

Abstract
In the cyanobacterium Anabaena sp. strain PCC 7120 (also known as Nostoc sp. strain PCC 7120), a zinc-responsive operon (all4725-all4721) has been described, which contains 4 distinct promoters. The two most upstream ones bind Zur with high affinity, whereas the other two do not or do so with a very low affinity. In this paper, a detailed characterization of the four promoters is presented, showing that all four were induced by metal depletion, and they were constitutively derepressed in a zur mutant, despite the two downstream promoters not being direct targets for this regulator. Crucially, induction by metal depletion of the two downstream promoters was abrogated when transcription initiated at the upstream promoters was interrupted by a polar insertion midway in the operon. In contrast, insertion of a nitrogen-responsive promoter at a roughly similar position provoked the two downstream promoters to adopt a regulatory pattern mimicking that of the inserted promoter. Thus, regulation of the two downstream promoters is apparently influenced by transcription from promoters upstream. Evidence is presented indicating that the activity of the two downstream promoters is kept basal in Anabaena by repression. A regulatory model compatible with these results is proposed, where promoters controlled by repression in bacterial operons may be subjected to a hierarchical regulation depending on their position in the operon. According to this model, internal promoters may respond to stimuli governing the activity of promoters upstream by an indirect regulation and to specific stimuli by a direct regulation.
INTRODUCTION
Gene operons are transcriptional units existing in prokaryotes and eukaryotes that gather genes, which are often functionally related and require a common expression pattern (1–3). In the simplest instance of a bacterial operon, coordinated expression of the genes is achieved by their transcription in a single polycistronic mRNA initiated at a promoter located upstream of the first gene. Control of the unique promoter allows a coordinated regulation of all genes. However, in many cases, the architecture and regulation of bacterial operons are rather more complex, involving many regulatory elements and multiple internal promoters that may be distinctly regulated. Sophisticated regulatory mechanisms have evolved to ensure that the relative expression level of each individual gene meets the necessities of the cell, which may vary under different conditions (4–6).
In the cyanobacterium Anabaena sp. strain PCC 7120 (here Anabaena), a zinc-responsive operon (all4725-all4721) was recently described (7). The five genes in the operon do not show an obvious functional relationship: all4725 encodes a putative porphobilinogen synthase, all4724 encodes a putative flavin adenine dinucleotide (FAD)-dependent oxidoreductase, all4723 encodes a threonyl-tRNA synthetase, all4722 encodes a putative metallochaperone, and all4721 encodes a putative GTP cyclohydrolase. This operon is part of the Anabaena Zur regulon, controlled by Zur, a regulator of the Fur family that senses the availability of zinc (8). Zur functions mostly as a repressor, binding to DNA when the concentration of zinc is high and its regulatory metal coordination sites are occupied (9, 10). Conversely, a decrease in the concentration of zinc determines that Zur detaches from DNA, releasing its target genes from repression. Four promoters were mapped in the all4725-all4721 operon by a 5′ rapid amplification of cDNA ends (5′-RACE) procedure suitable for the distinction of true 5′ mRNA ends with a triphosphate group from other 5′ ends carrying a monophosphate group which result from processing of a longer transcript (7, 11). One promoter mapped just upstream of the first gene in the operon, and three internal ones were localized upstream of all4723, all4722, and all4721, respectively. Anabaena Zur was demonstrated to bind with high affinity and in a zinc-dependent manner to DNA fragments of the two most upstream promoters, containing sequences that were identified as target sequences for Anabaena Zur. All five genes showed a similar regulatory profile, with their expression levels being basal under standard growth conditions and very high when the culture was depleted of zinc. Real-time PCR assays demonstrated that the level of induction provoked by zinc limitation was 50- to 100-fold, and Northern assays showed that all five genes produced a smeared hybridization signal, interpreted as hybridization with long polycistronic transcripts, superposed to discrete bands, which may result from processing of transcripts or initiation at internal promoters (7). Although the overall regulation of this operon has been well characterized, the particular contribution of individual promoters to the expression of the operon has not been analyzed in detail, and the possibility of the promoters being distinctly regulated has not been investigated.
We present evidence here showing that all four promoters of the Anabaena all4725-all4721 operon respond to metal deficiency and are regulated by Zur. Whereas the two most upstream promoters are direct targets for repression by Zur, the two most downstream ones are regulated by Zur through an indirect mechanism, perhaps involving the removal of an unknown repressor(s) from DNA. This mode of regulation would resemble a domino effect, where transcription initiated at a particular promoter in the operon would induce the activity of promoters located downstream.
MATERIALS AND METHODS
Organisms and growth conditions.
Wild-type (WT) Anabaena and derivative strains were cultured under standard growth conditions in BG11 medium (12) at 30°C under illumination with a light intensity of 75 microeinsteins m−2 s−1 and aerated with CO2-enriched air (1% [vol/vol] CO2) buffered with 10 mM NaHCO3. In experiments where ammonium was used as a nitrogen source, cells were cultured in BG110 medium, which lacks nitrate, supplemented with 5 to 10 mM ammonium chloride, and buffered with 10 mM TES {N-[tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid}-NaOH (pH 7.5). Difco agar (1%) was added for solid media. When needed, the medium was supplemented with antibiotics at the following concentrations: streptomycin at 2 to 5 μg ml−1, spectinomycin at 2 to 5 μg ml−1, and neomycin at 50 to 70 μg ml−1. The nonspecific divalent metal chelator TPEN [N,N,N′,N′-tetrakis(2-pyridilmethyl)ethylenediamine] was prepared in dimethyl sulfoxide (DMSO) and added to the cultures where indicated, at a final concentration of 20 μM. Whenever a culture was treated with TPEN, all other cultures of the experiment were supplemented with DMSO to a similar final concentration (0.1%).
Escherichia coli strain DH5α or XL1-Blue was routinely used for cloning and plasmid constructions and was grown in Luria-Bertani medium (13) supplemented with antibiotics, when required, at the following concentrations: ampicillin at 50 μg ml−1, kanamycin at 25 μg ml−1, streptomycin at 25 μg ml−1, spectinomycin at 100 μg ml−1, and chloramphenicol at 30 μg ml−1.
Construction of Anabaena-derived strains.
Plasmid construction is described in Fig. S2 in the supplemental material. Insertional inactivation of all4723 was carried out by construction of a plasmid (pCMN4) where the C.S3 gene cassette, conferring resistance to streptomycin and spectinomycin (14), was inserted at the internal HindIII site of all4723 in the same orientation as the open reading frame (ORF). The insert of this plasmid was transferred into the conjugation-mobilizable vector pRL278, yielding plasmid pCMN8, which was introduced into Anabaena by triparental mating conjugation (15). Integration of the plasmid was selected in solid BG11 medium supplemented with streptomycin and spectinomycin. Double recombinants were selected by sonication of single recombinants to obtain short filaments that were grown in solid BG11 medium supplemented with streptomycin, spectinomycin, and sucrose to counterselect the sacB marker gene present in the vector portion of pCMN8. Colonies were analyzed by PCR for segregation of the mutation (i.e., all chromosomes in all cells of all filaments would contain the mutation), and a segregated strain, named MN8, was selected for further studies.
all4723 deletion strain MN42 was constructed as follows. First, a 3.5-kb DNA fragment containing the 3′ region of all4724, the entire all4723 ORF, and the 5′ region of all4722 was amplified by PCR using primers 24STREP-1F and 22DEL-1R and cloned into the pBluescriptSK+ vector (Novagen), yielding plasmid pCMN32. Plasmid pCNM41 was constructed by deleting all4723 by PCR amplification of pCMN32 with divergent primers 22DEL-1F and 24DEL-1R, flanking the all4723 ORF, and religation of the PCR product. The insert of pCMN41 was transferred into the conjugative vector pRL278, yielding plasmid pCMN42, which was transferred into Anabaena by conjugation. Double recombinants were selected as described above, except that antibiotics were omitted in the last selection step in the presence of sucrose.
