

Abstract
The activity of DinB is governed by the formation of a multiprotein complex (MPC) with RecA and UmuD. We identified two highly conserved surface residues in DinB, cysteine 66 (C66) and proline 67 (P67). Mapping on the DinB tertiary structure suggests these are noncatalytic, and multiple-sequence alignments indicate that they are unique among DinB-like proteins. To investigate the role of the C66-containing surface in MPC formation, we constructed the dinB(C66A) derivative. We found that DinB(C66A) copurifies with its interacting partners, RecA and UmuD, to a greater extent than DinB. Notably, copurification of RecA with DinB is somewhat enhanced in the absence of UmuD and is further increased for DinB(C66A). In vitro pulldown assays also indicate that DinB(C66A) binds RecA and UmuD better than DinB. We note that the increased affinity of DinB(C66A) for UmuD is RecA dependent. Thus, the C66-containing binding surface appears to be critical to modulate interaction with UmuD, and particularly with RecA. Expression of dinB(C66A) from the chromosome resulted in detectable differences in dinB-dependent lesion bypass fidelity and homologous recombination. Study of this DinB derivative has revealed a key surface on DinB, which appears to modulate the strength of MPC binding, and has suggested a binding order of RecA and UmuD to DinB. These findings will ultimately permit the manipulation of these enzymes to deter bacterial antibiotic resistance acquisition and to gain insights into cancer development in humans.
INTRODUCTION
Upon exposure to both endogenous and exogenous damaging agents, the genome accumulates a variety of lesions that result in replication fork stalling, a potentially lethal event (1, 2). Cells employ a range of high-fidelity repair mechanisms, such as nucleotide excision repair (NER), base excision repair (BER), and homologous recombination (HR), to excise the damaged DNA and/or restore the original template. However, if the damage is extensive and overwhelms these repair processes, cells induce error-prone DNA damage tolerance systems to avert DNA replication stalling and permit survival (1). Translesion synthesis (TLS) is one such system, in which specialized DNA polymerases insert a nucleotide opposite a template lesion and extend past the adducted base (1, 3, 4). In TLS, lesions persist in the template strand and stall subsequent rounds of DNA replication unless they are removed by a high-fidelity DNA repair pathway. TLS DNA polymerases misincorporate deoxynucleotide triphosphates (dNTPs) on undamaged templates more frequently, likely as a result of decreased geometric base pair checking and a relatively open active site, than high-fidelity replicative DNA polymerases (3, 5).
In the model organism Escherichia coli, there are three translesion DNA polymerases. Pol II is a B-family polymerase (6), while Pol IV and Pol V belong to the Y family (7). dinB encodes DNA Pol IV (DinB), and the umuDC operon encodes DNA Pol V (UmuD′2C). These two DNA polymerases bypass distinct DNA lesions, but because DinB is the most abundant DNA polymerase in the cell upon DNA damage (∼2,500 molecules) (8), the lesions that DinB bypasses in vivo are likely to be prevalent. DinB proficiently and accurately bypasses N2-dG adducts generated by the damaging agents nitrofurazone (NFZ) and 4-nitroquinoline 1-oxide (4-NQO) in vivo (9–12). Expression of DinB is also critical for survival in methyl methanesulfonate (MMS), ethyl methanesulfonate (EMS), and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), indicating it is required for bypass of DNA lesions (such as 3-methyl-adenine) generated by alkylating agents (9, 13). Eukaryotic homologues of DinB have also been shown to bypass 3-deaza-3-methyl-adenine, a stable analog of the primary fork-stalling lesion occurring upon treatment with MMS, in vitro (14).
Interestingly, the majority of DNA damage-induced mutagenesis is attributed to the TLS activity of UmuD′2C (Pol V) of E. coli, despite the fact that intracellular levels of UmuC are considerably limited (about 200 molecules upon DNA damage) (15, 16). Pol V is responsible for the bypass of cis-syn T-T photodimers and 6-4 T-T or T-C photoproducts generated by UV irradiation (5, 17) and of abasic sites (3, 18).
In E. coli, Y-family DNA polymerases are highly regulated (1–3). UmuC requires and preferentially binds to a dimer of UmuD′, the processed form of the accessory protein UmuD, to be catalytically active (19–21). Recently, it has been found that DinB's activity is modulated by the formation of a ternary complex with RecA and UmuD2 (22). Notably, the formation of this multiprotein complex (MPC) decreases DinB-dependent −1 frameshifts in vitro and enhances its catalytic activity on undamaged DNA. The striking alteration of DinB's catalytic properties by binding of RecA and UmuD2 indicates that its enzymatic activity is strongly regulated by its interacting partners (22).
Studying the protein-protein interactions governing DinB's activity is critical to understanding the mechanisms modulating the activities of translesion polymerases in vivo. Both Pol IV and Pol V are evolutionarily conserved, and DinB's primary sequence, in particular, is considerably conserved across all domains of life (7). The human homologues of Pol IV and Pol V, Pol κ and Pol η, respectively, exhibit similar functions, lesion specificities, and general structures (3, 9, 10, 23). Determining how these bacterial enzymes are regulated will allow a better description of the regulation of TLS polymerase activity in other systems. Furthermore, TLS polymerases have been implicated in the evolution of antibiotic resistance in bacteria and cancer in humans (24–27). Deregulation of TLS polymerases, therefore, has a variety of deleterious consequences in both prokaryotic and eukaryotic organisms.
While it has been clearly shown in vitro that UmuD2 and RecA bind DinB, the MPC has not yet been isolated directly from cells, and the binding order of the two DinB interactors has not yet been established. To elucidate the mechanism of MPC formation, it is critical to determine whether the binding of UmuD2 enhances the binding of RecA or vice versa. In this vein, mutant MPC components, particularly those of DinB, may prove useful.
An in silico model for the stable ternary complex has been proposed (22) and suggests multiple exposed surfaces of DinB that may be crucial for formation of the MPC. Peptide array mapping has indicated several residues necessary for the binding of DinB to the accessory protein UmuD (22). These residues are not only highly conserved in the protein's primary sequence, but are also localized to a single DinB interface. One example is phenylalanine 172 (F172), a surface residue of DinB exhibiting significant conservation and shown to disrupt MPC formation when mutated to an alanine (22). It would be of great interest to discern which additional residues are vital for the formation and stability of the MPC, for TLS activity, and for interaction with the template or other unknown interacting partners.
Here, we have identified an interacting surface of DinB, which includes the residues cysteine 66 (C66) and proline 67 (P67). We find that these residues are not only highly conserved, but also unique among DinB-like proteins. We concentrated our efforts on understanding the function of DinB C66 in MPC formation and therefore generated the site-specific mutant DinB(C66A). The mutant protein DinB(C66A) copurifies with its interacting partners and with intact ternary complex to a greater extent than the wild-type enzyme, suggesting an important function for this unique protein interface. Study of this DinB derivative has revealed a key interface that appears to modulate the strength of MPC binding and has suggested a binding order of RecA and UmuD to DinB. The analysis of this binding interface is therefore critical, as alteration of the protein-protein interactions will ultimately allow manipulation of these proteins' activities.
MATERIALS AND METHODS
Construction of dinB(C66A) strains.
The dinB(C66A), dinB(P67A), dinB(C66A P67A), and dinB(C66A D103N) mutations were first generated in pYG768 (28). Site-directed mutagenesis, using the GeneTailor Kit (Invitrogen, Carlsbad, CA), was performed according to the manufacturer's instructions. dinB(D103N) was previously generated (29). DE192 (30) is the parental strain used in all experiments. RW86 is isogenic to the parental strain but contains a deletion of the umuDC operon (31). To eliminate differences in the levels of induction of the SOS gene network, all strains employed for in vivo assays are lexA deficient. All plasmid-borne alleles of dinB were introduced into DE192 ΔdinB or RW86 ΔdinB by transformation.
dinB(C66A) was recombined into the chromosome of the parental strain via SOE-Lred (32); this was confirmed by PCR of the dinB open reading frame (ORF) and sequencing. A deletion of umuDC was introduced into the dinB(C66A) strain by P1 transduction. recA and umuDC deletions were also generated in BL21-AI strains by P1 transduction. Gene deletions were confirmed by PCR. All strains used in this study are listed in Table 1. All primers used in the generation of site-specific mutants and in strain construction are listed in Table 2.
Table 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| DE192 | lexA51(def) sulA211 thi-1 Δ(lac-gpt)5 ilv(Ts) mtl-1 rpsL31 | 30 |
| RW86 | lexA51(def) sulA211 thi-1 Δ(lac-gpt)5 ilv(Ts) mtl-1 rpsL31 ΔumuDC595::cat | 31 |
| TMC423 | DE192 ΔdinB pWSK29 | This work |
| TMC425 | Like TMC423, but with pdinB+ (pYG768) | This work |
| TMC743 | Like TMC423, but with pdinB(C66A) (pWSK29 backbone) | This work |
| TMC111 | Like TMC423, but with pdinB(P67A) (pWSK29 backbone) | This work |
| TMC114 | Like TMC423, but with pdinB(C66A P67A) (pWSK29 backbone) | This work |
| TMC117 | Like TMC423, but with pdinB(D103N) (pWSK29 backbone) | This work |
| TMC1110 | Like TMC423, but with pdinB(C66A D103N) (pWSK29 backbone) | This work |
| TMC431 | RW86 ΔdinB pWSK29 | This work |
| TMC433 | Like TMC431, but with pdinB+ (pYG768) | This work |
| TMC747 | Like TMC431, but with pdinB(C66A) (pWSK29 backbone) | This work |
| TMC1113 | Like TMC431, but with pdinB(P67A) (pWSK29 backbone) | This work |
| TMC1116 | Like TMC431, but with pdinB(C66A P67A) (pWSK29 backbone) | This work |
| TMC1119 | Like TMC431, but with pdinB(D103N) (pWSK29 backbone) | This work |
| TMC1122 | Like TMC431, but with pdinB(C66A D103N) (pWSK29 backbone) | This work |
| TMC762 | DE192 dinB(C66A) | This work |
| TMC771 | RW86 dinB(C66A) | This work |
| TMC1144 | BL21-AI ΔdinB pET11T encoding native wild-type DinB | This work |
| TMC1148 | Like TMC1144, but encoding native DinB(C66A) | This work |
| RWB2630 | Like TMC1144, but encoding native DinB(D103N) | This work |
| TMC922 | Like TMC1144, but encoding C-terminally hexahistidine-tagged wild-type DinB | This work |
| TMC931 | Like TMC1144, but encoding C-terminally hexahistidine-tagged DinB(C66A) | This work |
| TMC1237 | BL21-AI ΔdinB ΔrecA pET11T encoding C-terminally hexahistidine-tagged wild-type DinB | This work |
| TMC1240 | Like TMC1237, but encoding C-terminally hexahistidine-tagged DinB(C66A) | This work |
| TMC967 | BL21-AI ΔdinB ΔumuDC pET11T encoding native wild-type DinB | This work |
| TMC970 | Like TMC967, but encoding native DinB(C66A) | This work |
| TMC1055 | Like TMC1237, but encoding native DinB | This work |
| TMC1058 | Like TMC1237, but encoding native DinB(C66A) | This work |
| CAG12204 | KL227 btuB3192::Tn10kan | 42 |
Table 2.
