

Abstract
Horizontally transferred DNA acquired through transformation and recombination has the potential to contribute to the diversity and evolution of naturally competent bacteria. However, many different factors affect the efficiency with which DNA can be transformed and recombined. In this study, we determined how the size of both homologous and nonhomologous regions affects transformation and recombination efficiencies in Xylella fastidiosa, a naturally competent generalist pathogen responsible for many emerging plant diseases. Our experimental data indicate that 96 bp of flanking homology is sufficient to initiate recombination, with recombination efficiencies increasing exponentially with the size of the homologous flanking region up to 1 kb. Recombination efficiencies also decreased with the size of the nonhomologous insert, with no recombination detected when 6 kb of nonhomologous DNA was flanked on either side by 1 kb of homologous sequences. Upon analyzing sequenced X. fastidiosa subsp. fastidiosa genomes for evidence of allele conversion, we estimated the mean size of recombination events to be 1,906 bp, with each event modifying, on average, 1.79% of the nucleotides in the recombined region. There is increasing evidence that horizontally acquired genes significantly affect the genetic diversity of X. fastidiosa, and DNA acquired through natural transformation could be a prominent mode of this horizontal transfer.
INTRODUCTION
Homologous recombination is an important mechanism by which horizontally transferred DNA is incorporated into the genome of the recipient organism. In bacteria, DNA can be horizontally acquired through three main mechanisms: transformation, conjugation, and transduction. Transformation is a likely route of DNA acquisition by naturally competent taxa, which are found in a wide range of phyla (1). Once inside the cell, DNA can be incorporated into the genome or degraded; it has been hypothesized that natural competence originally evolved as a nutrient uptake system (2). However, there is potential for horizontally acquired DNA to alter the recipient's fitness and phenotype if the DNA is recombined into the genome. Horizontal gene transfer through natural transformation can increase the rate of adaptation of pathogens to new environments (3). Natural transformation increased the pathogenicity of a Ralstonia solanacearum isolate when it recombined with DNA from a highly virulent strain (4). Furthermore, it has been shown that adding a single gene to Vibrio fischeri is sufficient to alter its host range (5). Thus, horizontal transfer of even small segments of DNA can have significant evolutionary effects.
Several factors can limit horizontal gene transfer through natural transformation and recombination. Gene transfer between organisms can be hampered by geographic separation. The lack of DNA uptake sequences, short DNA sequences recognized and preferentially bound by competent cells, in donor DNA can prevent the recipient from efficiently binding and transporting the DNA across the membrane (6). Restriction enzymes can degrade unmethylated or improperly methylated DNA in the cytoplasm before it is recombined into the genome (7, 8). In addition, the sequence similarity and size of transforming DNA can strongly regulate recombination efficiency. In several organisms across domains, recombination efficiency decreases exponentially with sequence divergence (9–13). However, in cases where sexual isolation is caused by the difficulty of strand invasion of highly divergent sequences, the presence of flanking regions of identity can remove most recombinatorial barriers (10). The minimal efficient processing segment (MEPS), or the shortest length of sequence homology necessary for efficient recombination, can vary greatly, depending on the organism, the recombination pathway used, and other factors. In Escherichia coli, efficient recombination has been observed with as little as 23 bp of sequence homology (13). The MEPS requirements for eukaryotes has been studied extensively, revealing great variation in this process: recombination efficiency in Trypanosoma brucei requires 50 bp of homology for efficient recombination, while essentially no recombination was observed in Saccharomyces cerevisiae with fewer than 248 bp of flanking homology (14, 15). The total size of transforming DNA can also affect recombination efficiency; R. solanacearum can naturally transform and recombine 90 kb of DNA, but efficiencies were 3 orders of magnitude lower than when the transforming DNA was 1 kb in length (16).
Xylella fastidiosa is a plant-pathogenic bacterium that colonizes the xylem vessels of its host plants and inhibits the flow of xylem sap, resulting in symptoms such as leaf scorching and stunted growth (17). It is the causative agent of several economically important diseases, such as Pierce's disease of grapevine, citrus variegated chlorosis, and coffee leaf scorch (18). Recently, this bacterium was shown to be naturally competent and able to homologously recombine acquired DNA into its genome in laboratory environments (19). Multilocus sequence typing (MLST) studies have also shown evidence of recombination between different strains of X. fastidiosa, with horizontally acquired sequences potentially playing a greater role in generating diversity than point mutations (20–22). The different strains and subspecies of X. fastidiosa share a highly conserved core gene pool containing genes likely responsible for adaptation to life in the plant xylem but also have a highly diverse flexible gene pool, potentially responsible for its wide host range (23). However, it is also possible that different alleles, as opposed to unique genes, are responsible for the varied phenotypes of X. fastidiosa: altering gene expression in one strain of X. fastidiosa increased its virulence to a different host plant (24). Recently emerged pathogenic strains of X. fastidiosa may have resulted from the horizontal transfer of elements in the flexible gene pool present in endemic populations to strains introduced from new geographic regions (23).
