

Abstract
Large numbers of bacteria coexist in the oral cavity. Streptococcus sanguinis, one of the major bacteria in dental plaque, produces hydrogen peroxide (H2O2), which interferes with the growth of other bacteria. Streptococcus mutans, a cariogenic bacterium, can coexist with S. sanguinis in dental plaque, but to do so, it needs a means of detoxifying the H2O2 produced by S. sanguinis. In this study, we investigated the association of three oxidative stress factors, Dpr, superoxide dismutase (SOD), and AhpCF, with the resistance of S. sanguinis to H2O2. The knockout of dpr and sod significantly increased susceptibility to H2O2, while the knockout of ahpCF had no apparent effect on susceptibility. In particular, dpr inactivation resulted in hypersensitivity to H2O2. Next, we sought to identify the factor(s) involved in the regulation of these oxidative stress genes and found that PerR negatively regulated dpr expression. The knockout of perR caused increased dpr expression levels, resulting in low-level susceptibility to H2O2 compared with the wild type. Furthermore, we evaluated the roles of perR, dpr, and sod when S. mutans was cocultured with S. sanguinis. Culturing of the dpr or sod mutant with S. sanguinis showed a significant decrease in the S. mutans population ratio compared with the wild type, while the perR mutant increased the ratio. Our results suggest that dpr and sod in S. mutans are involved in coexistence with S. sanguinis, and PerR is associated with resistance to H2O2 in regulating the expression of Dpr.
INTRODUCTION
The oral cavity is colonized by large numbers of bacteria, i.e., the indigenous microflorae (1, 2). The oral microflorae are considered to act as components of an innate immune system, which protects the host from infection by exogenous pathogenic bacteria. Oral bacteria, however, can sometimes cause infectious diseases, such as tooth decay, periodontitis, and aspiration pneumonitis. Among the oral bacteria, each bacterial species competes and/or coexists with other bacteria. To compete, bacteria produce antibacterial agents, such as bacteriolytic enzymes, bacteriocins, and hydrogen peroxide (3–5). In oral streptococci, especially Streptococcus sanguinis and Streptococcus gordonii, the production of hydrogen peroxide (H2O2) is well known as an antibacterial agent against other bacterial species (5–7).
H2O2 is one of the reactive oxygen species (ROS), which also include the superoxide anion (O2−) and hydroxyl radicals (•OH) (8, 9). These ROS cause serious damage to cellular macromolecules such as proteins and DNA. Although the toxicity of H2O2 is relatively weak compared with that of other ROS, H2O2 is converted nonenzymatically to the highly toxic hydroxyl radical via the Fenton pathway (H2O2 + Fe2+ → •OH + −OH + Fe3+) in the presence of iron (Fe). Generally, aerobes and facultative anaerobes have efficient mechanisms for protection against ROS during aerobic respiration or encounters with oxidative stress due to neutrophils or bacteria. Several factors for protection against ROS, such as catalase, superoxide dismutase (SOD), Dps-like protein, alkylhydroperoxide reductase (AhpCF), glutathione reductase, and thiol reductase, have been identified in many bacterial species (9–14). Among them, catalase, AhpCF, and Dps-like protein are considered to be primarily responsible for resistance to H2O2 (Fig. 1) (15). Catalase and AhpCF directly decompose H2O2, and Dps-like protein inhibits the reaction of the Fenton pathway by capturing free Fe.
Fig 1.
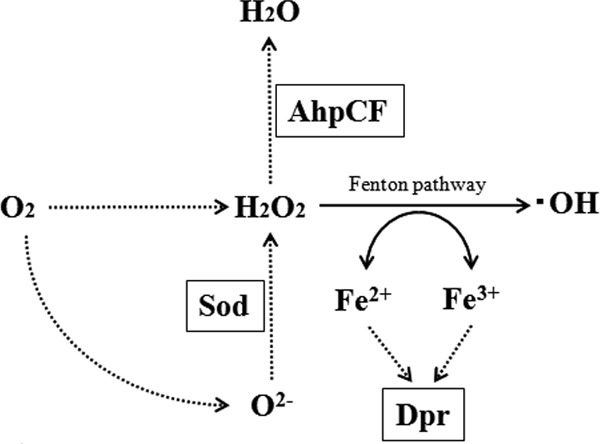
Function of Sod, AhpCF, and Dpr in S. mutans. Sod, superoxide dismutase; AhpCF, alkylhydroperoxide reductase; Dpr, Dps-like peroxidase resistance protein (15).
Streptococcus mutans, one of the commensal bacteria in the oral cavity, is known to be a cariogenic pathogen in humans (16, 17). S. mutans is able to attach to the smooth surfaces of teeth and to form biofilm known as dental plaque with other oral bacteria. Although S. sanguinis is a dominant bacterium in dental plaque, S. mutans coexists there with S. sanguinis. S. sanguinis is known to produce large amounts of H2O2, causing it to have antibacterial activity against several bacteria (5, 7, 18–20). Thus, to persist in dental plaque, S. mutans requires a resistance mechanism against H2O2 produced by S. sanguinis. To date, AhpCF and Dps-like protein (Dpr) have been identified and characterized in the resistance of S. mutans to ROS (11, 19, 21–23). In particular, Yamamoto et al. reported previously that Dpr was a major factor in resistance to oxidative stress during respiration (24). However, no comparative analysis of the contribution of the factors responsible for resistance to oxidative stress by S. sanguinis has been documented for S. mutans. Furthermore, the regulatory mechanism(s) for the expression of these factors has not been determined for S. mutans. To date, the peroxide regulator, PerR, has been demonstrated to be associated with resistance to oxidative stress in several bacteria, including other streptococci (25–28).
In this study, we investigated the contribution of three factors, Dpr, Sod, and AhpCF, to resistance against H2O2 produced by S. sanguinis and also identified a regulatory factor responsible for Dpr expression. Furthermore, we demonstrated that two factors in S. mutans are critical for coexisting with S. sanguinis in vivo by using an established coculture method.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
S. mutans strains used are listed in Table 1. S. mutans and S. sanguinis GTC217 were grown in Trypticase soy broth (TSB; Becton Dickinson Microbiology Systems, Cockeysville, MD) or Trypticase soy agar (TSA) at 37°C with 5% CO2. Erythromycin (EM) (10 μg ml−1) and spectinomycin (SPC) (600 μg ml−1) were added when necessary for S. mutans.
Table 1.