Oligonucleotides used in this study
| Nucleotide sequence (5′ to 3′) | Purpose | Source |
|---|---|---|
| ATGGCGCTCAAATTAGCCCCACATCTCACCTTG | dinB(C66A) mutagenesis forward primer | This work |
| ATTAAACTCGCGGTAGGGACAGCCGTATCG | dinB(C66A) mutagenesis reverse primer | This work |
| GCGCTCAAATTATGCGCACATCTCACCTTG | dinB(P67A) mutagenesis forward primer | This work |
| CGTATTAAACTCGCGGTAGGGACAGCC | dinB(P67A) mutagenesis reverse primer | This work |
| ATGGCGCTCAAATTAGCCGCACATCTCACCTTG | dinB(C66AP67A) mutagenesis forward primer | This work |
| ATTAAACTCGCGGTAGGGACAGCCGTATCG | dinB(C66AP67A) mutagenesis reverse primer | This work |
| CTGGTGCAAAAGCTGGATAAGCAGCAGGTGCTTTCGCAGCGAACGCGTTAATGAGCGATTGTGTTAGGCTGG | SOE-Lred primer 1; adds upstream chromosomal homology to cat | (32) |
| CTGGTAAAGTATACAGTGATTTCAGGGTTTGAGAAATGCGTAAAGATTCAGCATGCCATGGTCCATATGAATATCCTCC | SOE-Lred primer 2; adds dinB promoter homology to cat | (32) |
| ATGCTGAATCTTTACGCATTTCTCAAACCCTGAAATCACTGTATACTTTACCAGTGTTGAGAGGTGAGCAATGC | SOE-Lred primer 3; amplifies dinB with native promoter | (32) |
| GACCGATTTTTCAGCGAGAATTCGATGCATACAGTGATACCCTCATAATAATGCACACCAGAATATACATAATAGTATAC | SOE-Lred primer 4; amplifies dinB with downstream chromosomal homology | (32) |
| GCTCGTCAGACGATTTAGAGTCTGCAGTG | Undamaged control template for extension assay | This work |
| GCTCGTCAGACG/3-deaza-3-methylA/TTTAGAGTCTGCAGTG | Lesion-containing template for extension assay | This work |
| /HEX/CACTGCAGACTCTAAA | Fluorescently labeled primer for standing-start extension | This work |
Multiple-sequence alignment of DinB, epitope mapping, and in silico modeling.
A multiple-sequence alignment (MSA) of DinB was performed using CLC Bio Genomics Workbench software (CLC Bio, Aarhus, Denmark). Genomic entries for DinB-like and UmuC-like proteins were obtained from NCBI and were hand curated according to the following criteria: DinB sequences contained the signature catalytic residues SLDE (101 to 104) and were at least 300 amino acids in length, while UmuC sequences contained the signature catalytic residues SIDE (102 to 105) and were at least 400 amino acids in length. Slight variations of the signature catalytic motif were permitted, except at the second amino acid position and at no more than 1 residue. A total of 316 DinB-like sequences from over 100 diverse species and 1,425 UmuC-like sequences from over 600 species were examined. DinB entries were also aligned with UmuC sequences. A list of the organisms included in the MSA, and the number of sequences per organism, is provided in Tables S1 and S2 in the supplemental material. A list of accession numbers can be found in the supplemental material. The conservation of select residues was examined using CLC Bio Genomics Workbench software. Epitope mapping was performed as detailed by Godoy et al. (22). All in silico models of DinB and the MPC (22) were rendered using PyMol.
Protein purification of DinB(C66A) and immunoblotting.
Recombinant pET11T plasmids (33) carrying dinB and dinB(C66A) were constructed to include a C-terminal hexahistidine tag and were introduced by transformation into BL21-AI One Shot cells (Life Technologies, Carlsbad, CA) that we had previously made ΔdinB by P1 transduction. Gene products of interest were overexpressed by autoinduction (34). Briefly, strains were first grown in minimal noninducing medium (MDG) with 100 μg ml−1 ampicillin (Amp). A 10-ml culture of inducing medium containing both glucose and l-arabinose (ZYM-5052) with Amp was inoculated with a 1:1,000 dilution of the noninducing minimal-medium-grown cells. These cultures were incubated at 20°C with agitation for approximately 48 h. Cell lysis and protein purification were performed either mechanically, using the Maxwell 16 instrument (Promega, Madison, WI) and its corresponding Polyhistidine Protein Purification Kit, or manually, using BugBuster Protein Extraction Reagent (Novagen/EMD, Darmstadt, Germany), Lysonase Bioprocessing Reagent (Novagen/EMD), and a Dynabeads His Tag Isolation and Pulldown Kit (Life Technologies). All reagents and kits were used according to the manufacturers' instructions.
For large-scale purification of hexahistidine-tagged DinB and DinB(C66A), 1 liter of cells was induced as described above. A cleared lysate was prepared as previously described (22), except that imidazole and NaCl were added to the cleared lysate at final concentrations of 40 mM and 500 mM, respectively. The lysate was loaded onto a series of two 1-ml HisTrap FF columns (GE Healthcare, Uppsala, Sweden). The columns were washed with buffer containing increasing amounts of imidazole (40 mM imidazole, followed by 80 mM) and eluted with buffer containing 200 mM imidazole. All buffers contained 50 mM HEPES, pH 7.5, 500 mM NaCl, and 2 mM β-mercaptoethanol. A flow rate of 1 ml min−1 was used throughout the purification.
To purify native DinB(C66A), DinB, and DinB(D103N), recombinant pET11T plasmids (33) carrying dinB, dinB(C66A), and dinB(D103N) were constructed without a hexahistidine tag and were introduced by transformation into isogenic BL21-AI One Shot ΔdinB, ΔdinB ΔumuDC, or ΔdinB ΔrecA cells (Table 1). Proteins were overexpressed by autoinduction, as described above (34), and lysed using a French press disruptor (Standsted SPCH-10; Harlow, United Kingdom). A cleared lysate was prepared, and the proteins were purified by cation-exchange chromatography (CEC), as previously described (22). Pooled CEC fractions containing DinB were further purified by hydrophobicity interaction chromatography (HIC), as described by Godoy et al. (22).
The purified proteins were separated by SDS-PAGE and characterized by immunoblotting (35). Pooled fractions obtained by CEC were serially diluted, combined with an equal volume of 2× Laemmli sample buffer (Sigma-Aldrich, St. Louis, MO), and heated at 95°C for 10 min. Proteins were separated on NuPAGE Novex 4 to 12% Bis-Tris gels using MOPS (morpholinepropanesulfonic acid) SDS running buffer (Life Technologies) and transferred to Immobilon-FL polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA) at 20 to 25 V for 16 to 18 h at 4°C. The blots were blocked with 0.2% I-Block (Life Technologies) in Tris-buffered saline with Tween 20 (TBST) (35). Rabbit polyclonal anti-UmuD and anti-RecA antibodies were obtained from Abcam (Cambridge, MA) and used at dilutions of 1:2,500 and 1:30,000, respectively. Affinity-purified polyclonal rabbit anti-DinB antibodies were generated in house (36) and used at 1:500 dilution. Primary antibodies were diluted in TBST containing 0.1% I-Block. Cy5-conjugated goat anti-rabbit IgG (Abcam) was used at a dilution of 1:2,500. Secondary antibodies were prepared in TBST containing 0.8% I-Block. Bands were visualized using a Typhoon 8600 Imager. The amounts of DinB, RecA, and UmuD detected in each set of pooled fractions were determined by generating a standard curve in which known concentrations of each protein were transferred to membranes and probed with the respective antibody. The signal intensity was determined using ImageJ software (NIH). The total protein concentrations of elution fractions were determined by performing a Bradford assay; Coomassie Plus Protein Assay Reagent (Thermo Scientific, Rockford, IL) was used according to the manufacturer's instructions.
In vitro extension and pulldown assays.
Purified native DinB, DinB(C66A), or DinB(D103N) was obtained by the purification methods described above. E. coli DNA polymerase I was obtained from New England BioLabs (NEB) (Ipswich, MA). A common standing-start primer containing a 5′ hexachlorofluorescein (HEX) label and an extension assay template containing an internal 3-deaza-3-methyl-adenine were purchased from Integrated DNA Technologies (IDT) (Coralville, IA). Undamaged control template was purchased from Eurofins MWG Operon (Huntsville, AL). The common primer was independently annealed to each of the templates. Primers and templates were diluted to 10 μM in TE buffer (10 mM Tris HCl, pH 7.9, 100 mM NaCl, 1 mM EDTA, pH 8.0), mixed in equal volumes, and annealed in a thermocycler (95°C for 10 min, 55°C for 30 min, 20°C for 1 h). Extension reaction mixtures contained a total volume of 10 μl, and reactions were performed in a low-salt buffer [final concentration, 20 mM Tris-HCl, pH 8.75, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, 0.1 mg ml−1 bovine serum albumin (BSA); Agilent, Santa Clara, CA]. Reaction mixtures contained a final concentration of 25 nM primer-template, 0.5 mM dNTP mixture, and 1.25 μM DinB, DinB(C66A), or DinB(D103N) or 2 × 10−3 units of DNA polymerase I. Reactions were initiated by the addition of enzyme, heated to 37°C, and quenched by the addition of 2 μl of stop/loading dye (35) after 10 min. The separated extension products were visualized using the Typhoon 8600 Imager.