There is increasing evidence that horizontally acquired DNA affects the evolution of X. fastidiosa, with significant ecological consequences, and that natural transformation is a likely route for this to occur. This study examined how the characteristics of transforming DNA affect the ability of X. fastidiosa to naturally transform and recombine it into its genome. We examined this process in two different contexts: the integration of novel DNA experimentally and its occurrence in natural populations, as evident from genome sequence comparisons. The first context provides insight into how the flanking homologous region affects the recombination efficiency and how much novel DNA can be inserted by recombination, neither of which has been studied extensively in bacteria, while the second shows the extent of the effects of recombination in a population of X. fastidiosa.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The X. fastidiosa subsp. fastidiosa strain Temecula (25) was used in this study. Cells were grown in either periwinkle wilt Gelrite (PWG) medium (26) or in modified X. fastidiosa medium (XFM) (19). Where appropriate, kanamycin was added to a final concentration of 30 μg/ml. We used E. coli strain EAM1 (8), which expresses an X. fastidiosa methylase, to propagate plasmids. Previous work has shown that transformation and recombination efficiencies are higher for methylated plasmids than for unmethylated plasmids (8, 19).
Plasmid construction.
Plasmids for testing the effects of both flanking homology length and insert length were created by using the pGEM-5zf(+) vector backbone (Promega, Madison, WI), which cannot independently replicate in X. fastidiosa. Flanking homology length plasmids were created by amplifying genomic DNA from an rpfF mutant (27) using primers annealing approximately 26, 35, 50, 96, 200, 508, 760, 1,000, 2,000, or 4,000 bp upstream and downstream of the kanamycin resistance marker with a SacI restriction site engineered into the 5′ end of each primer (Table 1). PCR constructs and the vector backbone were digested with SacI and ligated to create p26, p35, p50, etc. (Fig. 1). Plasmids were transformed into E. coli strain EAM1 for propagation as previously described (19).
Table 1.
Primers used to construct plasmids
| Primer use and name | Sequence (5′–3′) |
|---|---|
| Homology length testing | |
| 26fwd | CATGAGCTCCGTATCAGGTCACAA |
| 26rev | CCGGAGCTCTACCATTACGGAGA |
| 35fwd | TGTGAGCTCTCCTTACGGCGTATC |
| 35rev | ATAGAGCTCCGACCGGACTACCAT |
| 50fwd | ATCGAGCTCAATAATGCTTCACGC |
| 50rev | AAAGAGCTCCGTCCGCAACAT |
| 96fwd | TAAGAGCTCAGCATGGAACGCATA |
| 96rev | GCAGAGCTCGCACATAGAATCAAGT |
| 200fwd | TACGAGCTCCCTTCTTCAGCTACG |
| 200rev | ATAGAGCTCGACCGCCCTATTCC |
| 508fwd | ACAGAGCTCGTTCGGTGATGC |
| 508rev | GAAGAGCTCAATGCAGTGACGC |
| 760fwd | ATTGAGCTCTGTGGTGGTAAAGCG |
| 760rev | ATAGAGCTCGTATCCCAGATTGGCA |
| 1kfwd | AAAGAGCTCCAGGTGTTCGATCC |
| 1krev | CGAGAGCTCCCTGGTACATCAGTC |
| 2kfwd | ATAGAGCTCCTCCTTGAAGGAGGTGA |
| 2krev | TTAGAGCTCAGTGTGGCACCACTTC |
| 4kfwd | ATTGAGCTCTCAACCTATGCTGCCT |
| 4krev | ATAGAGCTCCAACGCCAAGAACAC |
| Insert length testing | |
| F1 fwd SphI | ATAGCATGCCAGGTGTTCGATCC |
| F1 rev NcoI | AACCATGGACGGGCTGTCTCTTATAC |
| F2 fwd SalI | TAAGTCGACGTACAGCGGACATTTATTG |
| F2 rev SacI | CGAGAGCTCCCTGGTACATCAGTC |
| Insert rev NcoI | ATCCCATGGTAGAACAACCATTTATCG |
| 1k fwd NotI | TATAGCGGCCGCATGACAGTCCATGAAG |
| 2k fwd NotI | TATAGCGGCCGCTCTATTGATGGCTAGG |
| 3k fwd NotI | TATAGCGGCCGCTCTGCGGATAAAGGTA |
| 5k fwd NotI | TATAGCGGCCGCCAGTGATGGTGG |
Fig 1.