S. mutans strains used in this study
| Strain | Descriptionb | Reference |
|---|---|---|
| UA159 | Laboratory strain | 41 |
| Δdpr | SMU.540a deletion mutant; Emr | This study |
| Δsod | SMU.629 deletion mutant; Emr | This study |
| ΔahpCF | SMU.764–765 deletion mutant; Emr | This study |
| ΔperR | SMU.593 deletion mutant; Emr | This study |
| ΔperR::perR complement | perR complemented in ΔperR; Emr Spcr | This study |
| ΔΔperR dpr | perR dpr double deletion mutant; Emr Spcr | This study |
| ΔΔperR sod | perR sod double deletion mutant; Emr Spcr | This study |
| ΔΔperR ahpCF | perR ahpCF double deletion mutant; Emr Spcr | This study |
The GenBank locus tag was from the S. mutans UA159 genome at the NCBI database.
Emr, erythromycin resistance; Spcr, spectinomycin resistance.
Construction of perR, dpr, sod, and ahpCF single deletion mutants and perR dpr, perR sod, and perR ahpCF double deletion mutants.
We constructed the perR, dpr, sod, and ahpCF single deletion mutants via an overlapping-extension PCR approach, as described previously (29). To generate the dpr deletion mutant, two fragments corresponding to approximately 500 bp of the upstream and downstream sequences of this gene were generated by PCR using KOD Plus (Toyobo, Tokyo, Japan) with primer pairs for the upstream fragment Δdpr-F1 and the downstream fragment Δdpr-F2 (Table 2). Each of the F1 reverse and F2 forward primers incorporated 11 bases that were complementary to the erythromycin resistance cassette Emr, which was cloned by using pBlueScript-Em (Stratagene, La Jolla, CA) (30). The Emr gene was amplified by PCR using primers for Emr. All PCR amplicons were purified by using the Qiagen PCR purification kit (Qiagen KK, Tokyo, Japan), and the corresponding upstream and downstream amplicons were mixed in a 1:1:1 ratio with the Emr PCR product. The amplicon mixture was then used as a template for a second PCR using appropriate forward Δdpr-F1 and reverse Δdpr-F2 primers. The resulting PCR products were then transformed into UA159, as described previously (30). Additionally, the fragments of ΔΔperR-F1 and ΔΔperR-F2 were mixed in a 1:1:1 ratio with the Spcr PCR product cloned from pBSSK-Spc (29). This mixture was then used as a template for a second PCR using appropriate forward ΔΔperR-F1 and reverse ΔΔperR-F2 primers. The resulting PCR products were also transformed into the dpr, sod, and ahpCF single deletion mutants to construct double deletion mutants. The mutation was verified by PCR.
Table 2.
primers used in this study
| Primer | Description | Sequencea |
|
|---|---|---|---|
| Forward | Reverse | ||
| Δdpr-F1 | Upstream of dpr for Δdpr | AAGCCTATCCACAGTGT | CAGTCGAGGATCCTCCTTAAAAAATATTCTT |
| Δdpr-F2 | Downstream of dpr for Δdpr | GCTGACCTAGTAAAAGACAATCTGGATGTT | CATCTTCCAAAATATTGGT |
| Δsod-F1 | Upstream of sod for Δsod | GTCTTCAGGGAGAAGATG | CAGTCGAGGATTCATTTCCTCTTTTCTTTT |
| Δsod-F2 | Downstream of sod for Δsod | GCTGACCTAGTCTGTTGCTCGTCTTTATG | AACGAGCTATTCCATTCT |
| ΔahpCF-F1 | Upstream of ahpC for ΔahpCF | GAATTGTGGAAAGTCGC | CAGTCGAGGATACACTTGTCCTCCTTCT |
| ΔahpCF-F2 | Downstream of ahpF for ΔahpCF | GCTGACCTAGTGCTTTAGGTGCCTTTGA | TCGGAGTTCTCTCAACT |
| ΔperR-F1 | Upstream of perR for ΔperR | TTGAGTGCTTGGTTGAAT | CAGTCGAGGATTTGACTCCTTCGTTATTC |
| ΔperR-F2 | Downstream of perR for ΔperR | GCTGACCTAGTATTTGTCCAGACTGTCAA | GCCAATGTTCATGCTTTT |
| perR-compl.-F1 | perR complementation | TGCGGATCCTTGATGATGCTAGGCAC | TCTTTGTTTCTTACCTCCTATTTCCCATAT |
| Emr | Erythromycin resistance gene for deletion | ATCCTCGACTGGAAGCAAACTTAAGAGTG | ACTAGGTCAGCTTATTTCCTCCCGTTAAA |
| Spcr | Spectinomycin resistance gene for complementation, double deletion | ATCCTCGACTGATCGATTTTCGTTCGTGA | ACTAGGTCAGCTTCCACCATTTTTTCAATTT |
| ftf-N-terminal | Complementation | ATCTCGAGTTTACTAAGTTCAACAATGG | AAGTCGACCCACCAATAACATTCCAAT |
| ftf-C-terminal | Complementation | AAGAAACAAAGAAAGCTCATCATGTTTCAAC | CGGCCGCGGTTCGTCTTGTTTCTCTCA |
| ΔΔperR-F1 | Upstream of perR for double deletion | TTGAGTGCTTGGTTGAATb | CAGTCGAGGATTTGACTCCTTCGTTATTCb |
| ΔΔperR-F2 | Downstream of perR for double deletion | GCTGACCTAGTATTTGTCCAGACTGTCAAb | GCCAATGTTCATGCTTTTb |
| dpr | Primers for quantitative PCR for dpr | GTGGTTCAGGCTTCCTTTAT | ACTGTTTCTTCAAGTCTGGA |
| sod | Primers for quantitative PCR for sod | GTTTTGGCTCAGGTTGGGCT | ATAGTTTGGACGAACATTAC |
| ahpC | Primers for quantitative PCR for ahpC | TGGTTTAGCACAACGTGGAA | AGGGCAAACTTCTCCTGGAT |
| sloR | Primers for quantitative PCR for sloR | ACTGTCTCTGATGTGTTTGT | TCAACAAATATACTCCCATC |
| gyrA | Primers for quantitative PCR for gyrA | TCTCGCTGGACTTGTCACTG | CATCTAGGCGCATCACTTTG |
Restriction enzyme sequences are underlined.
Same primer as ΔperR-F1 or ΔperR-F2.
Construction of complement strains.