Strain MN52 was obtained as follows. Plasmid pCMN20 was constructed by cloning a DNA fragment containing the all4723 ORF amplified with primers THRS2-1F and THRS2-1R in the NdeI and XhoI sites of the pCMN28b expression vector. pCMN28b is a derivative of pET28b (Novagen) constructed by replacing the hexahistidine-encoding NcoI-NdeI fragment of pET28b with a 67-bp linker encoding a StrepTag II sequence. The linker was obtained by annealing oligonucleotides STREP-TAG-1F and STREP-TAG-1R. Fusion proteins expressed in this vector contain the N-terminal sequence MASTSHPQFEKGALEVLFQGPH. A PCR fragment amplified from Anabaena genomic DNA with primers NIR-2F and NIR-1R was cloned into the BglII and NcoI sites of the pCNM20 plasmid, producing the new plasmid pCNM49, whose insert was transferred into the pRL278 conjugal vector, yielding plasmid pCMN52, which was transferred into Anabaena by conjugation. Further steps were performed as described above.
Northern assays.
RNA was extracted from 40- to 50-ml cultures in the exponential phase by use of a procedure described previously (16). For each sample, 10 μg RNA was resolved in a formaldehyde-containing agarose gel, transferred onto Genescreen Plus (PerkinElmer) nylon membranes, and hybridized to radioactive DNA probes labeled with [α-32P]dCTP by using the Ready-To-Go labeling kit (GE Healthcare). Images of hybridized membranes were obtained with a Cyclone Storage Phosphor system (PerkinElmer).
Primer extension.
Twenty micrograms of RNA was denatured for 10 min by heating at 85°C; annealed to 2 pmol 32P-labeled primer 25R, 23R, or 22R during 1 h at 55°C; and extended with Superscript III enzyme (Invitrogen) in the presence of deoxynucleoside triphosphates (dNTPs) for 1 h at 55°C. When primer 21R was used, the annealing step was carried out at 50°C, and the extension step was carried out at 47°C. Extension products were resolved in 6% urea-containing acrylamide gels next to a sequencing ladder as a size marker.
In vitro transcription.
Runoff transcription assays were performed by using recombinant Anabaena RNA polymerase (RNAP), reconstituted and purified as previously described (17). Reactions were set up in a total volume of 15 μl containing 40 mM Tris-HCl (pH 8.0), 50 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 0.5 mg/ml bovine serum albumin (BSA), 5% glycerol, 30 to 60 fmol DNA fragment, and 150 nM Anabaena RNA polymerase. Complexes were allowed to form by incubation for 5 min at 30°C or 37°C, and transcription was started by the addition of 1.5 μl of a solution containing 0.15 mM (each) ATP, GTP, and UTP; 20 μM CTP; and 3 μCi [α-32P]CTP and was allowed to proceed for 30 min at 30°C or 37°C. After incubation, reactions were stopped by the addition of 6 μl phenol, 10 mM EDTA, and 1.5 μl of 200 ng/μl glycogen in 3 M sodium acetate (pH 5.2). After ethanol precipitation, the products were separated by electrophoresis on 6% acrylamide gels containing 8 M urea. Linear DNA templates with wild-type sequences of promoters P5, P3, P2, and P1 were generated by PCR with primer pairs ALL4725_3F and ALL4725_3R, ALL4723_3F and ALL4723_10R, ALL4722_6F and ALL4722_8R, and ALL4721_4F and ALL4721_5R, respectively. The fragment containing the sequence of the PpsbA promoter from Amaranthus hybridus, used as a control, was generated by using primers CK3-1 and CK3-2 and plasmid pRL278 as a PCR template (14). Mutant DNA templates with altered sequences of the −10 or −35 boxes were obtained by generating two overlapping PCR fragments, each amplified with one terminal primer and a mutagenic primer. The terminal primers were those used to amplify the wild-type fragments (see above), and the mutagenic primers were complementary and contained the sequence to be introduced. In a second round of PCR, the products of the first round were mixed in equimolar amounts and subjected to PCR with the terminal primers. Mutagenic primers used to mutate the −10 or the −35 sequences were ALL4725_BOX35_F, ALL4725_BOX35_R, ALL4725_BOX10_F, and ALL4725_BOX10_R for P5; ALL4723_BOX35_F, ALL4723_BOX35_R, ALL4723_BOX10_F, and ALL4723_BOX10_R for P3; ALL4722_BOX35_F, ALL4722_BOX35_R, ALL4722_BOX10_F, and ALL4722_BOX10_R for P2; and ALL4721_BOX35_F, ALL4721_BOX35_R, ALL4721_BOX10_F, and ALL4721_BOX10_R for P1.
Luminescence detection.
DNA fragments containing P2 or P1 were amplified by PCR using primers pair 4722LUX-1F and 4722LUX-1R or ALL4721_3F and ALL4721_4R, respectively, and cloned into the XhoI and BamHI sites of plasmid pSB377_1 (18) containing the luxCDABE genes from Photorhabdus luminescens in tandem. The orientation of P2 and P1 was the same as that of the lux genes in both constructions. Recombinant plasmids were transferred into Escherichia coli DH5α by transformation, and bacterial luminescence was detected by using a Chemi Doc XRS imaging system (Bio-Rad).
RESULTS
Internal promoters of the all4725-all4721 operon are transcribed by the Anabaena RNAP in vitro.
In the all4725-all4721 operon of Anabaena, promoters were mapped upstream of all4725, all4723, all4722, and all4721, which are here named P5, P3, P2, and P1, respectively (Fig. 1A and B) (7). To corroborate the existence of these promoters, PCR fragments encompassing each promoter were tested by in vitro transcription assays with purified recombinant Anabaena RNAP. Despite repeated efforts, no runoff product was obtained when a fragment containing the P5 promoter was used as the template, suggesting that an element required for transcription of this promoter was probably missing in our assay. In contrast, bands with the size expected for the runoff products were observed with templates of P3, P2, and P1 (Fig. 1C). Mutation of the putative −10 or −35 boxes of these three promoters had a strong negative impact on transcription assays in vitro, further confirming the localization of P3, P2, and P1 at the positions previously mapped by 5′-RACE.
Fig 1.