To assay for DinB-RecA binary complex formation, in vitro pulldown assays were performed. Purified RecA, containing a hexahistidine tag at the N terminus, was used as the bait. A constant concentration of RecA-His (0.5 μM; New England BioLabs) was incubated with increasing concentrations of pure, native recombinant DinB or DinB(C66A) (0.25 to 2 μM). To test whether bait and interacting proteins were in the linear range, the concentration of bait was decreased, while the molar ratios of bait to interacting proteins were preserved, as indicated above. The binding of DinB and DinB(C66A) to UmuD was also assessed but utilized purified hexahistidine-tagged DinB as the bait. A constant concentration of DinB-His or DinB(C66A)-His was used per reaction (0.5 μM), while pure, native UmuD (Walker Laboratory, MIT) was titrated from 0.5 to 2 μM. To assay for ternary complex formation, a constant concentration of RecA-His (0.3 μM) was used per reaction and was incubated with increasing concentrations of both pure UmuD (0.3 to 2 μM) and native recombinant DinB or DinB(C66A) (0.2 to 1.3 μM).
All pulldown assays were performed using 2× Pulldown Buffer (Life Technologies). Protein mixtures were incubated at room temperature for 1 h with gentle agitation to allow protein complex formation. Ten microliters of Dynabeads His Tag Isolation and Pulldown beads (Life Technologies), preequilibrated in 1× Pulldown Buffer, was then added in batch to each mixture and incubated for an additional hour to allow RecA-His or DinB-His and any interacting proteins to bind the beads. Magnetic beads were washed with 30 resin volumes three times (for a total of 90 resin volumes) with 1× Pulldown Buffer. They were then resuspended in 30 μl of 2× Laemmli buffer (Sigma) and heated at 95°C for 10 min. The resin-bound proteins were separated by SDS-PAGE. The intensities of monomeric UmuD (15-kDa) bands obtained in the DinB-UmuD binary assay were quantified using ImageJ (NIH). The intensities of bands corresponding to DinB (∼40 kDa), the DinB-RecA binary complex (70 kDa), UmuD (15 kDa), and UmuD2 (30 kDa) detected in the ternary complex reactions were also quantified. All quantified bands were normalized to the intensity of the constant band of the respective bait protein [DinB-His or DinB(C66A)-His for the DinB-UmuD binary complex assay; RecA-His for the ternary complex assay; both ∼40 kDa].
CD spectroscopy.
Purified DinB protein samples were concentrated using Amicon Ultra 0.5-ml, 10-kDa spin filters (Millipore) to a volume of 30 μl. The concentrated samples were then diluted in water to a final concentration of 100 μg ml−1 and loaded into a 100-μl circular-dichroism (CD) cuvette. The far-UV spectra were measured using a Jasco J-715 spectropolarimeter. The spectrum of each sample was determined six times from a wavelength of 240 nm to 200 nm, and the data were analyzed using the CDPro software package and by utilizing the CONTIN/LL algorithm (37).
In vivo assays.
The strains bearing various plasmid-borne alleles of dinB were treated with NFZ or MMS. Percent survival was calculated as the number of CFU obtained upon treatment divided by the CFU obtained for the same strain untreated. The same procedure was followed with the parental strain and the strain containing the chromosomal dinB(C66A) allele. Concentrations of 7.5 mM MMS (Acros Organics, Geel, Belgium) or 1.5 μg ml−1 NFZ (Sigma-Aldrich, St. Louis, MO) were used, since the parental strain was minimally affected. LB medium plates containing either MMS or NFZ were prepared immediately before use. Three biological replicates of all strains were plated and incubated at 37°C.
For UV survival assays, strains were serially diluted and plated on tetrazolium galactose (TG) agar (modified from reference 38), irradiated for increasing intervals of time (0 s to 55 s), and incubated at 37°C. Percent survival was determined for each culture by comparison to an untreated control culture of the same strain. Three biological replicates were assessed per strain.
NFZ-, MMS-, and UV-induced mutagenesis assays.
Strains were serially diluted and plated on LB medium plates containing either 7.5 mM MMS or 0.75 μg ml−1 NFZ. The protocol described by Benson et al. (9) was employed to identify mutant colonies. Briefly, DNA damage-induced mutants are screened as colonies that grow on LB rich medium but are unable to grow on glucose minimal medium. Any loss-of-function mutation (i.e., transitions, transversions, and indels) resulting in the inability to grow on minimal medium is detected with this protocol, ensuring the analysis of unlimited mutational changes in a large target. All mutants were confirmed by colony purification in both LB and M9 media.
To determine the levels of UV-induced mutagenesis, a TG agar-based assay was used (39; M. D. Norton and V. G. Godoy, unpublished data). Briefly, strains were serially diluted and plated on TG agar containing 1% d-galactose and 0.33 μg ml−1 of triphenyl tetrazolium chloride (TTC), the tetrazolium indicator dye. Each plate was UV irradiated at 56 J m−2 and incubated at 37°C for 16 to 18 h. Mutants were identified as dark pink or red colonies. The number of mutant colonies and total CFU were determined for cells exposed and not exposed to UV irradiation. The number of colonies assessed per strain in each treatment is indicated in the respective figure legend.
Homologous recombination assays.
Recombinogenicity was first assessed by P1 transduction. The ΔsrlD::kan P1 virulent phage was generated, and the titer was determined by standard techniques (40). The appropriate Keio deletion strain (41) was used as the host for phage preparation. Approximately 1 ml of saturated culture was used per transduction reaction at a multiplicity of infection (MOI) of 1. Kanamycin-resistant (Kanr) cells were selected on LB medium plates with 30 μg ml−1 of Kan. The total number of Kanr colonies was determined for each strain, and the frequency of recombination was calculated by dividing the number of Kanr trandsductants obtained by the total number of cells used per reaction.
Recombinogenicity was also assessed by conjugal mating (40). CAG12204 was used as the donor strain (42). The parental, ΔdinB, ΔumuDC, dinB(C66A), and dinB(C66A) ΔumuDC strains were used as the recipients. Saturated cultures of donor and recipient strains were diluted 1:50 and 1:20, respectively, in LB medium and grown for 2.5 h at 37°C, after which CFU were determined for all cultures. The cells were mixed and transconjugants selected for as previously described (40, 42).
RESULTS
Cysteine 66 is highly conserved among DinB-like proteins, but not in other Y-family TLS polymerases.
It was previously determined that residues critical for the interaction of DinB and UmuD, such as F172, were highly conserved in the DinB primary amino acid sequence (22) and were predicted to localize to a single exposed surface of the protein. It can be inferred that additional residues responsible for regulating the binding properties of DinB will also be highly conserved and can therefore be initially identified via multiple-sequence alignments. Indeed, an MSA of a mixture of 316 bona fide and predicted DinB-like sequences from over 100 species indicated C66 was an extensively conserved residue; 83% of sequences retained a cysteine at this position (Fig. 1A; see Table S1 in the supplemental material). In the remaining sequences, isoleucine, leucine, and valine were most commonly present. It was also noted that the immediately adjacent residue, P67, is highly conserved as well among the examined sequences (89%) (Fig. 1A). Furthermore, it was predicted that these residues would not alter the catalytic ability of DinB directly; when localized in the tertiary structure of DinB, they should be found exclusively at exposed surfaces and not in the catalytic pocket. Indeed, when mapped to an in silico model of DinB (22), both C66 and P67 are predicted to be on a single exposed surface of DinB (Fig. 1C). Notably, they are localized to the same surface as F172; while F172 is located in the thumb domain, C66 and P67 are located in the finger domain of DinB (Fig. 1C). Both residues were found to be highly conserved, noncatalytic residues in DinB.
Fig 1.
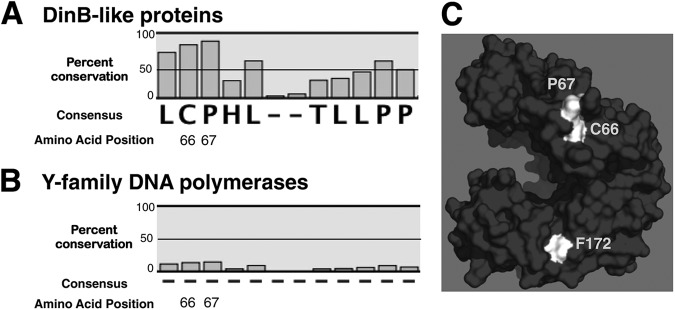
C66 and P67 are highly conserved noncatalytic residues in DinB-like proteins only. An MSA reveals extensive conservation of C66 (83%) and P67 (89%) in the primary sequence of DinB-like proteins, but not in all Y-family DNA polymerases (15% and 16%, respectively). (A) MSA of predicted and bona fide DinB-like sequences. (B) MSA of predicted and bona fide Y-family DNA polymerase sequences (DinB-like and UmuC-like). The dashes indicate a lack of consensus in the amino acid sequence. (C) DinBC66 and DinBP67 are surface residues, do not pertain to the active site, and are located at the same surface as F172. C66, P67, and F172 are highlighted in white. The in silico model of DinB was rendered using PyMol.