For genetic complementation, we constructed a DNA fragment to insert the Spcr and perR genes into the ftf gene, encoding fructosyltransferase. First, the plasmid and fused fragment were prepared. To prepare the plasmid, the ftf N-terminal fragment was transferred into the N terminus of the Spcr gene in pBSSK-Spc using XhoI and SalI sites (pBBSK-Spc::ftf-N-terminal). To prepare the fused fragment, the perR-compl.-F1 fragment and ftf-C-terminal fragment were generated by PCR using KOD Plus (Toyobo) with the primer pairs (Table 2). The perR-compl.-F1 reverse primer and the ftf-C-terminal forward primer added an extra 12 nucleotides to anneal each PCR fragment, and the perR-compl.-F1 forward primer and the ftf-C-terminal reverse primer contained BamHI and SacII restriction enzyme sites. The perR-compl.-F1 and the ftf-C-terminal fragments were mixed at a 1:1 ratio, and the mixture was then used as a template for a second PCR using the appropriate perR-compl.-F1 forward primer and ftf-C-terminal reverse primer. The BamHI- and SacII-digested amplified fragment was ligated with pBBSK-Spc::ftf-N-terminal digested with BamHI and SacII and then transformed into Escherichia coli XL-II (obtained plasmid, pBBSK-Spc::ftf-N-terminal+perR+ftf-C-terminal). The DNA fragment to insert the Spcr and perR genes into the ftf gene was obtained from pBBSK-Spc::ftf-N-terminal+perR+ftf-C-terminal using the ftf-N-terminal forward primer and the ftf-C-terminal reverse primer. The resulting PCR product was then transformed into the perR deletion mutant to construct the perR complement strain. By selecting for erythromycin and spectinomycin resistance, complementation strains were isolated. Finally, in the strain obtained, Spcr and perR gene insertion into the ftf gene was verified by PCR.
Quantitation of H2O2 concentration in culture medium.
The concentration of H2O2 was measured by using a method described previously (31). A small portion (108 cells) of an S. sanguinis culture grown overnight was inoculated into 10 ml TSB and then incubated at 37°C with 5% CO2. The culture supernatant at the appropriate phase was prepared by centrifugation (10,000 × g for 5 min) of the bacterial culture. Each culture supernatant (0.2 ml) was reacted with the reaction solution (0.8 ml) and incubated at 37°C for 20 min. The components of the reaction solution were 10 mM sodium phosphate buffer (pH 7.4), 0.16 mM o-dianisidine, 1.2 μg ml−1 horseradish peroxidase, and 0.02% Triton X-100. The absorbance at 570 nm was measured, and the concentration was calculated from a standard curve prepared from an experiment using various concentrations of H2O2.
Quantitative analysis of gene expression by quantitative PCR.
A small portion of S. mutans cells cultured overnight was inoculated into fresh TSB. S. mutans cells were then grown at 37°C with 5% CO2, and bacterial cells in the exponential phase were collected. Total RNA was extracted from bacterial cells with a FastRNA Pro Blue kit (MP Biomedicals, Cleveland, OH), according to the manufacturer's protocol. Total RNA (1 μg) was reverse transcribed to cDNA by using a first-strand cDNA synthesis kit (Roche, Tokyo, Japan). Using the cDNA as a template, quantitative PCR was performed by using the MyiQ system (Bio-Rad, Tokyo, Japan). The primers used are shown in Table 2. Since the DNA gyrase A subunit (gyrA) was stably expressed and used as the internal control for quantitative PCR (32, 33), the amount of gyrA was used as an internal control in this study. Three independent experiments were performed, and the means ± standard deviations (SD) were calculated. Data were analyzed for statistically significant differences from the UA159 control by a two-way analysis of variance (ANOVA) followed by Dunnett's post hoc tests.
Competition assay on agar plates.
To investigate the growth inhibition ability of S. sanguinis GTC217 against S. mutans, a competition assay was performed, as described previously (19). Briefly, 10 μl of an S. sanguinis culture grown overnight was spotted onto TSA as the pioneer colonizer. After incubation for 16 h at 37°C with 5% CO2, 10 μl of an S. mutans culture grown overnight, as the competitor, was spotted at a distance where the bacterial cells almost touched each other. After incubation overnight, the growth inhibition of S. mutans was evaluated. For the control, 10 μl of each S. mutans strain was spotted onto TSA without S. sanguinis to confirm the growth of each strain. This assay was performed under anaerobic conditions by using the GasPack system (Mitsubishi Gas Chemical Company Inc., Tokyo, Japan). When necessary, before spotting, the competitor strain with catalase solution (100 μg ml−1) was spotted at the same site where the competitor strain was spotted.
H2O2 was also used for evaluating susceptibility. An H2O2 solution (0.35 or 0.5%) was spotted onto TSA, and a bacterial culture of S. mutans was then spotted near the H2O2 solution. After incubation overnight, the growth inhibition of S. mutans was evaluated. We preliminarily used various concentrations of H2O2 and found that 0.35% H2O2 was the minimal concentration which did not inhibit the growth of the wild type and that 0.5% H2O2 was the concentration which showed 50% growth inhibition of the wild type.
Assay to determine the antibacterial activity of H2O2.
S. mutans strains at the appropriate phase were harvested, washed with phosphate-buffered saline (PBS), and suspended in 10 mM sodium phosphate buffer (PB). The bacterial suspension was diluted to 107 cells ml−1 with PB, and 10 μl of the bacterial suspension (105 cells) was inoculated into 500 μl of TSB with or without 0.04% H2O2 and incubated for 30 min at 37°C with 5% CO2. We preliminarily investigated the effect of various concentrations of H2O2 on susceptibility to H2O2 and found that the wild type showed 50% survival when exposed to 0.04% H2O2. Dilutions of the reaction mixture (100 μl) were plated onto agar medium and incubated at 37°C overnight. The CFU were determined as the total number of colonies identified on each plate. The antibacterial effect was calculated as the ratio of the number of surviving cells (percent survival rate) to the total number of bacteria incubated in TSB without 0.04% H2O2. Also, the dilutions of the mixture without 0.04% H2O2 prior to incubation were plated onto TSA and incubated at 37°C overnight. The CFU of each strain were determined as the total cell numbers prior to the reaction. Three independent experiments were performed, and the mean ± SD was calculated. Data were analyzed for statistically significant differences from the UA159 control by a two-way ANOVA followed by Dunnett's post hoc tests.
Coculture of S. mutans with S. sanguinis.