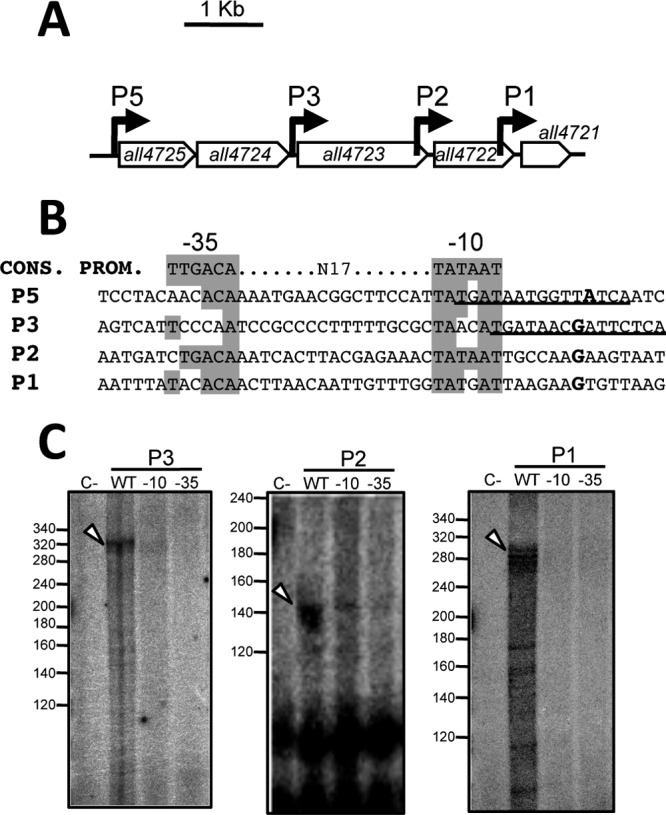
Promoters of the all4725-all4721 operon. (A) The positions of the P5, P3, P2, and P1 promoters, depicted as bent arrows, are indicated. (B) Sequence features of P5, P3, P2, and P1 promoters. The sequence of a consensus promoter (Cons. Prom.) is shown at the top. Conserved nucleotides of putative −10 and −35 boxes of the P5 to P1 promoters are shadowed. The nucleotide corresponding to the transcriptional start site is shown in boldface type. Zur binding sequences are underlined. (C) In vitro transcription of wild-type and mutant promoters. A total of 50 fmol linear DNA templates containing the sequences of the P3, P2, and P1 promoters or mutant versions where the −35 box was changed into CCTGAG or the −10 box was changed into GCGCGC was incubated with 150 nM recombinant Anabaena RNAP in the presence of dNTPs and [α-32P]CTP. A control reaction mixture (C−) containing a wild-type DNA template and all components except RNAP is shown in the left lane of each panel. The expected position for runoff transcripts is indicated with an arrowhead.
The two downstream promoters of the all4725-all4721 operon are regulated by metal availability.
P5 and P3 contain binding sequences for Zur and bind this regulator with high affinity in a zinc-dependent manner (7). However, it is not known whether the P2 and P1 promoters are regulated. The all4725-all4721 operon was shown previously to be highly induced by treatment with TPEN, a membrane-permeable metal chelator that induces Zn, Fe, and Cu deficiency in Anabaena cells (7). To analyze the possible responsiveness of P2 and P1 to metal deficiency induced by TPEN, we carried out primer extension experiments using RNA from cells cultured under standard conditions or incubated with TPEN for 6 or 24 h. When primers 22R and 21R, annealing with the 5′ region of all4722 and all4721, respectively (Fig. 2A and B), were used, extension products were observed at the exact positions of the transcription start sites previously mapped by 5′-RACE (7). The abundance of the extension products was low under standard growth conditions and increased 20- to 30-fold (Fig. 2B, left) or 20- to 50-fold (right) after treatment of the culture for six or more hours with the TPEN chelator, providing evidence that P2 and P1 are induced by metal deficiency and suggesting regulation by Zur or another metal-sensing regulator. A primer extension experiment performed with the same RNA preparations and a primer annealing to the 5′ region of the all4727 gene, used as a control (Fig. 2, bottom), demonstrated that TPEN does not provoke a general effect on transcription.
Fig 2.
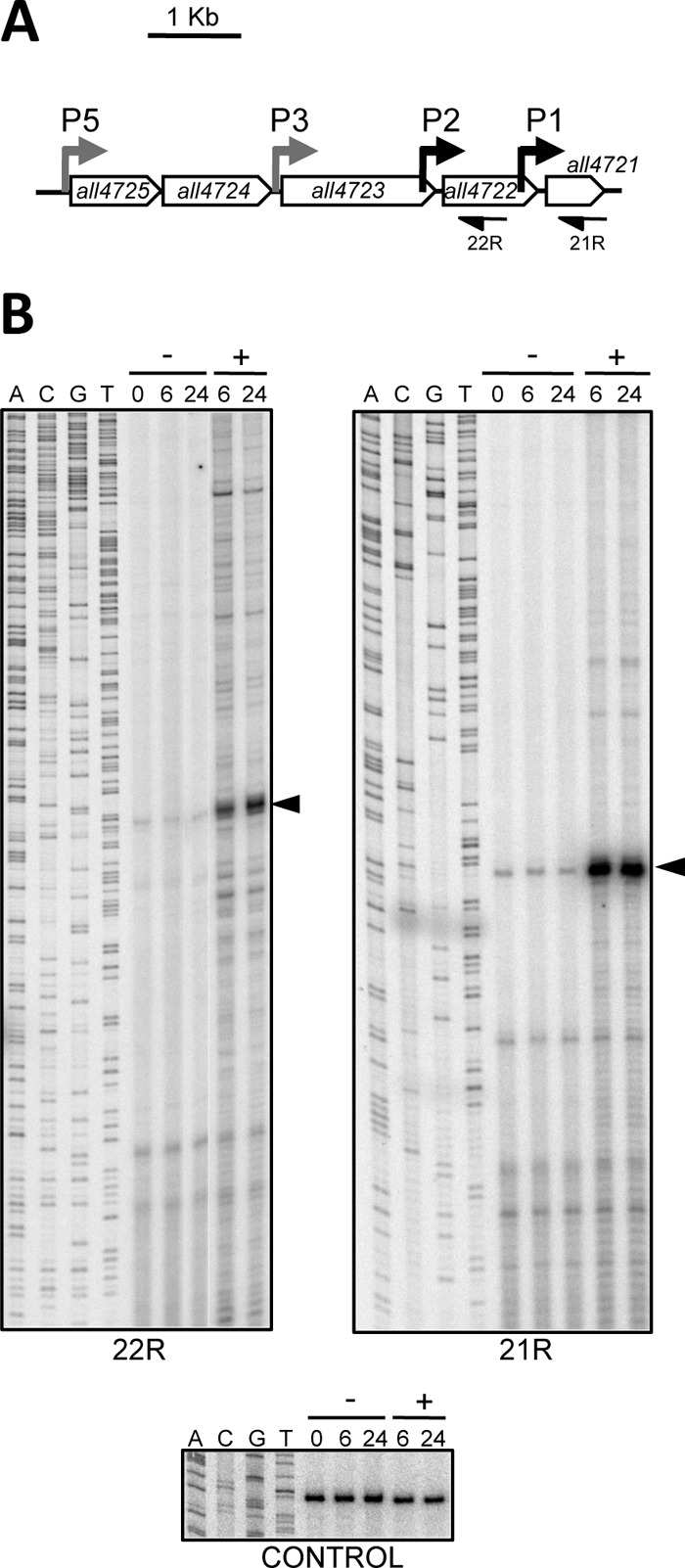
P2 and P1 are induced by metal deficiency. (A) Schematic representation of the all4725-all4721 operon. The localization of the promoters is indicated by bent arrows, with those that were analyzed by primer extension in panel B shown in black. Primers used for primer extension in panel B are also depicted. (B) RNA from cells not treated (−) or treated (+) with 20 μM TPEN for the period of time indicated at the top (in hours) was subjected to primer extension analysis with primer 22R (left) or 21R (right). A sequencing reaction generated with the same oligonucleotide used for primer extension is shown at the left of each panel for sizing. Arrowheads point to extension products at the expected positions of transcripts initiating at P2 (left) and P1 (right). The bottom panel corresponds to a primer extension control experiment performed with the same RNA preparations and primer ALL4727_2R.