Recombination efficiencies of plasmids with flanking regions varying from 96 to 4,000 bp (A) and from 26 to 1,000 bp (B; note that the x axis in panel B is on a log scale). Solid lines and squares show the recombination efficiencies (number of recombinants per total number of cells present) after normalization for the total number of transforming DNA units present. Dotted lines and empty diamonds show overall recombination efficiencies. Recombination efficiencies peaked with approximately 1,000 bp of flanking region and then plateaued. Recombinants were recovered with as few as 96 bp of homology, but rates were essentially 0. Images on the right depict the regions cloned into the vector backbone pGEM-5zf(+) to create plasmids with various flanking regions. Homologous DNA was amplified from the rpfF region of X. fastidiosa.
To construct plasmids with different lengths of nonhomologous DNA, we first used primers F1 fwd SphI and F1 rev NcoI (Table 1) to amplify the kanamycin resistance cassette and approximately 1 kb of DNA upstream of the KanR insertion site within the rpfF mutant. This was digested with SphI and NcoI and ligated into pGEM-5zf(+) to create pS1. Approximately 1 kb of flanking DNA immediately downstream of the kanamycin resistance marker in the rpfF mutant was amplified by using F2 fwd SalI and F2 rev SacI. We then digested this fragment and pS1 with SalI and SacI and ligated the two together to create pS2. Nonhomologous insert DNA fragments ranging in size from 1 to 5 kb with an NcoI site at one end and a NotI site at the other were amplified from cDNA from a single-stranded positive-sense RNA plant virus with no homology to any region of the X. fastidiosa genome (GenBank accession number JQ655296.1). All fragments were amplified from the region of approximately bp 3100 to 8100 by using the primers listed in Table 1. PCR products and pS2 were digested with NcoI and NotI and ligated together to form pS2-1k, pS2-2k, pS2-3k, and pS2-5k (Fig. 2B). Plasmids were transformed into E. coli strain EAM1 for propagation.
Fig 2.
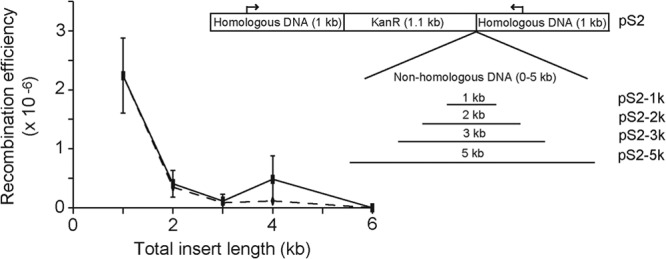
Recombination efficiencies of plasmids with nonhomologous inserts of different lengths ranging from 1 to 6 kb. Solid lines and squares show the recombination efficiencies (number of recombinants per total number of cells present) after normalization for the total number of transforming DNA units present. Dotted lines and empty diamonds show the overall recombination efficiencies. No recombinants were recovered when the total insert length was 6 kb. The image on the right depicts the region cloned into the vector backbone pGEM-5zf(+) to create the plasmids. The flanking region size was kept constant at approximately 1 kb on either side. The nonhomologous insert region consisted of a kanamycin resistance cassette (approximately 1.1 kb) and 0 to 5 kb of DNA amplified from a plant virus with no regions of homology to the X. fastidiosa genome (the total insert size for pS2 is approximately 1 kb, that for pS2-1k is approximately 2 kb, etc.). Arrows over the homologous DNA regions show the locations of primers rpfF-fwd and rpfF-rev, which were used to screen antibiotic-resistant colonies for proper insertion of the kanamycin resistance cassette and nonhomologous DNA.
Transformation protocol.