To examine whether C66 (and P67) are also conserved in UmuC-like sequences, the multiple-sequence alignment was expanded to include 1,425 bona fide and predicted UmuC-like sequences from over 600 species (Fig. 1B; see Tables S1 and S2 in the supplemental material). Remarkably, when the MSA was thus expanded, the extreme conservation of C66 and P67 was no longer apparent (Fig. 1B). These residues are therefore not conserved in all Y-family DNA polymerases but are unique to DinB-like proteins.
ΔdinB strains containing the plasmid-borne dinB(C66A) and dinB(P67A) alleles are active for TLS in vivo, while those harboring dinB(C66A D103N) are rescued from lethality.
We generated site-specific mutants to investigate the functions of C66 and P67, replacing these residues with alanines. To assess whether the point mutations at C66 and P67 altered the catalytic abilities of DinB, we constructed the mutant alleles on a low-copy-number plasmid, placing them under the control of the native dinB promoter, and introduced them into both ΔdinB and ΔdinB ΔumuDC strains by transformation. To eliminate differences in levels of SOS network induction, all strains are lexA deficient. Cell survival in DNA-damaging agents that produce DinB cognate lesions (e.g., NFZ) depends on the in vivo activity of DinB (9, 10). As expected, ΔdinB and ΔdinB ΔumuDC strains expressing DinB(C66A), DinB(P67A), and DinB(C66A P67A) survived as well as the dinB+ strain in both NFZ and MMS (Fig. 2A and B), indicating that DinB(C66A), DinB(P67A), and DinB(C66A P67A) have no detectable catalytic deficiency.
Fig 2.
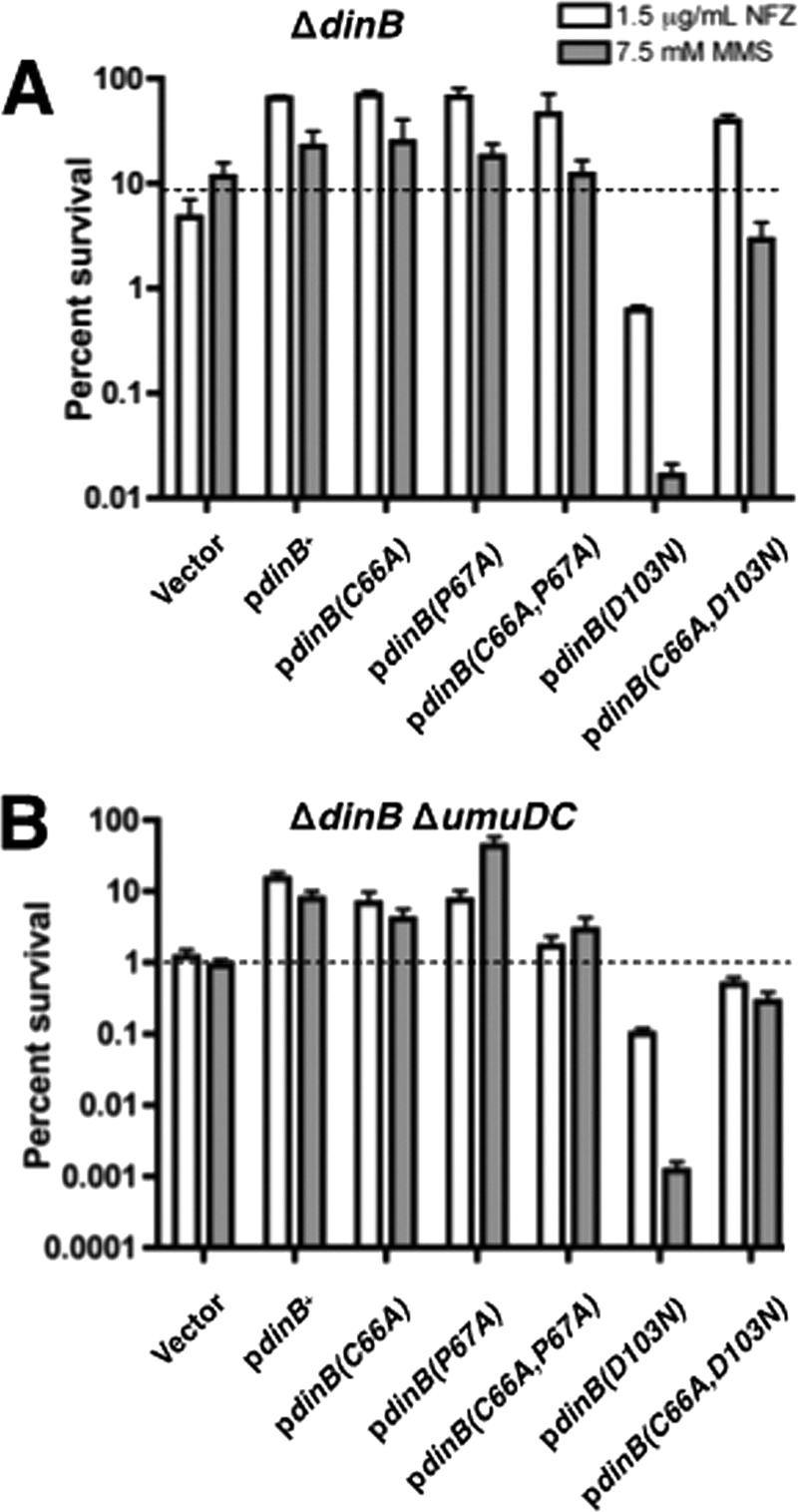
DinB site-directed derivatives are proficient for lesion bypass in vivo, and a C66A mutation eliminates hypersensitivity of the catalytically inactive dinB(D103N) strain. Survival of the ΔdinB strains (A) and ΔdinB ΔumuDC strains (B) is shown. In both backgrounds, expression of DinB(C66A) or DinB(P67A) does not affect survival in the presence of either NFZ (1.5 μg ml−1) or MMS (7.5 mM). Cells expressing DinB(D103N) are highly sensitive to both DNA-damaging agents. However, the extreme sensitivity of cells expressing DinB(D103N) is suppressed in cells expressing the double mutant, DinB(C66A D103N), to levels similar to those of the vector-only strain, which is consistent with loss of catalytic activity. The NFZ survival shown for the DinB(C66A D103N) double mutant in the ΔdinB strain suggests that other DNA polymerases, e.g., DNA Pol V, might be responsible for the bypass; this is no longer the case in the ΔdinB ΔumuDC strain. The various dinB alleles are expressed from the native promoter in a low-copy-number vector (pYG768) (28). Three biological replicates were assessed for each strain. Means and 1 standard deviation (SD) are shown.
We (9) and others (10) have shown that expression of DinB(D103N), a catalytically inactive form of DinB (29), from a low-copy-number plasmid renders cells more sensitive to NFZ or MMS than isogenic cells with the vector alone. The precise mechanism resulting in DinB(D103N)-mediated hypersensitivity has not yet been elucidated. To test whether this hypersensitivity is solely due to DinB's catalytic defect or whether it might also be mediated by protein-protein interactions, we combined the C66A mutation with the D103N mutation. Notably, expression of the plasmid-borne dinB(C66A D103N) allele in a ΔdinB strain restores viability to levels comparable to those of cells with the vector alone. Viability is restored by 64-fold in NFZ (39.44% ± 5.33% versus 0.62% ± 0.06%) and by 180-fold in MMS (2.89% ± 1.36% versus 0.016% ± 0.005%) relative to the strain expressing dinB(D103N) (Fig. 2A). The increased survival of cells expressing the DinB(C66A D103N) derivative suggested either an enhancement or a loss of a critical noncatalytic function of DinB.
It is also possible, however, that other TLS polymerases, particularly Pol V, were able to access replication forks stalled by NFZ- or MMS-induced lesions. To determine whether the increased survival is due to the altered functions of DinB caused by the C66A mutation or to the activity of Pol V, the plasmid-borne dinB(C66A D103N) allele was introduced by transformation into a ΔdinB ΔumuDC strain (Fig. 2B). A substantial restoration of viability was also detected in this strain relative to the ΔdinB ΔumuDC strain harboring dinB(D103N) (0.49% ± 0.13% versus 0.1% ± 0.02% for NFZ; 0.28% ± 0.1% versus 0.0012% ± 0.0004% for MMS). This indicates that the rescue from NFZ- and MMS-induced hypersensitivity of strains expressing the dinB(C66A D103N) double mutant is likely due to the altered noncatalytic functions resulting from a C66A substitution.
DinB(C66A) protein copurifies with RecA and UmuD by ion metal affinity and cation-exchange chromatography.
Since proline residues are generally important to maintain a stable tertiary structure, we concentrated our efforts on C66. We predicted that mutation of the C66 conserved surface residue would disrupt DinB MPC formation, and expression of this allele from the chromosome would thereby result in strains with altered fidelity under conditions of replication stress. To determine the effect of a single point mutation on the ability of DinB to form MPCs, we first sought to purify the mutant protein and analyze it in vitro.
Wild-type DinB and DinB(C66A), both containing a C-terminal hexahistidine tag, were introduced into BL21-AI ΔdinB cells by transformation. Protein overexpression was induced by autoinduction (34), and the cleared lysate was separated by ion metal affinity chromatography (IMAC). The eluted fractions were examined via SDS-PAGE, resulting in the banding pattern shown in Fig. 3A (left). DinB(C66A) had a distinct purification pattern that was largely absent for wild-type DinB (Fig. 3A, left). DinB(C66A) fractions separated by SDS-PAGE revealed a single band in the 100-kDa size range, a doublet in the 40-kDa size range, and another single band in the 15-kDa size range. We predicted these proteins to be the intact ternary complex (∼107 kDa), DinB (39 kDa), RecA (38 kDa), and a monomer of full-length UmuD (15 kDa), which had previously been identified (22). Conversely, a single dominant band of approximately 40 kDa was detected for DinB, consistent with previous reports (22, 29).
Fig 3.