P2 and P1 promoters are constitutively induced in a zur mutant, but they are not direct targets for Zur.
Primer extension results in Fig. 3B show that P5 and P3 are derepressed in a zur mutant under noninducing conditions (i.e., cells not treated with TPEN) (Fig. 3B, lanes 3 and 7), providing further evidence for a direct repression of these promoters by Zur. Interestingly, P2 and P1 were also derepressed in cells of the zur mutant under noninducing conditions (Fig. 3B, lanes 11 and 15), closely resembling the regulatory pattern of P5 and P3. Direct repression of these promoters by Zur was tested by supplementing in vitro transcription assay mixtures with preparations of recombinant Anabaena Zur protein. The presence of Zur in the reaction mixtures did not alter the abundance of runoff products when templates of P2 and P1 or a Zur-independent control promoter were used (Fig. 3C). However, Zur provoked a 4-fold decrease in the abundance of runoff products with the P3 template (Fig. 3C), consistent with the previously reported interaction of Zur with sequences of this promoter located at positions compatible with repression (Fig. 1B) (7). Therefore, the control exerted by Zur on P2 and P1 deduced from their constitutive induction in a zur mutant (Fig. 3B) must occur by an indirect mechanism and not by a direct interaction of the regulator with these promoters.
Fig 3.
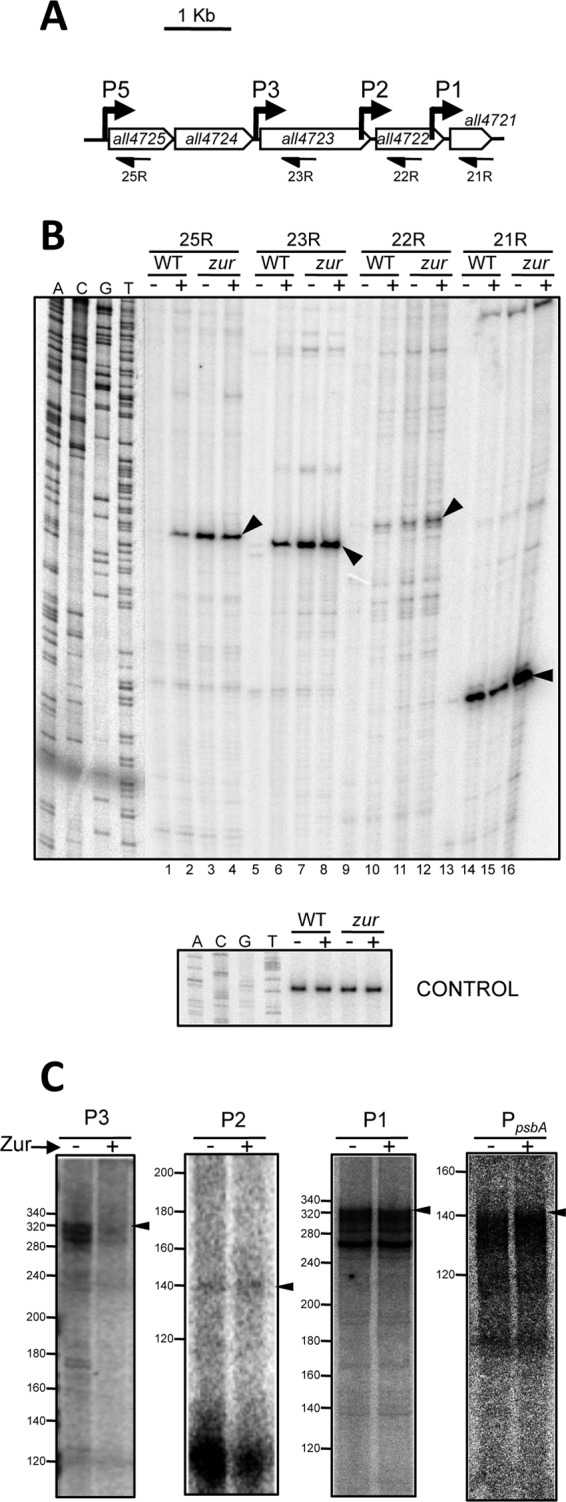
The four promoters of the all4725-all4721 operon are constitutively transcribed in a zur mutant. (A) Schematic representation of the all4725-all4721 operon showing the primers used for primer extension in panel B. (B) RNA from WT cells or the zur mutant not treated (−) or treated (+) with 20 μM TPEN for 24 h was subjected to primer extension with the primers indicated at the top. A sequencing reaction mixture generated with the P1 primer is shown at the left for sizing. Arrowheads point to extension products at the expected positions for transcripts initiating at P5, P3, P2, and P1. The bottom panel corresponds to a primer extension control experiment performed with the same RNA preparations and primer ALL4727_2R. (C) Impact of Zur on transcription of P3, P2, and P1 in vitro. A total of 25 fmol linear DNA templates containing the sequences of the P3, P2, and P1 promoters was incubated with 150 nM recombinant Anabaena RNAP, dNTPs, and [α-32P]CTP in the absence (−) or presence (+) of 5 pmol recombinant Zur protein. The expected position for runoff transcripts is indicated with an arrowhead.
A polar insertion in all4723 eliminates induction of P2 and P1.
To further characterize the regulation of P2 and P1, a mutant strain (MN8) was constructed, where the C.S3 cassette (14), containing genes conferring resistance to streptomycin and spectinomycin and a transcriptional terminator, was inserted within the all4723 gene (Fig. 4A). Interestingly, strain MN8 showed low levels of transcripts hybridizing with all4722 and all4721 under induction or noninduction conditions (i.e., cells treated or not with TPEN) (Fig. 4B). Consistent with this, in mutant strain MN8, the abundance of transcripts initiated at P2 and P1 was basal under both conditions (Fig. 4C, compare lanes 1 and 2 with 3 and 4). Thus, the low levels of transcripts hybridizing with all4722 and all4721 (Fig. 4B) are due to the combined effect of the interruption of transcription initiated at promoters upstream of the insertion (P5 and P3) and the elimination of the induction of the promoters located downstream (P2 and P1). A possibility to be tested was whether the product of the all4723 gene disrupted by the insertion in mutant strain MN8 could be required for activation of P2 and P1. all4723 encodes a threonyl-tRNA synthetase. Many aminoacyl-tRNA synthetases are moonlighting proteins, which, besides to their canonical role in aminoacyl-tRNA production, fulfill other functions, including transcriptional activation (19). To check this hypothesis, a deletion mutant lacking the all4723 ORF was constructed (strain MN42) (Fig. 4A). In sharp contrast to what was observed for the insertional mutant strain MN8, in the deletion strain MN42, transcripts hybridizing with all4722 or all4721 showed induction by TPEN treatment similar to that in the wild type (Fig. 4B). The P2 promoter could not be analyzed in mutant strain MN42 since it was deleted as part of the all4723 ORF. Primer extension with oligonucleotide 22R corroborated that in MN42, P2 is missing and P3 is placed immediately upstream of all4722 (Fig. 4C, top, lanes 5 and 6). Most importantly, in this mutant, P1 exhibited a regulation very similar to that in the wild type (Fig. 4C, bottom, compare lanes 1, 2, 5, and 6). Therefore, the lack of induction of P1 in mutant strain MN8 cannot be attributed to a requirement of All4723 for activation (Fig. 4C, compare lanes 2, 4, and 6).