Cultures of S. mutans (wild type and perR mutant) and S. sanguinis grown overnight were adjusted to an optical density at 660 nm (OD660) of 1.0 and diluted 10-fold. One hundred microliters of each bacterial culture (10- and 100-fold-diluted cultures for S. sanguinis and 10-fold-diluted culture for S. mutans) was then mixed well. For the control without coculture, 100 μl of S. mutans culture was mixed with 100 μl of TSB. A 20-μl aliquot of the nonmixed culture (S. mutans only) or the mixed culture was spotted onto 50% TSA plates. When necessary, 10 μl of catalase solution (100 μg ml−1) was also spotted after the bacterial solution was spotted. After 8 h of incubation at 37°C with 5% CO2, the bacterial colonies growing on the agar plate were scraped and suspended in 500 μl of PBS. Appropriate dilutions were plated onto TSA and TSA containing 32 μg ml−1 of bacitracin. After incubation at 37°C with 5% CO2 for 24 h, the CFU were counted, and the ratio of S. mutans with CFU from TSA containing bacitracin in the mixed culture/CFU from TSA in the nonmixed culture was then calculated. We also extracted total RNA from scraped cells and performed cDNA synthesis using the method described above, and gene expression analysis was conducted by quantitative PCR. Three independent experiments were performed, and the mean ± SD was calculated. The statistical difference between UA159 and the perR mutant was determined by two-sided Student's t test. A difference with a P value of <0.05 was considered significant.
Next, the coculture of S. mutans perR, dpr, sod, and ahpC mutants with S. sanguinis was investigated by using the method described above. The concentration of bacterial cells used in this assay was 108 cells ml−1 S. mutans mutant and 107 cells ml−1 S. sanguinis. For the coculture of S. mutans with S. sanguinis, TSA containing 32 μg ml−1 of bacitracin was used for S. mutans selection. Data were analyzed for statistically significant differences from the UA159 control by a two-way ANOVA followed by Dunnett's post hoc tests.
RESULTS
Susceptibility of sod, ahpCF, and dpr mutants to S. sanguinis and H2O2.
We first investigated whether the S. sanguinis strain used had the capability to produce H2O2. We found that S. sanguinis could produce H2O2 during growth (Fig. 2). Next, we investigated the association of three factors, dpr, sod, and ahpCF, which are known to be involved in resistance to oxidative stress. We constructed dpr, sod, and ahpCF mutants for further experiments. First, we checked the growth of the mutants and found that the dpr, sod, and ahpCF mutants grew well in TSB or on 50% TSA under anaerobic and aerobic conditions, although the dpr and sod mutants grew slowly compared to the wild type under aerobic conditions (data not shown). In a competition assay, the growth of S. mutans UA159 was partially inhibited by S. sanguinis (Fig. 3A), and this inhibition was abolished when the bacterial suspension was mixed with catalase (100 μg ml−1) before spotting onto the TSA plate (data not shown).
Fig 2.
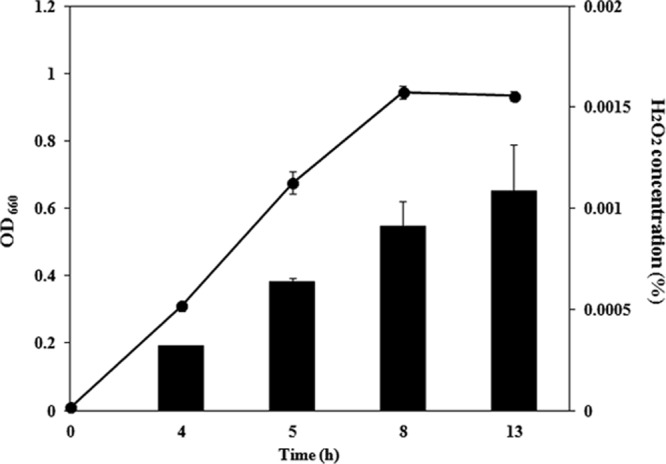
H2O2 production in S. sanguinis during growth. A small portion (108 cells) of an S. sanguinis culture grown overnight was inoculated into 10 ml TSB and then incubated at 37°C with 5% CO2. Culture supernatant at the appropriate phase was prepared, and the H2O2 concentration was measured as described in Materials and Methods (black bar). The line represents the growth curve of S. sanguinis.
Fig 3.
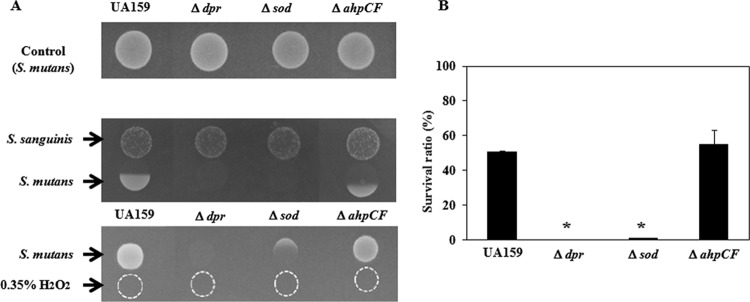
Susceptibility of S. mutans dpr, sod, and ahpCF mutants to S. sanguinis and H2O2. (A) Competition assay. A culture of S. sanguinis grown overnight (10 μl) was spotted onto TSA as the pioneer colonizer. After incubation for 16 h at 37°C with 5% CO2, 10 μl of a culture of S. mutans grown overnight as the competitor was spotted at a distance where both bacterial cells almost attached to each other. After incubation overnight, the growth inhibition of S. mutans was evaluated. Also, a 0.35% H2O2 solution was spotted onto TSA, and the S. sanguinis culture was then spotted using the same method as that described above. In the top panel, only S. mutans strains were spotted onto TSA and incubated for 16 h at 37°C with 5% CO2. (B) Quantitative assay. The bacterial suspension (105 cells) was inoculated into 500 μl of TSB with or without 0.04% H2O2 and incubated at 37°C with 5% CO2 for 30 min. Dilutions of the reaction mixture (100 μl) were plated onto agar medium and incubated at 37°C with 5% CO2 overnight. The CFU were determined as the total number of colonies identified on each plate. The antibacterial effect was calculated as the ratio of the number of surviving cells (percent survival rate) to the total number of bacteria incubated in control PB solution after exposure to H2O2. *, P < 0.005 compared to wild-type strain UA159, as determined by Dunnett's tests.