X. fastidiosa cells were harvested from PWG plates after approximately 5 to 7 days of growth and resuspended in 200 μl of XFM to a final optical density at 600 nm of approximately 0.01. After 2 days of growth at 28°C with constant shaking, we added the appropriate plasmids to a final concentration of 5 μg/ml. After an additional 24 h of growth, cultures were plated on PWG with kanamycin and an aliquot was frozen for quantification. We counted antibiotic-resistant colonies after approximately 14 days of growth. We confirmed the insertion of the antibiotic resistance marker and additional nonhomologous DNA (if appropriate) at the correct locus occurred by double recombination events through PCR analysis of a random sample of antibiotic-resistant colonies using primers rpfF-fwd and rpfF-rev (19), which anneal approximately 730 bp upstream and 350 bp downstream of the putative kanamycin resistance marker and nonhomologous DNA (if appropriate) insertion site. These primers produce an amplicon of about 1,200 bp in cells with the wild-type locus and one of 2 kb in cells with the kanamycin resistance cassette inserted in the proper location. Additional nonhomologous DNA from the plasmids was also amplified by these primers, with recombination with pS2-1k producing a fragment of approximately 3 kb, recombination with pS2-2k producing a fragment of 4 kb, etc. Cells were quantified by using quantitative PCR as previously described (19). Recombination efficiencies were calculated on the basis of the number of antibiotic-resistant colonies divided by the total number of cells present. Fifteen replicates for each plasmid were used to measure the effect of insert length on recombination efficiency; 6 to 15 replicates were used to test each plasmid with different homologous flanking regions. The estimated limit of detection was calculated as previously described (28).
Genome comparisons.
Homologous recombination events were inferred with the program ClonalFrame (29), which models bacterial evolution as the diversification of a clonal population with polymorphisms arising because of a combination of mutations that affect single nucleotides and allele conversions that affect contiguous regions of the chromosome and modify a small portion of the nucleotides in that region. The analysis included four publicly available chromosome sequences from X. fastidiosa subsp. fastidiosa: Temecula1 (GenBank accession no. AE009442.1), M23 (accession no. CP001011.1), GB514 (accession no. CP002165.1), and EB92.1 (accession no. AFDJ00000000.1). Chromosomes were aligned by using progressiveMauve (30) with default settings. Small aligned blocks (<5 kb) were removed with the program stripSubsetLCBs; small alignment blocks are less likely to represent segments of the ancestral chromosome, as reflected in the radically higher levels of polymorphism (data not shown). Coding regions conserved among X. fastidiosa subsp. fastidiosa, multiplex, and pauca were identified as reciprocal best BLAST hits among Temecula1, M12 (RefSeq accession no. NC_010513.1 [26 January 2012]), and 9a5c (accession no. NC_002488.3) by using the software DNAMaster.
RESULTS
Recombination efficiency depends on homologous sequence length.
We observed maximum recombination efficiency of a naturally transformed plasmid into the X. fastidiosa genome when the approximately 1-kb kanamycin resistance marker was flanked on both sides by 1 kb of homologous X. fastidiosa DNA. The maximum recombination efficiency (number of recombinants recovered per total number of cells present) was 5.62 × 10−5 (Fig. 1A). Overall recombination efficiencies decreased for plasmids with 2- and 4-kb flanking regions, but when efficiencies were normalized for the different plasmid sizes (thus accounting for the total number of plasmids added), there was no significant difference among the recombination efficiencies of plasmids with 1, 2, and 4 kb of homologous flanking regions (P = 0.025). An analysis of variance and a Tukey ad hoc test indicated that the recombination efficiencies of plasmids with 1 and 4 kb of flanking sequence homology were significantly higher than those of plasmids with 96, 200, 508, and 760 bp of flanking sequence homology (P < 1 × 10−4). When taking into account the size difference of the plasmids, recombination efficiency was reduced by an order of magnitude when the flanking region was decreased from 1,000 to 760 bp; a further decrease by an order of magnitude occurred between 760 and 508 bp. Recombination efficiency increased exponentially with the length of the flanking region in the range of 508 to 1,000 bp (r = 0.83, P < 1 × 10−5).
In separate trials, we tested the recombination efficiencies of plasmids with flanking regions ranging from 26 bp to 1,000 bp (Fig. 1B). A single instance of recombination mediated by 96 bp of flanking region was observed, but no other instance of recombination of plasmids with less than 508 bp of homology flanking the kanamycin resistance cassette was detected by the methods described here. The estimated limit of detection of recombination rates was approximately 6 × 10−8. Random samples of antibiotic-resistant colonies from each treatment were analyzed by PCR to confirm that additive integration at the correct locus occurred by a double recombination event. All amplicon lengths were as expected.
Recombination efficiency decreases exponentially with the size of the inserted nonhomologous DNA fragment.