Fig 4.
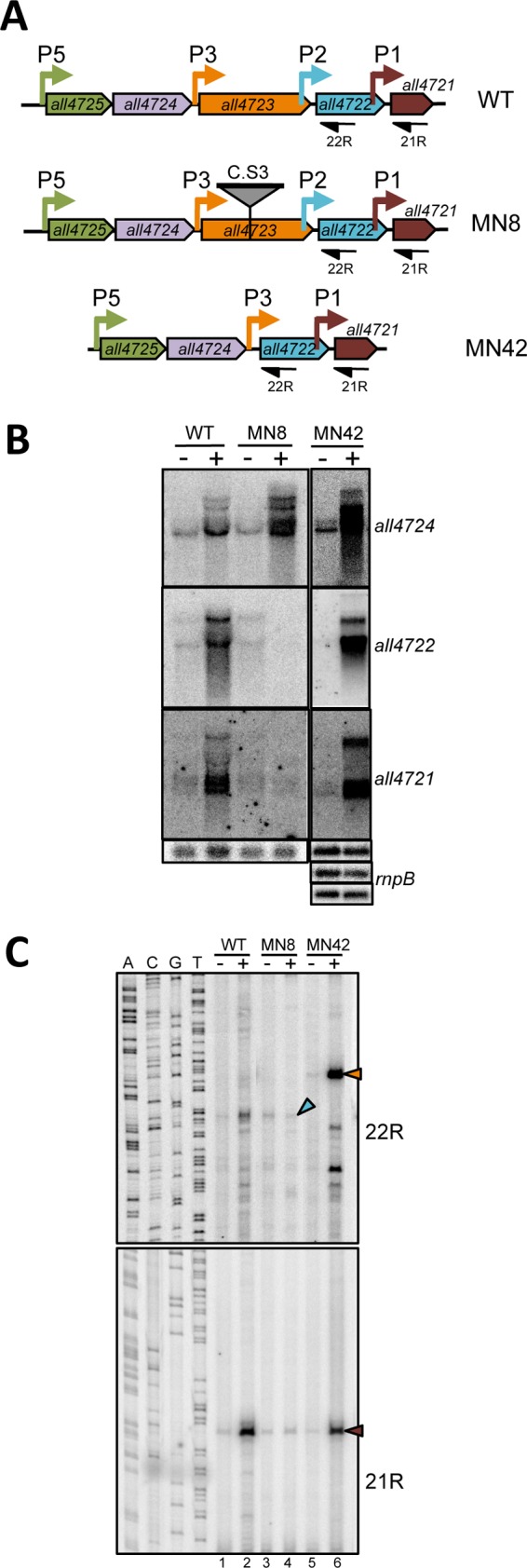
Regulation of P2 and P1 in all4723 mutants. (A) Schematic representation of the all4725-all4721 operon showing the structure of mutant strains MN8 and MN42 and the primers used for primer extension in panel C. (B) RNA from cells of the WT or mutant strain MN8 or MN42 cultured in the absence (−) or presence (+) of 20 μM TPEN for 24 h was subjected to a Northern assay with internal probes for the genes indicated at the right of each panel. Hybridization with the rnpB probe was used as a loading control. (C) RNA from cells of the WT or strain MN8 or MN42, not treated (−) or treated (+) with 20 μM TPEN for 24 h, was subjected to primer extension with the primers indicated at the right. Arrowheads point to extension products at the expected positions for transcripts initiating at P3 (orange), P2 (blue), or P1 (brown).
P2 and P1 adopt the regulatory pattern of a promoter inserted upstream.
Results presented above suggest that regulation of P2 and P1 is influenced by transcription from promoters upstream. To test this hypothesis, strain MN52 was constructed, where the Pnir promoter from the nirA-nrtABCD-narB operon of Anabaena was inserted upstream of all4723. Pnir was described previously to be responsive to nitrogen stimuli, showing higher transcriptional activity in nitrate- than in ammonium-containing medium (20). In strain MN52, transcripts hybridizing with genes downstream of the inserted Pnir promoter (all4723, all4722, and all4721) showed a 2.5-fold-higher level in nitrate than in ammonium, resembling the regulation of the nirA gene, in contrast to genes upstream of Pnir (all4724 and all4725), which showed basal levels under both conditions (Fig. 5A). P2, P1, and the Pnir promoter inserted upstream of all4723 were analyzed by primer extension using RNA from WT or MN52 cells cultured in ammonium- or nitrate-containing medium. The abundance of transcripts initiated at P2, which was low in the WT irrespective of the nitrogen source and in strain MN52 in ammonium, increased 4-fold in the latter strain grown in nitrate (Fig. 5B, blue arrowhead), closely resembling the regulation of Pnir (transcripts 3.6-fold more abundant in nitrate than in ammonium) (Fig. 5B, red arrowhead). The insertion of Pnir also had an effect on P1. In strain MN52, the abundance of transcripts initiated at this promoter was much higher in nitrate or ammonium than in the wild type (Fig. 5B, brown arrowhead). Although the regulation of P1 in strain MN52 does not fully match that of Pnir, such behavior was repeatedly observed in experiments using RNA extracted from independent cultures.
Fig 5.
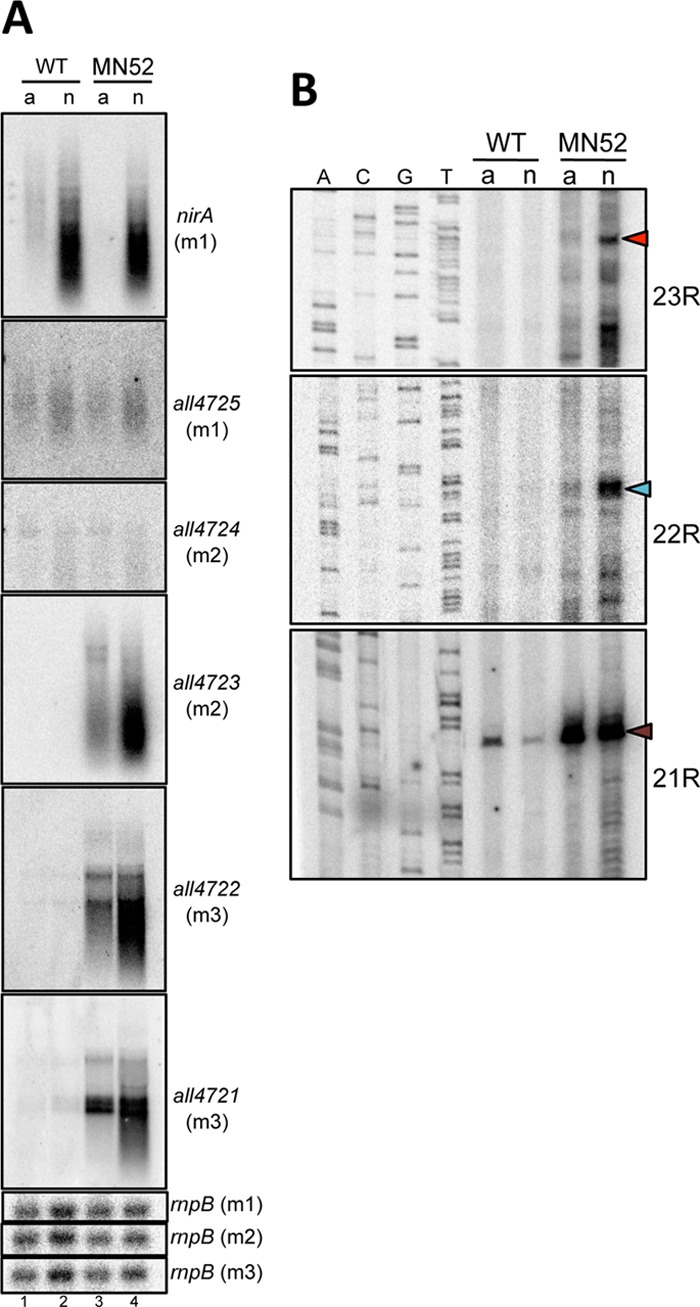
Regulation of P2 and P1 in mutant strain MN52. (A) Membranes m1, m2, and m3, containing RNA from cells of the WT or mutant strain MN52 cultured in medium containing ammonium (a) or nitrate (n) as a nitrogen source, as indicated, were hybridized with internal probes of the genes indicated at the right of each panel. Hybridization with rnpB was used as a control for RNA loading. (B) RNA from cells of the WT and strain MN52 cultured with ammonium or nitrate as a nitrogen source was subjected to primer extension with the primers indicated at the right. Arrowheads point to extension products at the expected positions for transcripts initiating at Pnir (red), P2 (blue), or P1 (brown).