We then performed the competition assay using the mutants (Fig. 3A). When the dpr, sod, and ahpCF mutants without S. sanguinis were spotted onto TSA, we confirmed that the growth of these mutants was similar to that of the wild type. The dpr and sod mutants increased the growth-inhibitory area compared with the wild-type strain. In fact, the dpr and sod mutants showed almost no growth against S. sanguinis. As a result of the competition assay using 0.35% H2O2, the dpr mutant showed almost no growth, while the sod mutant displayed slight growth compared with that of the dpr mutant (Fig. 3A). In a quantitative assay using TSB containing 0.04% H2O2, we investigated the susceptibility of the mutants to H2O2 (Fig. 3B). The results were almost the same as those of the competition assay. In the absence of H2O2, the cell numbers of the wild type and the dpr, sod, and ahpCF mutants were not changed during 30 min of incubation. In the presence of 0.04% H2O2, the wild-type strain demonstrated almost 50% survival, while the dpr and sod mutants showed 0% and 1.0% survival, respectively. The ahpCF mutant displayed susceptibility (55% survival) similar to that of the wild type.
Susceptibility of the perR mutant to H2O2.
The perR mutant showed a decrease in the growth-inhibitory zone compared with that of the wild type, while the growth of the perR complementation strain showed an inhibition zone similar to that of the wild type (Fig. 4A). Instead of S. sanguinis, an H2O2 solution (0.5%) was used for the competition assay, and a similar tendency was found. The perR mutant demonstrated decreased susceptibility compared with that of the wild type, while the perR complementation strain showed susceptibility similar to that of the wild type. In a quantitative assay for susceptibility to H2O2, the wild type and the complementation strain showed 50.8% and 57.5% survival against 0.04% H2O2, respectively, while the perR mutant displayed 83.3% survival (Fig. 4B).
Fig 4.
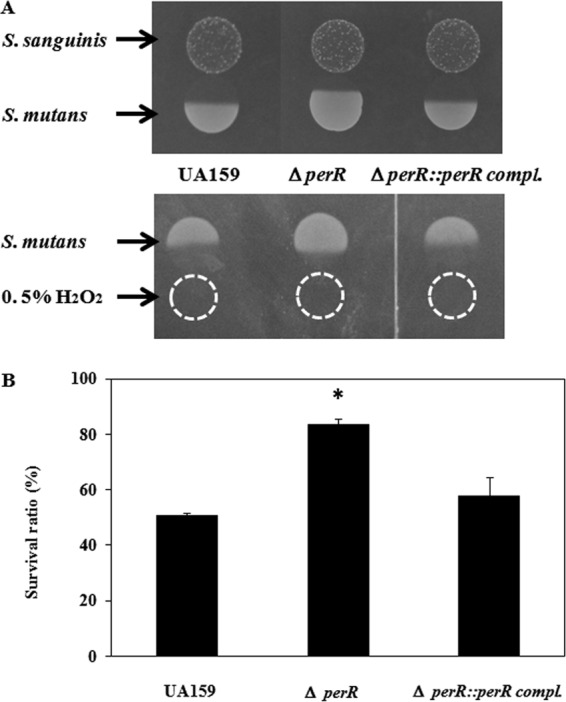
Susceptibility of the S. mutans perR mutant and its complemented strain to S. sanguinis and H2O2. Using wild-type S. mutans strain UA159 and the perR mutant and complemented strain, a competition assay (A) and a quantitative assay (B) were performed as described in Materials and Methods. *, P < 0.05 compared to wild-type strain UA159, as determined by Dunnett's tests.
Expression of factors responsible for resistance to oxidative stress in the perR mutant.
We investigated the expression of sod, ahpC, and dpr in the perR mutant (Fig. 5). The expression level of dpr in the perR mutant increased 2.5-fold compared with that of the wild type. The expression levels of sod and ahpC were increased 1.5- and 1.6-fold, respectively, compared with that of the wild type. In the perR-complemented strain, the expression level was similar to that of the wild type.
Fig 5.

Expression of dpr, sod, and ahpC in the perR mutant. A small portion of S. mutans cells cultured overnight was inoculated into fresh TSB. S. mutans cells were then grown at 37°C with 5% CO2, and bacterial cells at an OD660 of 0.3 were collected. After extraction of total RNA and cDNA synthesis, quantitative PCR was performed as described in Materials and Methods. The amount of gyrA was used as an internal control. *, P < 0.05 compared to wild-type strain UA159, as determined by Dunnett's tests.
We also investigated the perR box upstream of the dpr open reading frame (ORF), which has been reported to have a binding region for PerR. Based on the consensus sequence (NTANAANNATTNTAN) in Streptococcus pyogenes and Bacillus subtilis (25, 27), we found that a PerR box was found at 57 to 43 bases upstream of the dpr start codon, with the consensus sequence TTAGAATCGTTCTAA (data not shown). We also found a perR box at 121 to 107 bases upstream of the sod ORF, with the sequence TTAGAATTATTTTAC (data not shown). However, we found no consensus sequence upstream of the ahpC ORF (data not shown).
H2O2 susceptibility of the perR dpr, perR sod, and perR ahpCF double-knockout mutants.
We investigated the susceptibility of double mutants (perR dpr, perR sod, and perR ahpCF) to clarify the intrinsic factor for decreased resistance in the perR mutant. Before the competition assay, we confirmed that wild-type S. mutans strain UA159 and the double deletion mutants on TSA without S. sanguinis grew well. The perR dpr double-knockout mutant showed a phenotype very similar to that of the dpr mutant, with almost no growth in the competition assay (Fig. 6A). In the perR sod double mutant, the susceptibility to H2O2 decreased compared with that of the sod mutant, while the susceptibility of this double mutant to H2O2 increased compared with that of the perR single mutant. The perR ahpCF double mutant showed decreased susceptibility to H2O2 compared with that of the wild type. We also investigated the expression of dpr in the sod single mutant, ahpCF single mutant, perR sod double mutant, and perR ahpCF double mutant (Fig. 6B). For both of the double mutant strains, the dpr expression level increased compared with that of the single mutant.
Fig 6.
