobial peptide LL-37 with artificial biomembranes: epifluorescence and impedance spectroscopy approach. J. Phys. Condens. Matter 16:2413–2420 [Google Scholar]
- 50. Majerle A, Kidric J, Jerala R. 2003. Enhancement of antibacterial and lipopolysaccharide binding activities of a human lactoferricin peptide fragment by the addition of acyl chain. J. Antimicrob. Chemother. 51:1159–1165 [DOI] [PubMed] [Google Scholar]
- 51. Porter EA, Weisblum B, Gellman SH. 2002. Mimicry of host-defense peptides by unnatural oligomers: antimicrobial beta-peptides. J. Am. Chem. Soc. 124:7324–7330 [DOI] [PubMed] [Google Scholar]
- 52. Findlay B, Zhanel GG, Schweizer F. 2010. Cationic amphiphiles, a new generation of antimicrobials inspired by the natural antimicrobial peptide scaffold. Antimicrob. Agents Chemother. 54:4049–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Darville T, Jacobs RF. 2004. Management of acute hematogenous osteomyelitis in children. Pediatr. Infect. Dis. J. 23:255–257 [DOI] [PubMed] [Google Scholar]
- 54. Ellington JK, Harris M, Webb L, Smith B, Smith T, Tan K, Hudson M. 2003. Intracellular Staphylococcus aureus: a mechanism for the indolence of osteomyelitis. J. Bone Joint Surg. 85:918–921 [PubMed] [Google Scholar]
- 55. Krogstad P. 2004. Osteomyelitis and septic arthritis, p 713–736 In Feigin RD, Cherry JD, Demmler GJ, Kaplan SL. (ed), Textbook of pediatric infectious diseases, 5th ed Saunders, Philadelphia, PA [Google Scholar]
- 56. Proctor RA, Balwit JM, Vesga O. 1994. Variant subpopulations of Staphylococcus aureus as cause of persistent and recurrent infections. Infect. Agents Dis. 3:302–312 [PubMed] [Google Scholar]
- 57. von Eiff C, Becker K, Metze D, Lubritz G, Hockmann J, Schwarz T, Peters G. 2001. Intracellular persistence of Staphylococcus aureus small-colony variants within keratinocytes: a cause for antibiotic treatment failure in a patient with Darier's disease. Clin. Infect. Dis. 32:1643–1647 [DOI] [PubMed] [Google Scholar]
- 58. Bosse MJ, Gruber HE, Ramp WK. 2005. Internalization of bacteria by osteoblasts in a patient with recurrent, long-term osteomyelitis. A case report. J. Bone Joint Surg. Am. 87:1343–1347 [DOI] [PubMed] [Google Scholar]
- 59. Beck NK, Callahan K, Nappier SP, Kim H, Sobsey MD, Meschkei JS. 2009. Development of a spot-titer culture assay for quantifying bacteria and viral indicators. J. Rapid Methods Autom. Microbiol. 17:455–464 [Google Scholar]
- 60. Herigstad B, Hamilton M, Heersink J. 2001. How to optimize the drop plate method for enumerating bacteria. J. Microbiol. Methods 44:121–129 [DOI] [PubMed] [Google Scholar]
- 61. Ahmed S, Meghji S, Williams RJ, Henderson B, Brock JH, Nair SP. 2001. Staphylococcus aureus fibronectin binding proteins are essential for internalization by osteoblasts but do not account for differences in intracellular levels of bacteria. Infect. Immun. 69:2872–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ellington JK, Reilly SS, Ramp WK, Smeltzer MS, Kellam JF, Hudson MC. 1999. Mechanisms of Staphylococcus aureus invasion of cultured osteoblasts. Microb. Pathog. 26:317–323 [DOI] [PubMed] [Google Scholar]
- 63. Hamza T, Li B. 2012. Staphylococcus aureus internalization and associated osteoblast responses, paper 0919. Orthopaedic Res. Soc. (ORS) Annu. Meet., 4 to 7 February 2012, San Francisco, CA [Google Scholar]
- 64. Jevon M, Guo C, Ma B, Mordan N, Nair SP, Harris M, Henderson B, Bentley G, Meghji S. 1999. Mechanisms of internalization of Staphylococcus aureus by cultured human osteoblasts. Infect. Immun. 67:2677–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Testoni F, Montanaro L, Poggi A, Visai L, Campoccia D, Arciola CR. 2011. Internalization by osteoblasts of two Staphylococcus aureus clinical isolates differing in their adhesin gene pattern. Int. J. Artif. Organs 34:789–798 [DOI] [PubMed] [Google Scholar]