Evidence for control of P2 and P1 activity by repression.
Results presented above indicate that the transcription level from P2 and P1 is very low in cultures under standard growth conditions. Several features suggested that these promoters could be controlled in Anabaena by repression. For instance, P2 contained −10 and −35 sequences closely matching the signature sequences of sigma 70-dependent promoters, with only one mismatch in the −35 box and an optimal spacing (17 nucleotides) between the two boxes (Fig. 1B). Promoters adjusting to the consensus have been shown to be very efficiently transcribed in cyanobacteria (21–23). P1 does not so closely resemble the consensus promoter (with two mismatches in the −35 box and one in the −10 box), but it is very efficiently transcribed by the Anabaena RNAP in vitro. DNA fragments containing P2 or P1 were cloned into the pSB377_1 vector (18) upstream of the luxCDABE genes from Photorhabdus luminescens, so that transcription of the lux genes would be governed by the inserted promoters, and the resulting plasmids were introduced into E. coli by transformation. Compared to cells containing the empty vector, cells containing either construct showed elevated bioluminescence, indicative of high promoter activity (Fig. 6), which is in contrast to the low basal activity observed in Anabaena, suggesting that the putative repressor(s) that controls these promoters in Anabaena is absent in E. coli.
Fig 6.
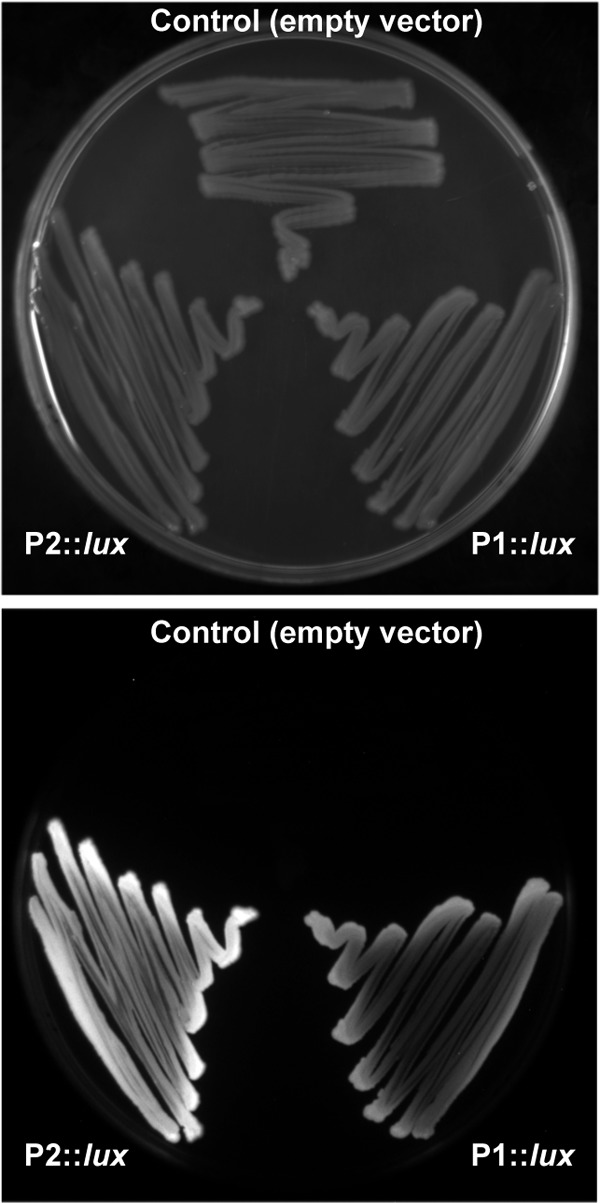
Promoter assay of P2 and P1 in E. coli by luminescence. A petri dish with E. coli cells containing the empty pSB377_1 vector or the vector containing DNA fragments of the P2 and P1 promoters upstream of the luxCDABE genes, as indicated, was photographed under white-light illumination (top) or in the dark (bottom).
DISCUSSION
In this work, the regulation of promoters in the all4725-all4721 operon of Anabaena is characterized in detail. The presence of three internal promoters previously mapped by 5′-RACE was corroborated here by in vitro transcription assays with wild-type and mutant DNA templates with altered −10 or −35 sequences. The P5 promoter, located at the 5′ end of the operon, and the three internal promoters P3, P2, and P1 are all induced by metal deficiency and derepressed in a zur mutant (Fig. 2 and 3). We previously reported that Zur binds in vitro with a very high affinity to DNA fragments of P5 and P3 and with a very low affinity to a DNA fragment of P1, and it did not interact with a DNA fragment of P2 (7). We show here that in vitro, Zur represses transcription of P3 but not of P2 and P1. It is also shown that in vivo, P1 is not responsive to metal deficiency induced by TPEN in insertional mutant strain MN8, indicating that it is not directly repressed by Zur. Thus, the previously reported weak in vitro interaction of Zur with P1 likely resulted from the high concentrations of protein used in those assays (10-fold higher than those used for P5 or P3) (7), probably exceeding the physiological concentrations of the regulator. Therefore, out of the four promoters mapped in the all4725-all4723 operon, the two upstream ones (P5 and P3) are directly repressed by Zur, and the two downstream ones (P2 and P1) are not directly repressed by Zur. This raises the question of how P2 and P1 are controlled with Zur excluded as a direct regulator. The involvement of a metal-sensing regulator other than Zur seems a remote possibility, because induction of P2 and P1 by TPEN would have been expected in mutant strain MN8. The possibility that the lack of induction of P2 and P1 in the MN8 mutant strain was due to the requirement of the disrupted all4723 gene does not seem plausible, as its deletion (strain MN42) had no effect on P1 (Fig. 4). In the MN42 deletion mutant strain, transcription initiated at the upstream promoters, P5 and P3, is not interrupted, which is in contrast with the MN8 insertional mutant strain. It appears that as long as transcription from the upstream promoters is allowed to proceed through the operon, regulation of P2 and P1 would imitate that of the upstream promoters. Indeed, an insertion midway in the operon of a Pnir promoter, which responds to nitrogen stimuli, has a strong impact on the regulatory profile of P2 and P1. Therefore, regulation of P2 and P1 seems to be influenced by transcription elongation complexes approaching from upstream, a phenomenon known as transcriptional interference of tandem promoters (24). The molecular mechanism underlying this type of regulation may be diverse, and in some cases, it may occur by the effect that RNAP transcribing from one promoter may have on the supercoiling state of neighbor promoters (25, 26). Advancement of RNAP during transcription produces the accumulation of positive supercoiling ahead of the elongation complex and negative supercoiling behind it (27, 28), and supercoiling-sensitive promoters whose activity is modulated in response to changes in the torsion level of DNA have been described (25, 29–31). Whether P2 and P1 are sensitive to the alterations in DNA topology that may provoke the advancement of RNAP from upstream promoters needs to be investigated further.