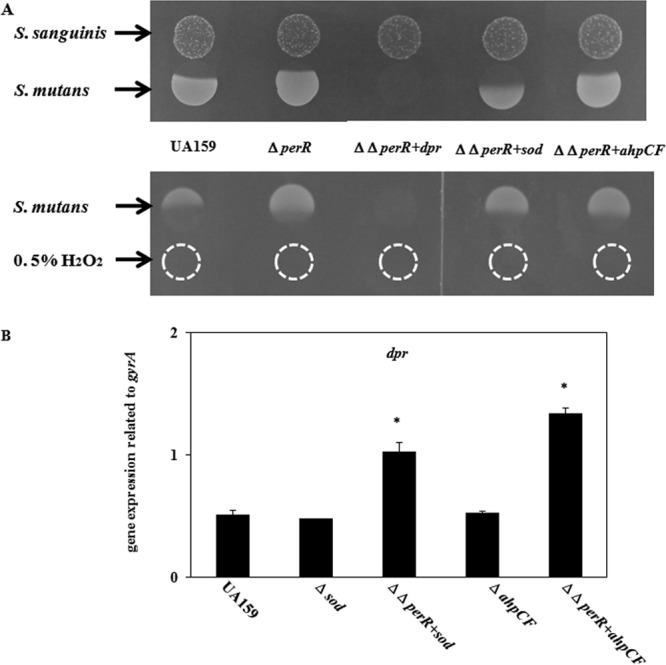
Susceptibility of S. mutans double mutants to S. sanguinis and H2O2. (A) Using wild-type S. mutans strain UA159, the perR mutant, and double mutants (perR with dpr, sod, or ahpCF), a competition assay was performed as described in Materials and Methods. (B) Expression of dpr in the double mutants was investigated as described in Materials and Methods. *, P < 0.005 compared to wild-type strain UA159, as determined by Dunnett's tests.
Coculture of S. mutans mutants with S. sanguinis.
Cocultures of the S. mutans wild type and mutants with S. sanguinis were analyzed. Figure 7A shows the percent ratios of the S. mutans population (wild type and the perR mutant) when various cell numbers (107, 106, and 105 cells) of S. sanguinis were mixed with 106 cells of S. mutans and spotted onto TSA plates, followed by incubation for 8 h at 37°C. For the mixed ratios of S. mutans with S. sanguinis at 1:10, 1:1, and 10:1, the population of the wild type accounted for approximately 0.3%, 3.4%, and 46.0%, respectively, while that of perR mutant accounted for approximately 0.8%, 15.3%, and 100%, respectively. When S. mutans was cocultured with S. sanguinis in the presence of catalase (100 μg ml−1), the population ratios of the S. mutans wild type and perR mutant were 100% and 99.7%, respectively.
Fig 7.

Coculture of S. mutans with S. sanguinis. Cultures of S. mutans and S. sanguinis grown overnight were adjusted to an OD660 of 1.0 and diluted 10- to 100-fold. One hundred microliters of S. mutans was then mixed with 100 μl S. sanguinis or 100 μl TSB. A 20-μl aliquot of the nonmixed culture and mixed culture was spotted onto 50% TSA. When necessary, 10 μl of catalase solution (100 μg/ml) was also spotted after the bacterial solution was spotted. After 8 h of incubation at 37°C with 5% CO2, the bacterial colonies growing on the agar plate were scraped and suspended in 1 ml TSB. Appropriate dilutions were plated onto TSA and TSA containing antibacterial agents. After 2 days of incubation, the CFU on the plates containing TSA and TSA containing antibiotics were counted, and the ratio of S. mutans with CFU from TSA containing bacitracin in mixed culture/CFU from TSA in nonmixed culture was determined. In addition, total RNA was extracted from the scraped cells, and cDNA synthesis was performed by using the method described above. Finally, a gene expression analysis was conducted by using quantitative PCR. (A) Percent ratio of the S. mutans population when mixed with various concentrations of S. sanguinis. *, P < 0.05 compared to wild-type strain UA159, as determined by t test for the percent ratio of the S. mutans population. (B) dpr expression of S. mutans UA1159 and the perR mutant. *, P < 0.05 compared to wild-type strain UA159, as determined by t test. (C) Percent ratio of the S. mutans population with or without the addition of catalase solution when mixed with S. sanguinis (1:0.1). *, P < 0.05 compared to wild-type strain UA159, as determined by Dunnett's tests.
We analyzed the expression of dpr under coculture conditions (Fig. 7B). The expression level of dpr in the perR mutant significantly increased (P < 0.05) compared with that in the wild type when S. mutans was cocultured with S. sanguinis at various ratios.
Next, we investigated the population ratio of S. mutans mutants cocultured with S. sanguinis (Fig. 7C). In the mixed S. mutans-S. sanguinis population at a ratio of 10:1, the populations of the wild type and the dpr, sod, and ahpCF mutants were 46.0%, 0.2%, 1.8%, and 40.2%, respectively. When S. mutans mutants were cocultured with S. sanguinis in the presence of catalase (100 μg ml−1), the population ratios of the S. mutans wild type and dpr, sod, and ahp mutants were 71.3%, 75.3%, 81.2%, and 81.4%, respectively, showing the increased population ratio of the mutants compared with that without catalase.
DISCUSSION
In this study, we investigated the roles of the oxidative stress-related factors Dpr, SOD, and AhpCF in resistance to H2O2. Although Dpr and AhpC have been reported to be involved in resistance to oxidative stress in S. mutans (22, 24), this is the first comprehensive analysis of these three factors against H2O2. Previous reports demonstrated that Dpr played a central role in resistance to ROS. Dpr binds to free Fe and suppresses the Fenton reaction pathway, which mediates the generation of •OH (24, 34). Notably, Sod was also significantly associated with H2O2 resistance, although the dpr mutant showed more susceptibility to H2O2 than did the sod mutant. As shown in Fig. 1, Sod is involved in the reaction that mediates the conversion of O2− to H2O2 (12). When sod is deleted, O2− accumulates in the cytoplasm. By the addition of H2O2 to the medium, the accumulated O2− in the sod mutant reacts with H2O2, causing the formation of •OH, which is considered to be highly toxic to bacteria (35). Thus, the sod mutation caused an increase in H2O2 susceptibility. In contrast, inactivation of ahpC, which decomposes H2O2 to H2O, had no effect on H2O2 susceptibility. Higuchi et al. also demonstrated previously that the ahpC deletion had no effect on the sensitivity of S. mutans to H2O2 (11). These results suggest that AhpC cannot decompose excess H2O2 completely. As a result, the presence of H2O2 in the cytoplasm has a cytotoxic effect on bacteria. In conclusion, Dpr and Sod are involved in H2O2 resistance in S. mutans.