The DinB(C66A) derivative copurifies with more RecA and UmuD than DinB. (A) SDS-PAGE of elution fractions for hexahistidine-tagged DinB and DinB(C66A) purified by IMAC (left) and native DinB and DinB(C66A) purified by CEC (right). The molecular masses shown are approximate. Seven micrograms of total protein was loaded for DinB-His purified by IMAC, and 1.3 μg of total protein was loaded for DinB(C66A)-His. For proteins purified by CEC, 6 μg of total protein was loaded for native DinB and 3 μg of total protein was loaded for native DinB(C66A). (B) Immunoblot of pooled elution fractions for DinB and DinB(C66A) purified by CEC. For all immunoblots, undiluted and serially diluted pooled elution fractions were loaded; 3 μg of total protein was loaded in the undiluted sample of DinB, while 1.5 μg of total protein was loaded in the undiluted sample of DinB(C66A). Less DinB(C66A) than DinB was present in the sample (300 ng versus 600 ng), consistent with less total protein loaded (approximately half); however, DinB(C66A) copurified with more RecA, UmuD, and intact putative MPC than DinB. The amounts of proteins per lane were determined by standard curves of blotted serially diluted proteins developed with the respective antibodies. The molecular masses shown are approximate. The amounts of copurified RecA and UmuD were determined to be less than 40 ng. (C) DinB epitope map for anti-DinB (αDinB) antibodies. Epitopes recognized by the affinity-purified αDinB antibody were determined by peptide array mapping, as indicated in Materials and Methods. Recognized epitopes were mapped onto an in silico model of DinB and are highlighted in white. The epitopes recognized by the polyclonal antibody localize to the exposed surfaces of DinB that are predicted to be bound by UmuD and RecA. The in silico DinB model was rendered using PyMol.
The purification of hexahistidine-tagged DinB(C66A) appeared to yield lower levels of purified protein than DinB. It is possible that the C-terminal hexahistidine tag altered the tertiary structure of DinB(C66A), resulting in either protein degradation or poor availability of the purification tag. To circumvent these issues, native DinB and its derivative, DinB(C66A), were purified by CEC, as previously described (Fig. 3A, right) (22). Purification of the native proteins resulted in increased yield, though approximately half as much DinB(C66A) was purified as DinB (Fig. 3A, right versus left). This suggested that DinB(C66A), though not degraded, might be folding differently and would therefore be less stable than the wild-type form. To further investigate these issues, we examined whether a change in the secondary structure of the purified native proteins could be detected by CD spectroscopy. We found no striking structural differences between DinB and DinB(C66A) (see Fig. S1 and Table S3 in the supplemental material).
When isolated by CEC, we could not easily recognize whether the mutant protein had copurified with known proteins of the MPC (Fig. 3A, right). Thus, we used pooled CEC fractions and immunoblotting to identify interacting proteins that could have copurified with DinB or DinB(C66A). We confirmed that DinB, RecA, and UmuD were present in the pooled CEC fractions by immunoblotting (Fig. 3B). RecA (39 kDa) was detected at its respective size, but also at about 100 kDa, the approximate predicted size of the intact MPC. UmuD was detected as a protein of 30 kDa, the size of an intact homodimer, as well as a large complex of about 100 kDa. While DinB and DinB(C66A) were both detected at about 40 kDa, we were unable to detect either DinB or DinB(C66A) in the putative ternary complex band (100-kDa band in Fig. 3B). However, we thought that the DinB epitopes might be obscured and therefore inaccessible to the antibodies as a result of MPC formation. We identified the epitopes recognized by the polyclonal DinB-specific antibodies using peptide mapping (43). The resulting epitopes, mapped to the in silico model of DinB, localized to the binding interfaces between DinB, RecA, and UmuD2 (Fig. 3C). Therefore, it is likely that the larger complexes contained DinB but were not detected by the DinB-specific antibody as a result of epitope masking by RecA and UmuD. Nevertheless, we found that both DinB and DinB(C66A) copurify with components of the MPC directly from cells.
We sought to quantify the relative amounts of RecA and UmuD in pooled CEC elution fractions. Overall, we found copurification of MPC-interacting proteins to be most efficient for the mutant. Approximately half as much total protein was loaded in the DinB(C66A) lanes (Fig. 3B), while RecA was detected as well or better for the mutant than for the wild-type protein (Fig. 3B). The ratio of copurified RecA to DinB is therefore higher for DinB(C66A) than for the wild-type enzyme. In addition, the putative intact ternary complex was only detected for DinB(C66A) with the RecA-specific antibody. Furthermore, DinB(C66A) copurified with UmuD2 and the intact ternary complex, as detected by the UmuD-specific antibody. In contrast, there was no detectable copurification of UmuD with DinB. A single amino acid substitution in DinB, therefore, allowed the enhanced copurification of UmuD and RecA. Both interacting proteins copurified at easily detectable levels; this is particularly impressive given that RecA and UmuD were not overexpressed.
Purification of DinB from ΔumuDC and ΔrecA strains reveals that DinB-RecA binary complexes may precede the formation of ternary complexes in vivo.
We wondered if the enhanced binding ability of DinB(C66A) could be utilized to elucidate the in vivo binding order of the MPC proteins. Therefore, purification by CEC was repeated in strains lacking one of DinB's interacting proteins. Native DinB and DinB(C66A) were purified from BL21-AI ΔdinB ΔumuDC strains (Fig. 4A), using the methods described previously. We predicted that DinB would bind RecA independently of UmuD. Remarkably, we found that copurification of RecA with DinB was somewhat better in the absence of UmuD (Fig. 3B versus 4B). At approximately the same protein load, RecA can be detected in the second dilution in the ΔumuDC strain, but not in the umuDC+ strain (compare Fig. 3B and Fig. 4B, first and second lanes for DinB). Similarly, the copurification of RecA with DinB(C66A) is moderately enhanced (Fig. 3A versus 4A and 3B versus 4B). Notably, the RecA antibody also detects a band of approximately 70 kDa only in the DinB(C66A) lanes (Fig. 4B). This is likely to be the intact DinB(C66A)-RecA binary complex. Thus, RecA appears to copurify better with DinB(C66A) than with DinB in the absence of UmuD.
Fig 4.

DinB-RecA binary complexes may form first, independently of UmuD. (A) SDS-PAGE of pooled CEC elution fractions obtained for native DinB and DinB(C66A) purified from a BL21-AI ΔumuDC strain. Approximately 4 μg of total protein was loaded per lane. (B) Immunoblot of proteins copurifying with DinB or DinB(C66A) isolated from an E. coli BL21 ΔumuDC strain. In the absence of the umuD gene, the copurification of RecA is enhanced for DinB and DinB(C66A) (compare Fig. 3B), though DinB(C66A) copurifies RecA better than DinB in the ΔumuDC strain. (C) SDS-PAGE of pooled CEC elution fractions obtained for DinB and DinB(C66A) purified from a BL21-AI ΔrecA strain. Approximately 3 μg of total protein was loaded per lane. (D) Immunoblot of proteins copurifying with DinB or DinB(C66A) isolated from an E. coli BL21 ΔrecA strain. In the absence of the recA gene, the amount of UmuD that copurifies with DinB(C66A) is reduced relative to protein copurified from a recA+ strain (Fig. 3B). The molecular masses shown are approximate. The amounts of copurified RecA and UmuD, when present, are less than 40 ng.
We then sought to determine whether UmuD would copurify with DinB or DinB(C66A) in the absence of RecA. If a binding order exists, then copurification of UmuD would be limited in the absence of the DinB-RecA binary complex. However, if DinB exhibits no preferred binding order, then UmuD would copurify from cells lacking RecA. Purification by CEC was therefore performed from a BL21-AI strain with recA deleted (Fig. 4C). We found that in the absence of RecA, UmuD copurified poorly with DinB (Fig. 4D). Furthermore, copurification of UmuD was reduced for the DinB(C66A) derivative (Fig. 4D) compared to the recA+ strain (Fig. 3B). While we were still able to detect UmuD2, we were no longer able to detect the presence of a larger complex using the UmuD-specific antibody. These results indicate that DinB is able to bind to UmuD in the absence of RecA, albeit not as well as in the recA+ strain, and suggests an order in MPC formation.
DinB(C66A) exhibits increased binding of RecA and UmuD in vitro.
DinB(C66A) copurified with MPC proteins when isolated by IMAC and CEC. To confirm the increased binding ability of DinB(C66A) to UmuD and RecA, we performed an in vitro pulldown assay using purified proteins.
We first examined the abilities of wild-type DinB and DinB(C66A) to form a binary complex with UmuD. For this assay, we employed hexahistidine-tagged DinB and DinB(C66A); to avoid carryover of RecA, we purified these proteins from ΔrecA strains (see Fig. S2 in the supplemental material). A constant concentration of DinB or DinB(C66A) (0.5 μM), containing a C-terminal hexahistidine tag, was incubated with increasing concentrations of pure UmuD (0.5 to 2 μM). These mixtures result in different molar ratios of the assayed proteins, as shown in Fig. 5A. UmuD was allowed to bind to DinB-His or DinB(C66A)-His, and protein mixtures were purified by IMAC and then separated by SDS-PAGE. While no UmuD was retrieved in the absence of the bait protein, we found that UmuD bound DinB and DinB(C66A) equally well (Fig. 5A). The intensities of the UmuD bands were quantified using ImageJ software (NIH) and normalized to the intensities of the respective bands of the bait protein in the same reaction. The adjusted relative intensities of copurified UmuD obtained per reaction are shown in Fig. 5B. Comparable levels of UmuD were retrieved for both DinB-His and DinB(C66A)-His (Fig. 5B), indicating no clear advantage in the ability to form DinB-UmuD2 binary complexes for either enzyme.
Fig 5.