In other cases, transcriptional interference of tandem promoters may occur through transcription factor dislodgement by advancing RNAP (32–34). This may also be the case for P2 and P1. Some evidences suggest that transcription from P2 and P1 may be controlled by repression. We find it plausible that induction of P2 and P1 may occur through dislodgement of a repressor sitting on these promoters by RNAP molecules proceeding from upstream promoters. Previous reports of RNAP colliding into a protein bound to a downstream position on DNA showed that in some cases, RNAP may be halted by the bound protein through a roadblock mechanism (35–38), whereas in others, the elongating complexes are able to bypass the roadblock, probably by dislodgement of the blocking protein (39–42). Two factors contribute to readthrough by the RNAP: (i) when multiple RNAP complexes transcribe in tandem, trailing elongation complexes cooperate with the leading complex to bypass the block (39, 43), and (ii) nascent RNA-translating ribosomes, which closely follow transcribing RNAP, provide a supplemental impelling force that facilitates readthrough of roadblocks in vivo (44, 45). In Anabaena, both factors may contribute to dislodgement of a putative repressor from P2 and P1 because, on the one hand, tandem arrays of RNAP engaged at P5 and P3 upon induction are expected to arrive at P2 and P1 and, on the other hand, the sequences of P2 and P1 overlap (although partially in the case of P1) with the translated region of the gene located upstream. Dislodgement of a putative repressor from P2 and P1 could allow the engagement of new RNAP molecules and the initiation of transcription at these promoters. The consequence of this would be that the regulation of P2 and P1 would imitate the regulation of upstream promoters, which is consistent with the observations presented here.
A plausible model for the regulation of promoters in the all4725-all4721 operon is presented in Fig. 7A. Under conditions of metal sufficiency, all four promoters would be off, P5 and P3 would be repressed by Zur, and P2 and P1 would be repressed by an unknown repressor. Under these conditions, no gene would be expressed (Fig. 7, empty arrows). Metal deficiency would induce all four promoters: P5 and P3 by Zur release and P2 and P1 through repressor dislodgement by advancing RNAP. Under these conditions, all genes of the operons would be expressed (Fig. 7, dark arrows).
Fig 7.

Model for the regulation of promoters in the all4725-all4721 operon. (A) The all4725-all4721 operon is depicted, showing as empty arrows the genes that are repressed and as dark-gray arrows those that are induced in each situation. The positions of promoters P5 to P1 are shadowed. Three different conditions, indicated in the center, are depicted. The status (on or off) of the four promoters under each of these conditions is indicated at the right. (B) Model for the regulation of promoters in the all4725-all4721 operon, including two distinct repressors for the regulation of P2 and P1.
Despite the consistency of this model, the existence of multiple promoters showing a similar regulatory pattern is intriguing. A putative role for the internal promoters could be to counteract the natural polarity of the operon, helping to maintain the expression level that the cell requires for each individual gene. Alternatively, this mode of regulation may help to specifically induce different regions of the operon in response to specific stimuli, as explained below. Regulation of P2 and P1 by a repressor responsive to a specific stimulus (“stimulus A” in Fig. 7) may allow independent induction of all4722 and all4721. For instance, if stimulus A is concomitant to metal sufficiency, P5 and P3 would be repressed by Zur, whereas P2 and P1 would be derepressed. Furthermore, since there is no reason to assume that P2 and P1 are controlled by the same repressor, there is still the possibility that all4721 could be independently induced if P1 was controlled by a distinct repressor responding to a specific stimulus (“stimulus B” in Fig. 7B). This mode of regulation would resemble a domino effect, where induction of one promoter in the operon would determine induction of promoters downstream. According to this model, genes in bacterial operons could be subjected to a hierarchical regulation dictated by their position and the localization and features of internal promoters, so that genes at the 5′ region would be induced together with all genes located downstream, whereas those at the 3′ region may also be induced by specific conditions. Thus, the more toward the 3′ region a gene is located, the more distinct stimuli it could be responsive to.
Multiple efforts are being made at present in synthetic biology to engineer, by the use of molecular toolkits, systems where genes need to be precisely controlled (46, 47). The regulatory model unveiled in this work may be helpful for engineering arrays of genes with a nested regulation by properly distributing internal promoters controlled by repression.
Supplementary Material
ACKNOWLEDGMENTS
This work was financed with grants BFU2010-19544 from the Ministerio de Ciencia e Innovación (Spain), cofinanced by the European Social Fund, and CVI2007-03167 from the Junta de Andalucía and FEDER. M.N. is a recipient of a predoctoral fellowship from the Junta de Andalucía.
Footnotes
Published ahead of print 11 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01488-12.