- 66. Wu JA, Kusuma C, Mond JJ, Kokai-Kun JF. 2003. Lysostaphin disrupts Staphylococcus aureus and Staphylococcus epidermidis biofilms on artificial surfaces. Antimicrob. Agents Chemother. 47:3407–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Armstead AL, Li B. 2011. Nanomedicine as an emerging approach against intracellular pathogens. Int. J. Nanomed. 6:3281–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Briones E, Colino CI, Lanao JM. 2008. Delivery systems to increase the selectivity of antibiotics in phagocytic cells. J. Control. Release 125:210–227 [DOI] [PubMed] [Google Scholar]
- 69. Hudson MC, Ramp WK, Nicholson NC, Williams AS, Nousiainen MT. 1995. Internalization of Staphylococcus aureus by cultured osteoblasts. Microb. Pathog. 19:409–419 [DOI] [PubMed] [Google Scholar]
- 70. Marriott I. 2004. Osteoblast responses to bacterial pathogens—a previously unappreciated role for bone-forming cells in host defense and disease progression. Immunol. Res. 30:291–308 [DOI] [PubMed] [Google Scholar]
- 71. Sendi P, Proctor RA. 2009. Staphylococcus aureus as an intracellular pathogen: the role of small colony variants. Trends Microbiol. 17:54–58 [DOI] [PubMed] [Google Scholar]
- 72. Sinha B, Fraunholz M. 2010. Staphylococcus aureus host cell invasion and post-invasion events. Int. J. Med. Microbiol. 300:170–175 [DOI] [PubMed] [Google Scholar]
- 73. Li H, Hamza T, Clovis NB, Smith S, Tidwell J, Li B. 2012. Antimicrobial properties of platelet-rich plasma—in vitro and in vivo studies, paper 0271. Orthopaedic Res. Soc. (ORS) Annu. Meet., 4 to 7 February 2012, San Francisco, CA [Google Scholar]
- 74. Chen X, Niyonsaba F, Ushio H, Okuda D, Nagaoka I, Ikeda S, Okumura K, Ogawa H. 2005. Synergistic effect of antibacterial agents human b-defensins, cathelicidin LL-37 and lysozyme against Staphylococcus aureus and Escherichia coli. J. Dermatol. Sci. 40:123–132 [DOI] [PubMed] [Google Scholar]
- 75. Barańska-Rybak W, Sonesson A, Nowicki R, Schmidtchen A. 2006. Glycosaminoglycans inhibit the antibacterial activity of LL-37 in biological fluids. J. Antimicrob. Chemother. 57:260–265 [DOI] [PubMed] [Google Scholar]
- 76. Hancock RE, Sahl HG. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551–1557 [DOI] [PubMed] [Google Scholar]
- 77. Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. 1998. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 273:3718–3724 [DOI] [PubMed] [Google Scholar]
- 78. Ciornei CD, Sigurdardottir T, Schmidtchen A, Bodelsson M. 2005. Antimicrobial and chemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and inhibition by serum of analogs of human cathelicidin LL-37. Antimicrob. Agents Chemother. 49:2845–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fukumoto K, Nagaoka I, Yamataka A, Kobayashi H, Yanai T, Kato Y, Miyano T. 2005. Effect of antibacterial cathelicidin peptide CAP18/LL-37 on sepsis in neonatal rats. Pediatr. Surg. Int. 21:20–24 [DOI] [PubMed] [Google Scholar]
- 80. Wang G, Epand RF, Mishra Lushnikova BT, Thomas VC, Kenneth W, Bayles KW, Epand RM. 2012. Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob. Agents Chemother. 56:845–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boyce BM, Lindsey BA, Clovis NB, Smith ES, Hobbs GR, Hubbard DF, Emery SE, Barnett JB, Li B. 2012. Additive effects of exogenous IL-12 supplementation and antibiotic treatment in infection prophylaxis. J. Orthop. Res. 30:196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li B, Jiang B, Boyce BM, Lindsey BA. 2009. Multilayer polypeptide nanoscale coatings incorporating IL-12 for the prevention of biomedical device-associated infections. Biomaterials 30:2552–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li B, Jiang B, Dietz MJ, Smith ES, Clovis NB, Rao KM. 2010. Evaluation of local MCP-1 and IL-12 nanocoatings for infection prevention in open fractures. J. Orthop. Res. 28:48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]