Recombination of pS2, which contained a kanamycin resistance marker flanked on either side by 1 kb of homologous DNA, occurred in approximately 1 out of every 2.24 × 10−6 cells (Fig. 2). Increasing the size of the nonhomologous region by 1 kb (by using plasmid pS2-1k) decreased the recombination efficiency by almost an order of magnitude (4.65 × 10−7). The recombination efficiencies of pS2-2k and pS2-3k, having an additional 2 or 3 kb of nonhomologous DNA in addition to the 1-kb kanamycin resistance marker, were 1.42 × 10−7 and 2.31 × 10−7, respectively. We did not detect recombination between pS2-5k, having a total nonhomologous insert size of approximately 6 kb, and the X. fastidiosa genome. The estimated limit of detection was approximately 9 × 10−8.
Recombination occurs in natural populations.
The software package ClonalFrame (29) was used to analyze four published X. fastidiosa subsp. fastidiosa genomes to detect recombination events resulting in allele conversion. The estimates of the ClonalFrame parameters indicated that the ratio of recombination events to mutation events (ρ/θ) was 0.48, while the ratio of the number of nucleotides changed by recombination to the number of nucleotides changed through mutation (r/m) was 15. This suggests that allele conversion was a major contributor to clonal diversification, occurring half as often as point mutations but contributing much more to the accumulation of polymorphisms in the core genome of this population. The average size of each allele conversion event was 1,906 bp, with 1.79% of the nucleotide sequence changed (95% credibility regions of 1,464 to 2,392 bp and 1.71 to 1.87%, respectively). To compare the diversities of possible donor DNA sequences, we found that the nucleotide sequence identity between the shared coding regions of X. fastidiosa subsp. fastidiosa and multiplex was 98.4% and sequence identity between the shared coding regions of X. fastidiosa subsp. fastidiosa and pauca was 97%.
DISCUSSION
Horizontal gene transfer plays a large role in generating genetic diversity in a wide range of bacterial species, and natural transformation can be an important way for organisms to acquire novel DNA sequences. The average import size of DNA acquired through transformation for a variety of naturally competent bacteria is in the range of 1 to 10 kb (31–33), although natural transformation and recombination of much larger segments of DNA have been demonstrated (4, 16). Recombination of shorter fragments may be inefficient because of degradation during uptake and processing, or such events could be undetectable. Our experimental data indicated that natural transformation in a population of X. fastidiosa is consistent with these size parameters, as was the average size of recombination events as determined by the ClonalFrame analysis. These data support our hypothesis that transformation is an important driver of horizontal gene transfer in X. fastidiosa.
To determine the size requirements of recombination events in natural isolates, we performed a ClonalFrame analysis of four genomes within X. fastidiosa subsp. fastidiosa. The small sample size resulted from the lack of full genome sequences available within the subspecies. We could not include genomes from outside X. fastidiosa subsp. fastidiosa, as there is evidence of nonuniform substitution rates between different subspecies (34), which violates one of the assumptions of ClonalFrame (29). In addition, including genomes from the entire species led to long branches between subspecies, decreasing the robustness of the results (data not shown). Lastly, large insertion/deletion events in single genomes would not be detected in this analysis, as these regions would not align properly (29). The average nucleotide replacement rate for each recombination event was 1.79%, suggesting that X. fastidiosa is routinely able to recombine with genomes with 98.2% similarity. On the basis of the sequence alignments of the shared coding regions of various X. fastidiosa genomes, we found that X. fastidiosa subsp. fastidiosa and multiplex share 98.4% sequence identity of their aligned regions, supporting conclusions from previous MLST studies (20–22) that recombination between different subspecies occurs at relatively high frequencies. Since ClonalFrame models recombined fragments as originating from a single population with the estimated level of divergence, it will have largely ignored recombination events originating from within X. fastidiosa subsp. fastidiosa, which would have altered very few nucleotides. As the biology of the different subspecies varies in terms of pathogenicity and host range (35), recombination could potentially result in the emergence of new strains with markedly different phenotypes.
A log-linear relationship has been established between sequence divergence and recombination efficiencies for a number of recombining bacteria, although the slope of the curve varies between species (9, 11, 36). Decreasing sequence similarity to approximately 90% reduces the recombination efficiency by 1 order of magnitude for Streptococcus pneumoniae (11) and 3 orders of magnitude for Bacillus subtilis (36). Although we did not determine the actual relationship between sequence similarity and recombination efficiency, the average replacement rate for recombination events in X. fastidiosa, which indicates that, on average, 1.79% of the donor DNA nucleotide sequence differs from the sequence of the recipient DNA, is consistent with previously reported data.