REFERENCES
- 1. Osbourn AE, Field B. 2009. Operons. Cell. Mol. Life Sci. 66:3755–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blumenthal T. 2004. Operons in eukaryotes. Brief. Funct. Genomic. Proteomic. 3:199–211 [DOI] [PubMed] [Google Scholar]
- 3. Fondi M, Emiliani G, Fani R. 2009. Origin and evolution of operons and metabolic pathways. Res. Microbiol. 160:502–512 [DOI] [PubMed] [Google Scholar]
- 4. Medina-Aparicio L, Rebollar-Flores JE, Gallego-Hernandez AL, Vazquez A, Olvera L, Gutierrez-Rios RM, Calva E, Hernandez-Lucas I. 2011. The CRISPR/Cas immune system is an operon regulated by LeuO, H-NS, and leucine-responsive regulatory protein in Salmonella enterica serovar Typhi. J. Bacteriol. 193:2396–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shin JH, Price CW. 2007. The SsrA-SmpB ribosome rescue system is important for growth of Bacillus subtilis at low and high temperatures. J. Bacteriol. 189:3729–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishihama A. 2010. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol. Rev. 34:628–645 [DOI] [PubMed] [Google Scholar]
- 7. Napolitano M, Rubio MA, Santamaría-Gómez J, Olmedo-Verd E, Robinson NJ, Luque I. 2012. Characterization of the response to zinc deficiency in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 194:2426–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee JW, Helmann JD. 2007. Functional specialization within the Fur family of metalloregulators. Biometals 20:485–499 [DOI] [PubMed] [Google Scholar]
- 9. Ma Z, Gabriel SE, Helmann JD. 2011. Sequential binding and sensing of Zn(II) by Bacillus subtilis Zur. Nucleic Acids Res. 39:9130–9138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shin JH, Jung HJ, An YJ, Cho YB, Cha SS, Roe JH. 2011. Graded expression of zinc-responsive genes through two regulatory zinc-binding sites in Zur. Proc. Natl. Acad. Sci. U. S. A. 108:5045–5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bensing BA, Meyer BJ, Dunny GM. 1996. Sensitive detection of bacterial transcription initiation sites and differentiation from RNA processing sites in the pheromone-induced plasmid transfer system of Enterococcus faecalis. Proc. Natl. Acad. Sci. U. S. A. 93:7794–7799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rippka R. 1988. Isolation and purification of cyanobacteria. Methods Enzymol. 167:3–27 [DOI] [PubMed] [Google Scholar]
- 13. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. (ed). 2010. Current protocols in molecular biology. Greene Publishing & Wiley-Interscience, New York, NY [Google Scholar]
- 14. Elhai J, Wolk CP. 1988. A versatile class of positive-selection vectors based on the nonviability of palindrome-containing plasmids that allows cloning into long polylinkers. Gene 68:119–138 [DOI] [PubMed] [Google Scholar]
- 15. Elhai J, Wolk CP. 1988. Conjugal transfer of DNA to cyanobacteria. Methods Enzymol. 167:747–754 [DOI] [PubMed] [Google Scholar]
- 16. Luque I, Contreras A, Zabulon G, Herrero A, Houmard J. 2002. Expression of the glutamyl-tRNA synthetase gene from the cyanobacterium Synechococcus sp PCC 7942 depends on nitrogen availability and the global regulator NtcA. Mol. Microbiol. 46:1157–1167 [DOI] [PubMed] [Google Scholar]
- 17. Camargo S, Valladares A, Flores E, Herrero A. 2012. Transcription activation by NtcA in the absence of consensus NtcA-binding sites in an Anabaena heterocyst differentiation gene promoter. J. Bacteriol. 194:2939–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Winson MK, Swift S, Hill PJ, Sims CM, Griesmayr G, Bycroft BW, Williams P, Stewart GS. 1998. Engineering the luxCDABE genes from Photorhabdus luminescens to provide a bioluminescent reporter for constitutive and promoter probe plasmids and mini-Tn5 constructs. FEMS Microbiol. Lett. 163:193–202 [DOI] [PubMed] [Google Scholar]
- 19. Guo M, Yang XL, Schimmel P. 2010. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 11:668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frias JE, Flores E, Herrero A. 2000. Activation of the Anabaena nir operon promoter requires both NtcA (CAP family) and NtcB (LysR family) transcription factors. Mol. Microbiol. 38:613–625 [DOI] [PubMed] [Google Scholar]
- 21. Elhai J. 1993. Strong and regulated promoters in the cyanobacterium Anabaena PCC 7120. FEMS Microbiol. Lett. 114:179–184 [DOI] [PubMed] [Google Scholar]
- 22. Huang HH, Camsund D, Lindblad P, Heidorn T. 2010. Design and characterization of molecular tools for a synthetic biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res. 38:2577–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferino F, Chauvat F. 1989. A promoter-probe vector-host system for the cyanobacterium, Synechocystis PCC6803. Gene 84:257–266 [DOI] [PubMed] [Google Scholar]
- 24. Palmer AC, Egan JB, Shearwin KE. 2011. Transcriptional interference by RNA polymerase pausing and dislodgement of transcription factors. Transcription 2:9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rhee KY, Opel M, Ito E, Hung S, Arfin SM, Hatfield GW. 1999. Transcriptional coupling between the divergent promoters of a prototypic LysR-type regulatory system, the ilvYC operon of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 96:14294–14299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fang M, Wu HY. 1998. A promoter relay mechanism for sequential gene activation. J. Bacteriol. 180:626–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu LF, Wang JC. 1987. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. U. S. A. 84:7024–7027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leng F, McMacken R. 2002. Potent stimulation of transcription-coupled DNA supercoiling by sequence-specific DNA-binding proteins. Proc. Natl. Acad. Sci. U. S. A. 99:9139–9144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peter BJ, Arsuaga J, Breier AM, Khodursky AB, Brown PO, Cozzarelli NR. 2004. Genomic transcriptional response to loss of chromosomal supercoiling in Escherichia coli. Genome Biol. 5:R87 doi:10.1186/gb-2004-5-11-r87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niehus E, Cheng E, Tan M. 2008. DNA supercoiling-dependent gene regulation in Chlamydia. J. Bacteriol. 190:6419–6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lim HM, Lewis DE, Lee HJ, Liu M, Adhya S. 2003. Effect of varying the supercoiling of DNA on transcription and its regulation. Biochemistry 42:10718–10725 [DOI] [PubMed] [Google Scholar]
- 32. Bird AJ, Gordon M, Eide DJ, Winge DR. 2006. Repression of ADH1 and ADH3 during zinc deficiency by Zap1-induced intergenic RNA transcripts. EMBO J. 25:5726–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martens JA, Wu PY, Winston F. 2005. Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev. 19:2695–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petruk S, Sedkov Y, Riley KM, Hodgson J, Schweisguth F, Hirose S, Jaynes JB, Brock HW, Mazo A. 2006. Transcription of bxd noncoding RNAs promoted by trithorax represses Ubx in cis by transcriptional interference. Cell 127:1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deuschle U, Gentz R, Bujard H. 1986. lac repressor blocks transcribing RNA polymerase and terminates transcription. Proc. Natl. Acad. Sci. U. S. A. 83:4134–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Belitsky BR, Sonenshein AL. 2011. Roadblock repression of transcription by Bacillus subtilis CodY. J. Mol. Biol. 411:729–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dole S, Nagarajavel V, Schnetz K. 2004. The histone-like nucleoid structuring protein H-NS represses the Escherichia coli bgl operon downstream of the promoter. Mol. Microbiol. 52:589–600 [DOI] [PubMed] [Google Scholar]
- 38. Choi SK, Saier MH., Jr 2005. Regulation of sigL expression by the catabolite control protein CcpA involves a roadblock mechanism in Bacillus subtilis: potential connection between carbon and nitrogen metabolism. J. Bacteriol. 187:6856–6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Epshtein V, Toulme F, Rahmouni AR, Borukhov S, Nudler E. 2003. Transcription through the roadblocks: the role of RNA polymerase cooperation. EMBO J. 22:4719–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horowitz H, Platt T. 1982. Regulation of transcription from tandem and convergent promoters. Nucleic Acids Res. 10:5447–5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lopez PJ, Guillerez J, Sousa R, Dreyfus M. 1998. On the mechanism of inhibition of phage T7 RNA polymerase by lac repressor. J. Mol. Biol. 276:861–875 [DOI] [PubMed] [Google Scholar]
- 42. Perez-Roger I, Macian F, Armengod ME. 1995. Transcription termination in the Escherichia coli dnaA gene is not mediated by the internal DnaA box. J. Bacteriol. 177:1896–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Epshtein V, Nudler E. 2003. Cooperation between RNA polymerase molecules in transcription elongation. Science 300:801–805 [DOI] [PubMed] [Google Scholar]
- 44. Proshkin S, Rahmouni AR, Mironov A, Nudler E. 2010. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 328:504–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Burmann BM, Schweimer K, Luo X, Wahl MC, Stitt BL, Gottesman ME, Rosch P. 2010. A NusE:NusG complex links transcription and translation. Science 328:501–504 [DOI] [PubMed] [Google Scholar]
- 46. Rothschild LJ. 2010. A powerful toolkit for synthetic biology: over 3.8 billion years of evolution. Bioessays 32:304–313 [DOI] [PubMed] [Google Scholar]
- 47. Fischbach M, Voigt CA. 2010. Prokaryotic gene clusters: a rich toolbox for synthetic biology. Biotechnol. J. 5:1277–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.