PerR is known as an oxidative stress-responsive repressor and has been demonstrated to be associated with resistance to oxidative stress in Gram-positive and -negative bacteria (25–28, 36, 37). In Streptococcus pyogenes, PerR regulates perR itself, ahpC, and mrgA (Dps-like peroxide resistance protein) and contributes to resistance to ROS (25). Also, Ricci et al. reported previously that the perR mutant in S. pyogenes caused reduced transcription of sodA, encoding superoxide dismutase, which is involved in resistance to superoxide anions (38). For Streptococcus suis, perR was reported to regulate two oxidative stress response factors, dpr and metQIN (27). From our results, in S. mutans, the perR deletion caused significantly increased expression levels of dpr and slightly increased sod and ahpC expression levels. We also investigated the perR box, based on the consensus sequence in S. pyogenes and Bacillus subtilis, upstream of the dpr ORF (25, 27); we found that a PerR box was found upstream of the dpr and sod start codons. However, no consensus sequence was found upstream of the ahpC ORF. Thus, we suggest that PerR regulates dpr and sod directly. As described above, PerR is considered to regulate many genes, mainly factors responsible for oxidative stress resistance and metal transporters (10, 39, 40). ABC-type metal ion transporters are responsible for efflux or incorporation of metal ions, Fe and Mn, which are associated with oxidative stress responses because PerR and other Fur-like proteins bind metal ions for activation (10, 26). Thus, the increased resistance of the perR mutant to H2O2 may be due to factors other than dpr. However, the analysis of double-knockout mutants revealed that the dpr deletion in the perR mutant showed almost the same (increased) susceptibility to H2O2 as the dpr single mutant, suggesting that Dpr is significantly associated with H2O2 resistance in the perR mutant. Also, the perR sod double mutant showed increased susceptibility to H2O2 compared with that of the perR single mutant, indicating that sod is also partially associated with H2O2 resistance. In this study, we established a method for the coculture of two bacterial species. When a 1/10 dilution of S. sanguinis was cocultured with the S. mutans wild-type strain, the ratio of S. mutans (81.6%) was reduced significantly. In contrast, when catalase was added to the mixed culture, the ratio of S. mutans was slightly higher (91.2%). This result indicates that H2O2 produced by S. sanguinis affects competition with S. mutans. Next, we investigated the population ratio of S. mutans mutants cocultured with S. sanguinis. The results showed the same tendency as that seen in the competition and quantitative experiments. The sod and dpr genes mainly functioned in resistance to H2O2 when S. mutans was cocultured with S. sanguinis. Also, because the perR deletion, which caused significantly increased dpr expression levels, showed lower sensitivity to H2O2, the perR mutant showed a larger population than the wild type. Kreth et al. previously investigated coexistence between S. mutans and S. sanguinis in biofilms and also highlighted the importance of mutacin and H2O2 in the coexistence of the strains (19). Together with this result, we suggest that H2O2 produced by S. sanguinis is one of the factors affecting the growth of S. mutans within multispecies bacterial communities. As for S. mutans cocultured with S. sanguinis, we found that the dpr or sod mutants drastically reduced the population compared with that of the wild type. To evaluate the coculture assay, we confirmed that the expression of nlmA, encoding mutacin IV, showed no change among UA159 and the dpr, sod, and ahpCF mutants, suggesting that mutacin IV from S. mutans had no effect on the result of survival ratios of individual strains. Thus, we conclude that dpr and sod are responsible for resistance to H2O2 in S. mutans cocultured with S. sanguinis.
In conclusion, we analyzed three factors in the oxidative stress response, dpr, sod, and ahpCF, and found that Dpr and Sod are involved in H2O2 resistance. We also demonstrated that these two factors had key roles in coexistence with S. sanguinis. Furthermore, we identified the factor, PerR, which negatively regulated the expression of oxidative stress factors and demonstrated that PerR was associated with resistance to H2O2 in S. mutans. Our findings may indicate the mechanism of the coexistence of S. mutans and S. sanguinis in dental biofilms.
ACKNOWLEDGMENT
This study was supported in part by grants-in-aid for young scientists (B) from the Ministry of Education, Culture, Sports, Sciences, and Technology of Japan.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Jenkinson HF, Lamont RJ. 2005. Oral microbial communities in sickness and in health. Trends Microbiol. 13:589–595 [DOI] [PubMed] [Google Scholar]
- 2. Pennisi E. 2005. A mouthful of microbes. Science 307:1899–1901 [DOI] [PubMed] [Google Scholar]
- 3. Baba T, Schneewind O. 1996. Target cell specificity of a bacteriocin molecule: a C-terminal signal directs lysostaphin to the cell wall of Staphylococcus aureus. EMBO J. 15:4789–4797 [PMC free article] [PubMed] [Google Scholar]
- 4. Dobson A, Cotter PD, Ross RP, Hill C. 2012. Bacteriocin production: a probiotic trait? Appl. Environ. Microbiol. 78:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ryan CS, Kleinberg I. 1995. Bacteria in human mouths involved in the production and utilization of hydrogen peroxide. Arch. Oral Biol. 40:753–763 [DOI] [PubMed] [Google Scholar]
- 6. Barnard JP, Stinson MW. 1999. Influence of environmental conditions on hydrogen peroxide formation by Streptococcus gordonii. Infect. Immun. 67:6558–6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. García-Mendoza A, Liébana J, Castillo AM, de la Higuera A, Piédrola G. 1993. Evaluation of the capacity of oral streptococci to produce hydrogen peroxide. J. Med. Microbiol. 39:434–439 [DOI] [PubMed] [Google Scholar]
- 8. Imlay JA. 2002. How oxygen damages microbes: oxygen tolerance and obligate anaerobiosis. Adv. Microb. Physiol. 46:111–153 [DOI] [PubMed] [Google Scholar]