Our experimental work determined the parameters needed for efficient additive integration, which required a double-crossover event to allow the insertion of novel DNA flanked by two regions of sequence homology. Homology-facilitated illegitimate recombination, where a single region of high sequence similarity initiates recombination that extends into areas of low sequence similarity, can also occur, although this generally happens at frequencies several orders of magnitude lower than for double-crossover events (1). However, since our PCR analysis of antibiotic-resistant colonies used primers that annealed to the homologous flanking region surrounding the insert, we were able to confirm that the entire nonhomologous region was inserted into the genome at the correct locus, strongly suggesting a double-crossover event.
We observed recombination of naturally transformed DNA with as little as 96 bp of flanking homology. However, it is possible that the MEPS for X. fastidiosa is lower than 96 bp but that recombination with these plasmids occurred at frequencies below our limit of detection (approximately 10−8). In comparison, the MEPS is 23 bp for E. coli, approximately 50 bp for R. solanacearum, and approximately 70 bp for B. subtilis (13, 37, 38). We observed an exponential relationship between flanking region length and recombination efficiency for X. fastidiosa up to 1 kb, after which recombination efficiency was insensitive to increases in flanking region length. As the X. fastidiosa species shares a conserved set of core genes, as well as more diverse flexible genes (23), the sequence similarity provided by the conserved core genes could facilitate the recombination of more variable genes responsible for host adaptation or pathogenicity. However, unlike with sequence similarity and recombination efficiency, there is no consistent relationship between homologous flanking region length and recombination efficiency among different organisms. In R. solanacearum, for example, there appears to be a logarithmic relationship between homologous flanking region length and recombination efficiency, while in B. subtilis and S. cerevisiae, the relationship is linear over the dynamic range (15, 37, 38). The differences among organisms could be based on the recombination pathway used or the efficiency of RecA, which detects homology between the donor and recipient DNAs (13).
Our experimental data also provide a sense of how much nonhomologous DNA can be inserted into the X. fastidiosa genome, which is relevant for assessing the potential of natural transformation and recombination to affect, for example, adaptation to novel environments. Previous work with X. fastidiosa has shown that altering a single gene can alter pathogenicity (24), illustrating how the insertion or replacement of as little as 1 kb of DNA can significantly affect the phenotype. In R. solanacearum, recombination efficiencies decreased exponentially with the length of the integrated nonhomologous DNA fragment (16). A similar relationship was observed in X. fastidiosa. No insertion of a DNA fragment longer than 4 kb was observed, but it is possible that the integration of longer fragments occurred at frequencies below our limit of detection. During the transformation process, donor DNA is typically fragmented and reduced to single-stranded DNA before entering the cytoplasm (39). Fragmentation of plasmids with long stretches of nonhomologous DNA is more likely to result in pieces containing only one homologous region, reducing recombination efficiencies. Likewise, for plasmids with long homologous flanking regions, fragmentation could trim the flanking region, possibly explaining why there was no difference in recombination efficiency for plasmids with 1 to 4 kb of flanking homology. Although our experiments only tested for recombination at a single locus in any given cell, studies with other naturally competent organisms have shown that an individual cell is capable of acquiring multiple independent strands of DNA at different loci by recombination (31, 40). It is possible that X. fastidiosa is also capable of such multiple cases of horizontal gene acquisition.
The analysis presented here illustrates the DNA size requirements necessary for efficient transformation and recombination in X. fastidiosa. We have also paired experimental data with in silico genome analysis to sample the effects of recombination on X. fastidiosa in natural environments. There is evidence that isolates of X. fastidiosa have been transferred between geographic regions (20, 23), and it has been hypothesized that the strain responsible for causing Pierce's disease in the United States diverged from an isolate introduced from Costa Rica (34). Understanding the DNA requirements for recombination could provide insights into how natural transformation affects the evolution of X. fastidiosa and other naturally competent bacteria.
ACKNOWLEDGMENTS
We thank Steve Lindow and our laboratory colleagues for providing helpful suggestions and discussions.
S.H.K. was supported by a USDA NIFA predoctoral fellowship. A.C.R. was supported by a postdoctoral fellowship from the Miller Institute for Basic Research in Science. Funding was also provided by the California Agricultural Experiment Station.