DinB(C66A) binds RecA and UmuD better than DinB in vitro. (A) In vitro pulldown assay to assess the abilities of DinB and DinB(C66A) to bind to UmuD. Molar ratios of DinB to UmuD2 are shown per reaction, in which the bait concentration [hexahistidine-tagged DinB or DinB(C66A)] is kept constant. In the absence of RecA, DinB(C66A) binds UmuD as well as DinB. (B) Quantification of the binding of UmuD to hexahistidine-tagged DinB or DinB(C66A) in the absence of RecA. (C) In vitro pulldown assay assessing the binding of DinB and DinB(C66A) to RecA-His. Molar ratios of DinB or DinB(C66A) to RecA are shown per reaction, in which the bait concentration (hexahistidine-tagged RecA) is kept constant. DinB(C66A) binds RecA better than DinB. (D) Adjusted quantification of the binding of DinB or DinB(C66A) to hexahistidine-tagged RecA. (E) Quantification of intact DinB-RecA binary complexes (70 kDa). Unlike DinB, DinB(C66A) forms highly stable binary complexes with RecA, which were unaffected by treatment with heat or denaturants. (F) In vitro pulldown assay to assess the abilities of DinB and DinB(C66A) to form ternary complexes with RecA and UmuD2. For the ternary complex, molar ratios of DinB, RecA, and UmuD2 are shown per reaction, in which the bait concentration (hexahistidine-tagged RecA) is kept constant. (G) Quantification of the binding of monomeric UmuD to DinB or DinB(C66A) in the presence of RecA. (H) Quantification of the binding of UmuD2 to DinB or DinB(C66A) in the presence of RecA. For all quantifications, the intensities of bands corresponding to the protein of interest were determined using ImageJ software. The intensity of each band in question was normalized to the intensity of the band corresponding to the constant bait protein in its respective lane and is therefore shown as adjusted relative density. The quantification of DinB or DinB(C66A) copurifying with RecA-His was adjusted to account for the fraction of prey protein interacting nonspecifically with the resin.
The ability of DinB and DinB(C66A) to bind RecA was also of interest. It is known that DinB and RecA form stable and catalytically active binary complexes and bind in a 1:1 stoichiometric ratio (22). Therefore, we performed a pulldown assay to assess binary complex formation but utilized hexahistidine-tagged RecA as the bait for these reactions. We incubated constant concentrations of RecA (0.1 or 0.5 μM) containing an N-terminal hexahistidine tag with increasing concentrations of purified native DinB or DinB(C66A). The native proteins were again purified from ΔrecA strains to avoid RecA carryover (see Fig. S3 in the supplemental material). We found that both DinB and DinB(C66A) copurified with RecA (Fig. 5C). Interestingly, while both DinB and DinB(C66A) were detected (∼40 kDa), reaction mixtures containing DinB(C66A) also revealed a 70-kDa band (Fig. 5C). This band contained both DinB and RecA by immunoblotting, indicating that DinB(C66A) generates a stable and strongly bound binary complex that was not disrupted by either heat or denaturants (see Fig. S4 in the supplemental material). This stable binary complex was disassembled, however, in the presence of 8 M urea (see Fig. S5 in the supplemental material).
We found, in addition, that native DinB and DinB(C66A) bound nonspecifically to the nickel resin in the absence of RecA-His bait and identified a number of clustered surface histidine residues that may have mediated this nonspecific interaction (see Fig. S6 in the supplemental material). We quantified the intensity of the DinB bands obtained via nonspecific binding and compared it to the intensities of the DinB bands in reaction mixtures containing the same concentrations of DinB or DinB(C66A) and the bait protein (RecA-His). In doing so, we found that approximately 80% of DinB and 40% of DinB(C66A) may have bound nonspecifically to the resin. Despite numerous attempts, it was not possible to reduce nonspecific interaction with the resin. Therefore, Fig. 5D depicts only the fraction of binding that is RecA-His specific. The adjusted data indicate that the ability of DinB(C66A) to bind to RecA is greater than that of DinB, as more DinB(C66A) was copurified by specific interaction with RecA than the wild-type enzyme (Fig. 5D). Quantification of the intact binary complex (70 kDa) is shown in Fig. 5E. The DinB(C66A)-RecA binary complex increased at higher molar ratios of DinB(C66A) (Fig. 5E) and appeared to be resistant to denaturing electrophoretic conditions. In contrast, the intact binary complex was not detected for DinB (Fig. 5C and E), indicating that it is sensitive to the electrophoresis denaturing conditions. These data suggest that DinB(C66A) has a higher binding affinity for RecA than DinB (Fig. 5C and E). We can also infer that this DinB interface is important for the binding of RecA.
We then assessed the abilities of DinB and DinB(C66A) to form ternary complexes using this in vitro pulldown assay (Fig. 5F). Again, RecA-His was used at a constant concentration per reaction (0.3 μM). DinB and its derivative, DinB(C66A), were added at increasing concentrations, as was pure UmuD. Since a dimer of full-length UmuD is required for ternary complex formation, the stoichiometric ratio of UmuD to DinB and RecA is 2:1:1, respectively (22). The molar ratios used per reaction are again shown above each panel in Fig. 5F. As observed in the DinB-RecA binary complex pulldown assay, both DinB and DinB(C66A) bound to RecA-His and were detected in the 40-kDa range by SDS-PAGE. The 70-kDa band corresponding to the DinB-RecA binary complex was observed here, as well, but only in reaction mixtures containing DinB(C66A) (Fig. 5F, right). UmuD also copurified with both DinB and DinB(C66A), but the patterns of copurification were strikingly different for the two proteins. DinB copurified with UmuD, which was solely observed as a free monomer of approximately 15 kDa (Fig. 5F, left; quantification is shown in Fig. 5G). DinB(C66A) copurified UmuD as a monomer and as a stable dimer of 30 kDa (Fig. 5F, right; quantifications are shown in Fig. 5G and H). Furthermore, DinB(C66A) allowed the copurification of UmuD monomer even at the lowest molar ratios of protein, an ability not seen for DinB (Fig. 5F and G). The ability to copurify UmuD was thus enhanced for DinB(C66A), but only in the presence of RecA.
These results indicate that DinB(C66A) has an increased ability to bind both MPC proteins. Most notably, the enhanced ability of DinB(C66A) to bind UmuD was dependent on RecA (Fig. 5A and B versus F and G).
DinB(C66A) is proficient for translesion synthesis in vitro.
We have provided evidence indicating that the in vivo translesion activity of DinB(C66A) is not altered compared to DinB (Fig. 2). We sought to confirm this in vitro, for which we used the purified proteins and a lesion-containing template in a fluorescence-based extension assay. We examined the extension of the fluorescently labeled primer, using either an undamaged oligonucleotide (Fig. 6A, top) or a lesion-containing oligonucleotide (Fig. 6A, bottom) as the template. The templates were identical in sequence but contained either dA (−) or 3-deaza-3-methyl-adenine (L), a DinB cognate lesion, at the primer-template junction (Fig. 6A). The abilities of wild-type DinB, DinB(C66A), the catalytically inactive derivative DinB(D103N), and DNA Pol I to synthesize DNA using either a lesion-containing or an undamaged template were thus compared.
Fig 6.
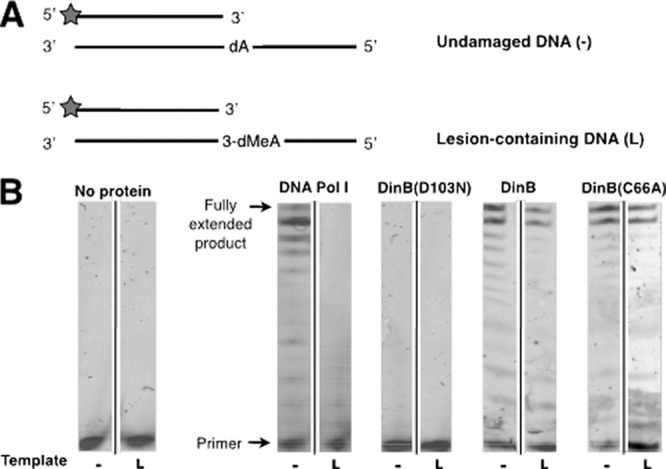
DinB(C66A) both synthesizes DNA and performs TLS of 3-deaza-3-methyl-adenine in vitro. (A) Schematic of undamaged substrate (−) and lesion-containing substrate (L) used for the extension assay. A common primer containing a 5′ hexachlorofluorescein label (HEX label; indicated by the gray stars) was annealed to either an undamaged control template (top) or a template containing 3-deaza-3-methyl-adenine (3-dMeA), a stable analog of the DinB cognate lesion generated by treatment with MMS. These substrates require insertion of a nucleotide directly opposite either dA (undamaged template) or 3-dMeA (lesion-containing template). (B) Standing-start extension of reaction mixtures containing 25 nM either undamaged control substrate (−) or the lesion-containing substrate (L) and 0.5 mM dNTP mixture. Reaction mixtures contained 1.25 μM DinB, DinB(C66A), or DinB(D103N) or 2 × 10−3 units of DNA Pol I.
As shown in Fig. 6B, DNA Pol I was able to extend the primer annealed to undamaged template (−) but unable to insert nucleotides when given the lesion-containing template (3-deaza-3-methyl-adenine) (L). The catalytically inactive derivative DinB(D103N) was not able to extend either substrate. Furthermore, wild-type DinB and the DinB(C66A) derivative generated comparable levels of fully extended product using undamaged and damaged templates (Fig. 6B). Therefore, we have shown that DinB(C66A) has DNA polymerase and TLS activities similar to those of DinB in vitro.
DinB(C66A) exhibits increased fidelity in the TLS of DinB cognate lesions in vivo.
In vitro analyses suggested that, contrary to our initial hypothesis, the DinB(C66A) variant copurifies readily with other MPC components compared to DinB. We thus sought to assess the effect of this single amino acid substitution in vivo. We hypothesized that DinB(C66A) would be preferentially found in an MPC in vivo. Since the MPC has been shown to decrease DinB-dependent frameshift events, strains expressing this derivative should exhibit accurate TLS of DinB cognate lesions (22). We constructed a strain in which dinB(C66A) is expressed from the native dinB promoter by recombining this allele on the chromosome.