- 9. Miller RA, Britigan BE. 1997. Role of oxidants in microbial pathophysiology. Clin. Microbiol. Rev. 10:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Faulkner MJ, Helmann JD. 2011. Peroxide stress elicits adaptive changes in bacterial metal ion homeostasis. Antioxid. Redox Signal. 15:175–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Higuchi M, Yamamoto Y, Poole LB, Shimada M, Sato Y, Takahashi N, Kamio Y. 1999. Functions of two types of NADH oxidases in energy metabolism and oxidative stress of Streptococcus mutans. J. Bacteriol. 181:5940–5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Imlay JA. 2008. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 77:755–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Storz G, Tartaglia LA. 1992. OxyR: a regulator of antioxidant genes. J. Nutr. 122:627–630 [DOI] [PubMed] [Google Scholar]
- 14. Vlamis-Gardikas A. 2008. The multiple functions of the thiol-based electron flow pathways of Escherichia coli: eternal concepts revisited. Biochim. Biophys. Acta 1780:1170–1200 [DOI] [PubMed] [Google Scholar]
- 15. Higuchi M, Yamamoto Y, Kamio Y. 2000. Molecular biology of oxygen tolerance in lactic acid bacteria: functions of NADH oxidases and Dpr in oxidative stress. J. Biosci. Bioeng. 90:484–493 [PubMed] [Google Scholar]
- 16. Hamada S, Slade HD. 1980. Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol. Rev. 44:331–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuramitsu HK. 1993. Virulence factors of mutans streptococci: role of molecular genetics. Crit. Rev. Oral Biol. Med. 4:159–176 [DOI] [PubMed] [Google Scholar]
- 18. Carlsson J, Edlund MB, Lundmark SK. 1987. Characteristics of a hydrogen peroxide-forming pyruvate oxidase from Streptococcus sanguis. Oral Microbiol. Immunol. 2:15–20 [DOI] [PubMed] [Google Scholar]
- 19. Kreth J, Merritt J, Shi W, Qi F. 2005. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J. Bacteriol. 187:7193–7203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kreth J, Zhang Y, Herzberg MC. 2008. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J. Bacteriol. 190:4632–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakayama K. 1992. Nucleotide sequence of Streptococcus mutans superoxide dismutase gene and isolation of insertion mutants. J. Bacteriol. 174:4928–4934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poole LB, Higuchi M, Shimada M, Calzi ML, Kamio Y. 2000. Streptococcus mutans H2O2-forming NADH oxidase is an alkyl hydroperoxide reductase protein. Free Radic. Biol. Med. 28:108–120 [DOI] [PubMed] [Google Scholar]
- 23. Yamamoto Y, Fukui K, Koujin N, Ohya H, Kimura K, Kamio Y. 2004. Regulation of the intracellular free iron pool by Dpr provides oxygen tolerance to Streptococcus mutans. J. Bacteriol. 186:5997–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamamoto Y, Higuchi M, Poole LB, Kamio Y. 2000. Role of the dpr product in oxygen tolerance in Streptococcus mutans. J. Bacteriol. 182:3740–3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brenot A, King KY, Caparon MG. 2005. The PerR regulon in peroxide resistance and virulence of Streptococcus pyogenes. Mol. Microbiol. 55:221–234 [DOI] [PubMed] [Google Scholar]
- 26. Mongkolsuk S, Helmann JD. 2002. Regulation of inducible peroxide stress responses. Mol. Microbiol. 45:9–15 [DOI] [PubMed] [Google Scholar]
- 27. Zhang T, Ding Y, Li T, Wan Y, Li W, Chen H, Zhou R. 2012. A Fur-like protein PerR regulates two oxidative stress response related operons dpr and metQIN in Streptococcus suis. BMC Microbiol. 12:85 doi:10.1186/1471-2180-12-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zuber P. 2009. Management of oxidative stress in Bacillus. Annu. Rev. Microbiol. 63:575–597 [DOI] [PubMed] [Google Scholar]
- 29. Okinaga T, Xie Z, Niu G, Qi F, Merritt J. 2010. Examination of the hdrRM regulon yields insight into the competence system of Streptococcus mutans. Mol. Oral Microbiol. 25:165–177 [DOI] [PubMed] [Google Scholar]
- 30. Kawada-Matsuo M, Shibata Y, Yamashita Y. 2009. Role of two component signaling response regulators in acid tolerance of Streptococcus mutans. Oral Microbiol. Immunol. 24:173–176 [DOI] [PubMed] [Google Scholar]
- 31. Pulliainen AT, Hytönen J, Haataja S, Finne J. 2008. Deficiency of the Rgg regulator promotes H2O2 resistance, AhpCF-mediated H2O2 decomposition, and virulence in Streptococcus pyogenes. J. Bacteriol. 190:3225–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Theis T, Skurray RA, Brown MH. 2007. Identification of suitable internal controls to study expression of a Staphylococcus aureus multidrug resistance system by quantitative real-time PCR. J. Microbiol. Methods 70:355–362 [DOI] [PubMed] [Google Scholar]
- 33. Mazda Y, Kawada-Matsuo M, Kanbara K, Oogai Y, Shibata Y, Yamashita Y, Miyawaki S, Komatsuzawa H. 2012. Association of CiaRH with resistance of Streptococcus mutans to antimicrobial peptides in biofilms. Mol. Oral Microbiol. 27(2):124–135 [DOI] [PubMed] [Google Scholar]
- 34. Yamamoto Y, Poole LB, Hantgan RR, Kamio Y. 2002. An iron-binding protein, Dpr, from Streptococcus mutans prevents iron-dependent hydroxyl radical formation in vitro. J. Bacteriol. 184:2931–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCormick ML, Buettner GR, Britigan BE. 1998. Endogenous superoxide dismutase levels regulate iron-dependent hydroxyl radical formation in Escherichia coli exposed to hydrogen peroxide. J. Bacteriol. 180:622–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun. 69:3744–3754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Vliet AH, Baillon ML, Penn CW, Ketley JM. 1999. Campylobacter jejuni contains two fur homologs: characterization of iron-responsive regulation of peroxide stress defense genes by the PerR repressor. J. Bacteriol. 181:6371–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ricci S, Janulczyk R, Björck L. 2002. The regulator PerR is involved in oxidative stress response and iron homeostasis and is necessary for full virulence of Streptococcus pyogenes. Infect. Immun. 70:4968–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brenot A, Weston BF, Caparon MG. 2007. A PerR-regulated metal transporter (PmtA) is an interface between oxidative stress and metal homeostasis in Streptococcus pyogenes. Mol. Microbiol. 63:1185–1196 [DOI] [PubMed] [Google Scholar]
- 40. Wu HJ, Seib KL, Srikhanta YN, Kidd SP, Edwards JL, Maguire TL, Grimmond SM, Apicella MA, McEwan AG, Jennings MP. 2006. PerR controls Mn-dependent resistance to oxidative stress in Neisseria gonorrhoeae. Mol. Microbiol. 60:401–416 [DOI] [PubMed] [Google Scholar]
- 41. Murchison HH, Barrett JF, Cardineau GA, Curtiss R., III 1986. Transformation of Streptococcus mutans with chromosomal and shuttle plasmid (pYA629) DNAs. Infect. Immun. 54:273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]