Footnotes
Published ahead of print 11 January 2013
REFERENCES
- 1. Thomas CM, Nielsen KM. 2005. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3:711–721 [DOI] [PubMed] [Google Scholar]
- 2. Redfield RJ. 1993. Genes for breakfast: the have-your-cake-and-eat-it-too of bacterial transformation. J. Hered. 84:400–404 [DOI] [PubMed] [Google Scholar]
- 3. Baltrus DA, Guillemin K, Phillips PC. 2008. Natural transformation increases the rate of adaptation in the human pathogen Helicobacter pylori. Evolution 62:39–49 [DOI] [PubMed] [Google Scholar]
- 4. Coupat-Goutaland B, Bernillon D, Guidot A, Prior P, Nesme X, Bertolla F. 2011. Ralstonia solanacearum virulence increased following large interstrain gene transfers by natural transformation. Mol. Plant Microbe Interact. 24:497–505 [DOI] [PubMed] [Google Scholar]
- 5. Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG. 2009. A single regulatory gene is sufficient to alter bacterial host range. Nature 458:215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lorenz MG, Wackernagel W. 1994. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 58:563–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kobayashi I. 2001. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29:3742–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsumoto A, Igo MM. 2010. Species-specific type II restriction-modification system of Xylella fastidiosa temecula1. Appl. Environ. Microbiol. 76:4092–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vulić M, Dionisio F, Taddei F, Radman M. 1997. Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobacteria. Proc. Natl. Acad. Sci. U. S. A. 94:9763–9767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Majewski J, Cohan FM. 1999. DNA sequence similarity requirements for interspecific recombination in Bacillus. Genetics 153:1525–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Majewski J, Zawadzki P, Pickerill P, Cohan FM, Dowson CG. 2000. Barriers to genetic exchange between bacterial species: Streptococcus pneumoniae transformation. J. Bacteriol. 182:1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Opperman R, Emmanuel E, Levy AA. 2004. The effect of sequence divergence on recombination between direct repeats in Arabidopsis. Genetics 168:2207–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen P, Huang HV. 1986. Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics 112:441–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barnes RL, McCulloch R. 2007. Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases. Nucleic Acids Res. 35:3478–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jinks-Robertson S, Michelitch M, Ramcharan S. 1993. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 13:3937–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coupat B, Chaumeille-Dole F, Fall S, Prior P, Simonet P, Nesme X, Bertolla F. 2008. Natural transformation in the Ralstonia solanacearum species complex: number and size of DNA that can be transferred. FEMS Microbiol. Ecol. 66:14–24 [DOI] [PubMed] [Google Scholar]
- 17. McElrone AJ, Sherald JL, Forseth IN. 2001. Effects of water stress on symptomatology and growth of Parthenocissus quinquefolia infected by Xylella fastidiosa. Plant Dis. 85:1160–1164 [DOI] [PubMed] [Google Scholar]
- 18. Chatterjee S, Almeida RPP, Lindow S. 2008. Living in two worlds: the plant and insect lifestyles of Xylella fastidiosa. Annu. Rev. Phytopathol. 46:243–271 [DOI] [PubMed] [Google Scholar]
- 19. Kung SH, Almeida RPP. 2011. Natural competence and recombination in the plant pathogen Xylella fastidiosa. Appl. Environ. Microbiol. 77:5278–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Almeida RPP, Nascimento FE, Chau J, Prado SS, Tsai CW, Lopes SA, Lopes JRS. 2008. Genetic structure and biology of Xylella fastidiosa strains causing disease in citrus and coffee in Brazil. Appl. Environ. Microbiol. 74:3690–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scally M, Schuenzel EL, Stouthamer R, Nunney L. 2005. Multilocus sequence type system for the plant pathogen Xylella fastidiosa and relative contributions of recombination and point mutation to clonal diversity. Appl. Environ. Microbiol. 71:8491–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan X, Morano L, Bromley R, Spring-Pearson S, Stouthamer R, Nunney L. 2010. Multilocus sequence typing of Xylella fastidiosa causing Pierce's disease and oleander leaf scorch in the United States. Phytopathology 100:601–611 [DOI] [PubMed] [Google Scholar]