We first examined whether there was increased sensitivity of the dinB(C66A) strain to MMS and NFZ. As shown in Fig. 7A, strains that expressed both dinB+ and dinB(C66A) survived well in the presence of both DNA-damaging agents. Strains expressing the dinB(C66A) chromosomal allele did not show altered in vivo TLS activities (Fig. 7A), consistent with what we had found previously with strains expressing the plasmid-borne allele (Fig. 2) and in agreement with what we had described in vitro (Fig. 6).
Fig 7.
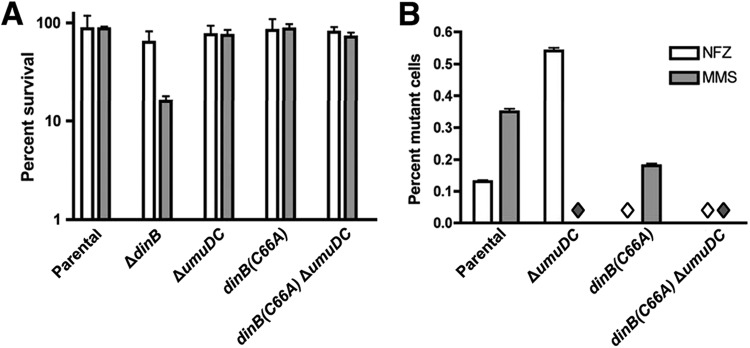
A dinB(C66A) strain is proficient for lesion bypass in vivo and gives rise to fewer DNA damage-induced mutants. (A) Percent survival of parental, ΔdinB, ΔumuDC, dinB(C66A), and dinB(C66A) ΔumuDC cells on LB medium containing 1.5 μg ml−1 NFZ or 7.5 mM MMS, relative to untreated cultures. Expression of the chromosomal dinB(C66A) allele does not result in increased sensitivity to NFZ or MMS compared to the parental dinB+ strain. The bars represent means and 1 SD of three replicate experiments. (B) Frequencies of NFZ- or MMS-induced mutagenesis in parental, ΔumuDC, dinB(C66A), and dinB(C66A) ΔumuDC strains. A mutagenesis assay was carried out as described by Benson et al. (9) at the concentrations of NFZ and MMS indicated in Materials and Methods. The bars represent means and 1 SD. The diamonds indicate that no mutants were detected. The numbers of colonies examined were 752 (NFZ) and 568 (MMS) for the parental strain, 744 (NFZ) and 643 (MMS) for the ΔumuDC strain, 617 (NFZ) and 562 (MMS) for the dinB(C66A) strain, and 789 (NFZ) and 572 (MMS) for the dinB(C66A) ΔumuDC strain. The mutational frequency was determined by a minimum of two independent experiments for each strain.
We then sought to quantify mutagenesis in both the parental and dinB(C66A) strains. To this end, we employed a methodology previously described by Benson et al. (9), in which mutants are identified as colonies that grow on rich medium but have lost the ability to grow on minimal medium. This allowed us to detect any loss-of-function mutations in a large variety of genes (i.e., a mutational target larger than 17 kb), including those for amino acid, vitamin, or nucleotide biosynthesis or carbon source uptake and utilization (9). In agreement with previous findings (9), we found modest levels of mutagenesis for the parental strain, with MMS treatment inducing more mutations than NFZ (Fig. 7B). Interestingly, the frequency of MMS- or NFZ-induced mutagenesis is significantly, though modestly, reduced for dinB(C66A) to nearly one-half the levels of mutagenesis detected in cells of the parental strain (Fig. 7B). Remarkably, it appears that mutagenesis by dinB(C66A) requires the umuDC gene products, as no NFZ-induced mutants are detected in the dinB(C66A) ΔumuDC strain (Fig. 7B). This result suggests that, unlike the DinB-RecA binary complex, the DinB(C66A)-RecA binary complex or free DinB(C66A) is not mutagenic in vivo.
Moreover, no MMS-induced mutagenesis was detected in the absence of umuDC for either the dinB+ or the dinB(C66A) strain. This demonstrates that while both Pol IV and Pol V may bypass alkylation damage, there is an appreciable difference in the fidelity of bypass for each polymerase.
Expression of the dinB(C66A) derivative from the chromosome thus resulted in a detectable decrease in the level of NFZ- and MMS-induced mutagenesis relative to the isogenic parental strain. Furthermore, we can infer that the MPC is important for TLS of NFZ-induced lesions in vivo and that changes in the attributes of the MPC also impact mutagenesis that is dependent on other polymerases (e.g., DNA Pol V).
Expression of dinB(C66A) causes decreased UV-induced mutagenesis.
Since our evidence suggests that DinB(C66A) exhibits an increase in both ternary complex formation in vitro and fidelity for DinB cognate lesions in vivo, we wondered what might be the evolutionary advantage of the conservation of this cysteine residue. Why would cells conserve a cysteine when replacement with an alanine seemed to be beneficial? We hypothesized that a trade-off might exist: cells conserved the cysteine not only to retain full protein stability, but also to modulate the strength of MPC interactions and thereby retain the functionality of a key cellular process. This process would be dependent on the components of the MPC but would not necessarily be Pol IV-mediated TLS.
To identify what this key process might be, we examined the various functions of DinB's interacting proteins. We first assessed the functionality of UmuD, an accessory protein required for the functions of both Pol IV and Pol V. Upon autocleavage, which is facilitated by the RecA nucleoprotein filament (RecA*), UmuD becomes UmuD′ and subsequently interacts with UmuC to become DNA Pol V (1, 2). UV irradiation generates lesions that in E. coli are specifically bypassed by Pol V. Therefore, we examined both the sensitivity and the level of UV-induced mutagenesis of the dinB(C66A) strain. Increased ternary complex formation in the dinB(C66A) strain may sequester the umuD gene products, making the protein unavailable for the catalytic subunit of Pol V, UmuC, and thus limiting the number of active DNA Pol V complexes.
The sensitivities of the parental and dinB(C66A) strains to UV irradiation were first examined. The parental strain exhibited the least sensitivity, while a ΔumuDC strain was more sensitive to UV exposure (Fig. 8A). We found that the dinB(C66A) and dinB(C66A) ΔumuDC strains are as sensitive to UV irradiation as their respective isogenic parental strains (Fig. 8A). Expression of dinB(C66A) from the chromosome thus did not result in enhanced sensitivity to UV irradiation.
Fig 8.
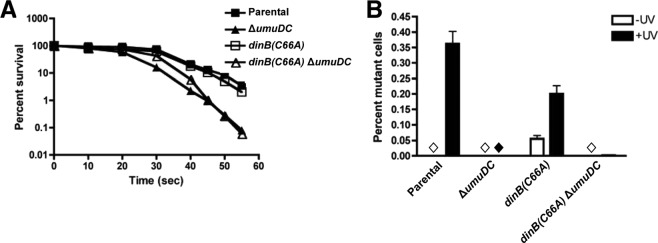
dinB(C66A) cells are not sensitive to UV irradiation but exhibit a decrease in UV-induced mutagenesis compared to isogenic dinB+. (A) Graph depicting survival of cells expressing dinB(C66A) from the chromosome compared to parental, ΔumuDC, and dinB(C66A) ΔumuDC strains. Means and 1 SD are shown. (B) Mutagenesis was measured as loss of function in a galactose mutation assay, as described in Materials and Methods. The dinB(C66A) allele is present on the chromosome and expressed from its native promoter. The bars represent means and 1 SD. The diamonds indicate no mutants were detected. The mutation frequency was assessed in three biological replicates. The total numbers of colonies examined were 7,190 (−UV) and 8038 (+UV) for the parental strain, 9,594 (−UV) and 34,770 (+UV) for the ΔumuDC strain, 9,012 (−UV) and 9,857 (+UV) for the dinB(C66A) strain, and 13,010 (−UV) and 37,729 (+UV) for the dinB(C66A) ΔumuDC strain.
ΔumuDC strains were deficient for mutagenesis, in agreement with the literature (Fig. 8B) (1–3, 44). Intriguingly, the mutation frequency for the dinB(C66A) strain was modestly reduced relative to the parental strain (0.2% ± 0.03% versus 0.4% ± 0.04%, respectively) (Fig. 8B). While the decrease in UV-induced mutagenesis is moderate, there is an observable deviation from the parental strain in cells expressing dinB(C66A) from the chromosome. We also observed an increase in the frequency of spontaneous mutagenesis in the dinB(C66A) strain, but we do not know whether this difference is physiologically relevant.
Cells expressing dinB(C66A) exhibit an increased ability to undergo homologous recombination.
We then examined the ability of the dinB(C66A) strain to undergo homologous recombination, which is largely mediated by RecA but is affected by the activities of TLS polymerases (15, 45, 46). To this end, we employed a P1 transduction assay, which has previously been shown to be effective in quantification of recombination events (47). In this assay, the number of transductants directly correlates with the number of recombination events. A modest level of recombination is detected in the parental strain (Fig. 9), while a larger number of transductants are obtained in the isogenic strain with a deletion of the umuDC operon. This agrees with previous reports, which suggest that Pol V inhibits RecA-mediated recombination by disassembling RecA* (15, 45). Interestingly, an increase in the number of recombination events was similarly observed for the dinB(C66A) strain (Fig. 9). Notably, the enhancement seems to be dependent on the umuDC gene products, since the dinB(C66A) ΔumuDC strain no longer shows enhanced recombination compared to the parental strain (Fig. 9). Thus, unlike the full DinB(C66A) MPC, the binary RecA-DinB(C66A) complex or free DinB(C66A) has no effect on the recombination functions within the cell.
Fig 9.