- 23. Nunes LR, Rosato YB, Muto NH, Yanai GM, Da Silva VS, Leite DB, Gonçalves ER, De Souza AA, Coletta-Filho HD, Machado MA, Lopes SA, De Oliveira RC. 2003. Microarray analyses of Xylella fastidiosa provide evidence of coordinated transcription control of laterally transferred elements. Genome Res. 13:570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Killiny N, Almeida RPP. 2011. Gene regulation mediates host specificity of a bacterial pathogen. Environ. Microbiol. Rep. 3:791–797 [DOI] [PubMed] [Google Scholar]
- 25. Van Sluys MA, de Oliveira MC, Monteiro-Vitorello CB, Miyaki CY, Furlan LR, Camargo LEA, da Silva ACR, Moon DH, Takita MA, Lemos EGM, Machado MA, Ferro MIT, da Silva FR, Goldman MHS, Goldman GH, Lemos MVF, El-Dorry H, Tsai SM, Carrer H, Carraro DM, de Oliveira RC, Nunes LR, Siqueira WJ, Coutinho LL, Kimura ET, Ferro ES, Harakava R, Kuramae EE, Marino CL, Giglioti E, Abreu IL, Alves LMC, do Amaral AM, Baia GS, Blanco SR, Brito MS, Cannavan FS, Celestino AV, da Cunha AF, Fenille RC, Ferro JA, Formighieri EF, Kishi LT, Leoni SG, Oliveira ARVER, Jr, Sassaki FT, Sena JA, de Souza AA, Truffi D, Tsukumo F, Yanai GM, Zaros LG, Civerolo EL, Simpson AJ, Almeida NF, Jr, Setubal JC, Kitajima JP. 2003. Comparative analyses of the complete genome sequences of Pierce's disease and citrus variegated chlorosis strains of Xylella fastidiosa. J. Bacteriol. 185:1018–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill B, Purcell AH. 1995. Acquisition and retention of Xylella fastidiosa by an efficient vector, Graphocephala atropunctata. Phytopathology 85:209–212 [Google Scholar]
- 27. Newman KL, Almeida RPP, Purcell AH, Lindow SE. 2004. Cell-cell signaling controls Xylella fastidiosa interactions with both insects and plants. Proc. Natl. Acad. Sci. U. S. A. 101:1737–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nielsen KM, Bones AM, Van Elsas JD. 1997. Induced natural transformation of Acinetobacter calcoaceticus in soil microcosms. Appl. Environ. Microbiol. 63:3972–3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175:1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147 doi:10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin EA, Zhang Levine X-SSM, Gill SR, Falush D, Blaser MJ. 2009. Natural transformation of Helicobacter pylori involves the integration of short DNA fragments interrupted by gaps of variable size. PLoS Pathog. 5:e1000337 doi:10.1371/journal.ppat.1000337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fornili SL, Fox MS. 1977. Electron microscope visualization of the products of Bacillus subtilis transformation. J. Mol. Biol. 113:181–191 [DOI] [PubMed] [Google Scholar]
- 33. Linz B, Schenker M, Zhu P, Achtman M. 2000. Frequent interspecific genetic exchange between commensal neisseriae and Neisseria meningitidis. Mol. Microbiol. 36:1049–1058 [DOI] [PubMed] [Google Scholar]
- 34. Nunney L, Yuan X, Bromley R, Hartung J, Montero-Astúa M, Moreira L, Ortiz B, Stouthamer R. 2010. Population genomic analysis of a bacterial plant pathogen: novel insight into the origin of Pierce's disease of grapevine in the U.S. PLoS One 5:e15488 doi:10.1371/journal.pone.0015488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hopkins DL, Purcell AH. 2002. Xylella fastidiosa: Cause of Pierce's disease of grapevine and other emergent diseases. Plant Dis. 86:1056–1066 [DOI] [PubMed] [Google Scholar]
- 36. Zawadzki P, Roberts MS, Cohan FM. 1995. The log-linear relationship between sexual isolation and sequence divergence in Bacillus transformation is robust. Genetics 140:917–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bertolla F, Gijsegem FV, Nesme X, Simonet P. 1997. Conditions for natural transformation of Ralstonia solanacearum. Appl. Environ. Microbiol. 63:4965–4968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khasanov FK, Zvingila DJ, Zainullin AA, Prozorov AA, Bashkirov VI. 1992. Homologous recombination between plasmid and chromosomal DNA in Bacillus subtilis requires approximately 70 bp of homology. Mol. Gen. Genet. 234:494–497 [DOI] [PubMed] [Google Scholar]
- 39. Chen I, Christie PJ, Dubnau D. 2005. The ins and outs of DNA transfer in bacteria. Science 310:1456–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mell JC, Shumilina S, Hall IM, Redfield RJ. 2011. Transformation of natural genetic variation into Haemophilus influenzae genomes. PLoS Pathog. 7(7):e1002151 doi:10.1371/journal.ppat.1002151 [DOI] [PMC free article] [PubMed] [Google Scholar]