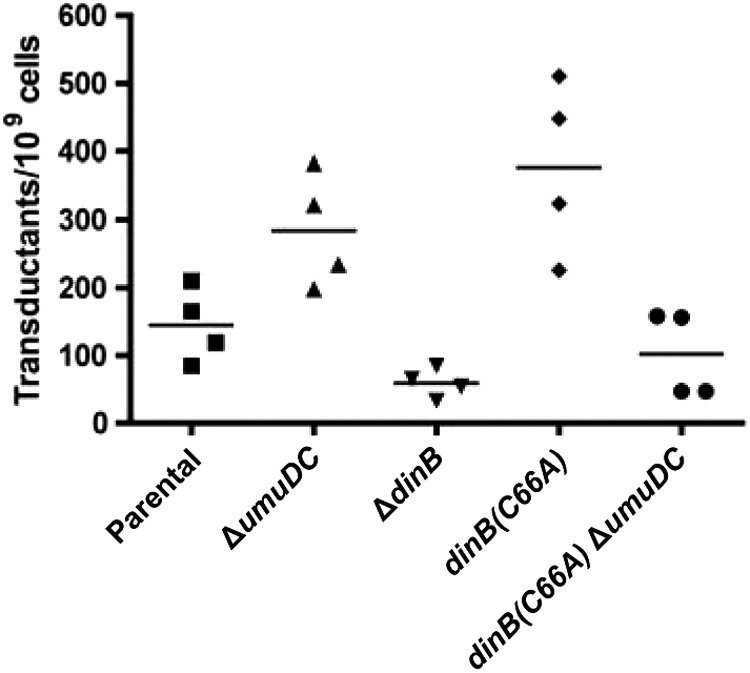
The dinB(C66A) strain has an elevated frequency of homologous recombination. The dinB(C66A) allele is present on the chromosome and is expressed from its native promoter. Cells were transduced with ΔsrlD::kan P1 virulent phage at an MOI of 1. In this assay, the number of transductants is directly correlated with the number of recombination events. Transductants were identified as kanamycin-resistant (Kanr) colonies. The number of transductants was normalized to the total number of cells used per transduction. Horizontal lines indicate the mean for each sample.
We sought to validate our results by performing an independent measurement of recombination proficiency via a conjugation assay. Hfr conjugation generates single-stranded DNA and also initiates RecA-mediated homologous recombination (48, 49). As shown in Table 3, the greatest number of transconjugants was obtained for the dinB(C66A) strain. These cells exhibit a 3-fold increase in the efficiency of Hfr-initiated homologous recombination relative to the parental strain (2.73 × 106 ± 3.93 × 105 transconjugants ml−1 versus 9.00 × 105 ± 8.49 × 104 transconjugants ml−1, respectively). The increase in homologous recombination is modest, however, once umuDC is deleted from cells expressing DinB(C66A) (1.13 × 106 ± 4.20 × 105 transconjugants ml−1). Consistent with the results obtained by the P1 transduction assay, these data suggest that an increase in the frequency of homologous recombination results from expression of dinB(C66A) from the chromosomal allele; however, this increase in recombinogenicity is dependent upon DinB(C66A) ternary complex formation.
Table 3.
Expression of DinB(C66A) enhances the frequency of conjugal recombination
| Strain | No. of transconjugants (CFU/ml) |
|---|---|
| Parental strain | None |
| Parental strain + Hfr | 9.00 × 105 ± 8.49 × 104 |
| ΔdinB + Hfr | 1.41 × 106 ± 4.83 × 105 |
| ΔumuDC + Hfr | 2.25 × 106 ± 2.57 × 105 |
| dinB(C66A) + Hfr | 2.73 × 106 ± 3.93 × 105 |
| dinB(C66A) ΔumuDC + Hfr | 1.13 × 106 ± 4.20 × 105 |
DISCUSSION
DinB is a highly evolutionarily conserved Y-family DNA polymerase present in all domains of life (7, 22). Here, we identified extensively conserved residues C66 and P67, which are present only in DinB-like proteins and are localized to a single binding surface on E. coli DinB. The absence of C66, P67, and F172, previously shown to interact with UmuD (22), in UmuC-like proteins suggests that the gene products of recA (RecA) and umuD (UmuD and UmuD′) bind competitively to unique residues/motifs (50, 51), which are specific to each Y-family DNA polymerase. This is consistent with the diverse cellular functions of Y-family DNA polymerases, despite their similar structures (7). Thus, protein-protein interaction appears to be a critical mechanism by which highly conserved TLS polymerases are regulated in various organisms (22, 52).
To investigate the roles of these residues in the regulation of DinB, we constructed the novel derivative dinB(C66A). The data suggest that both the catalytic and TLS abilities of the dinB(C66A) allele are not impaired in vivo; there is proficient bypass of MMS- and NFZ-induced lesions (Fig. 2 and 7A), as has been observed with DinB (9, 10, 14). In vitro extension assays show for the first time that E. coli DinB and the novel DinB(C66A) derivative are proficient for the bypass of 3-deaza-3-methyl-adenine in vitro (Fig. 6). Together, these data confirm that this single amino acid substitution does not affect the catalytic or TLS ability of DinB.
It has been postulated that expression of DinB(D103N) results in extreme sensitivity to NFZ and MMS, because the catalytically inactive protein may enhance or limit a number of protein-protein interactions; while some of these interactions have been investigated, the precise mechanism has not yet been elucidated (29, 53). The increased survival of DinB(C66A D103N) to levels that are typical of ΔdinB strains may be indicative of a deviation from the normal repertoire of protein-protein interactions and suggests the altered interactions are the direct result of the C66A substitution.
The affinity of DinB(C66A) for its interacting partners appears to be greater than that of DinB. Purification of DinB(C66A) not only results in the copurification of more RecA and UmuD, but also results in the retrieval of more putatively intact ternary complex (Fig. 3), as shown by IMAC and CEC (Fig. 3A and B). It is indeed impressive that both DinB and DinB(C66A) copurify with the other MPC proteins, since RecA and UmuD are not equally overproduced. Taking these results together, we have provided evidence for the existence of the MPC in vivo and the isolation of the complex directly from cells and have demonstrated that the purification of the MPC can be enhanced by the introduction of a single amino acid substitution.
We noted in many cases the persistence of stable binary and ternary protein complexes (Fig. 3, 4, and 5). Indeed, the MPC proteins remained bound in the complex when examined by SDS-PAGE and appeared to be undisturbed by heat, detergent, and the reducing agent β-mercaptoethanol. This is a well-documented phenomenon: kinetically stable proteins are described as requiring an unusually high energy input to dissociate and unfold (54). Though we have not determined the kinetic binding properties of DinB(C66A), the lack of MPC disruption by reducing conditions strongly suggests that DinB(C66A) has increased binding affinity for RecA relative to wild-type DinB (Fig. 3, 4, and 5). This could either be because the interaction at the C66-containing interface has been strengthened or because subtle changes in structure caused by the change from C to A result in enhancement of the interacting protein binding somewhere else on DinB.
By studying this site-specific mutant, we have begun to elucidate the binding order of MPC proteins in vivo. When the DinB proteins were purified from ΔdinB ΔumuDC cells to test whether the binary RecA-DinB complex could be detected, we found that there was enhanced RecA copurification for both DinB and DinB(C66A) compared to the proteins copurified from ΔdinB cells (Fig. 4B versus 3B). The ability of DinB and DinB(C66A) to form DinB-RecA binary complexes is thus not limited in the absence of UmuD, but is improved. These complexes may be further stabilized by the binding of UmuD. This may explain the intermediate level of catalytic activity detected in vitro for DinB-RecA complexes on undamaged DNA (22). When DinB proteins were purified from cells lacking recA, however, the copurification of UmuD was reduced for both DinB and DinB(C66A), and no larger complexes were detected for DinB(C66A). The binding of UmuD may therefore depend on RecA in vivo (Fig. 4). Thus, the C66-containing binding surface appears to be critical to modulate interaction with UmuD, and particularly with RecA.
Using controlled amounts of purified proteins, we also demonstrated that DinB(C66A) copurified more efficiently with RecA and UmuD in vitro and, interestingly, that UmuD copurification depends on formation of the binary DinB-RecA complex. It is possible that when bound, RecA stabilizes DinB(C66A) and/or induces a conformational change, which in turn allows increased ternary complex formation (Fig. 5). These data are consistent with the existence of a binding order: binding of RecA precedes that of UmuD in vivo. It is possible that DinB associates with RecA most of the time in vivo; the intracellular concentration of RecA is high enough (∼1 μM) (22) to permit this without limiting its functions.
Increased ternary complex formation in cells expressing DinB(C66A) presumably produces a greater number of DinB MPCs in vivo, resulting in higher TLS fidelity than in cells expressing the wild-type enzyme upon treatment with reagents that cause DinB cognate lesions. The decrease in NFZ- and MMS-induced mutagenesis detected for the dinB(C66A) strain (Fig. 7B) agrees with this hypothesis. Interestingly, dinB+ cells exhibit a higher frequency of mutagenesis in the absence of UmuD, indicating that this accessory protein is required to limit NFZ-induced mutagenesis (Fig. 7B). In contrast, we did not detect dinB(C66A)-dependent NFZ-induced mutagenesis in the absence of UmuD. This indicates that the DinB(C66A)-RecA binary complex is less mutagenic than DinB-RecA. We also noted that MMS-induced mutagenesis was largely dependent on umuDC in both dinB+ and dinB(C66A) cells (Fig. 7B). This suggests that while both Pol IV and Pol V may perform TLS of alkylation lesions, the majority of mutagenesis appears to be Pol V dependent (13). Therefore, the decrease in MMS-induced mutagenesis observed for dinB(C66A) cells may be due to the increased activity of Pol IV and the reduced activity of Pol V.
We also examined the role of DinB(C66A) in homologous recombination, which is largely mediated by RecA but is influenced by both Pol IV and Pol V in E. coli. We observed a detectable and consistent increase in the frequency of homologous recombination for DinB(C66A) (Fig. 9 and Table 3). It is possible that DinB(C66A) may sequester free UmuD from the cellular pool, which would mimic a deletion of the umuDC operon, previously shown to enhance homologous recombination (15, 45, 55, 56). Thus, if increased MPC formation mediated by DinB(C66A) effectively decreases the concentration of free UmuD, then the increase in homologous recombination is consistent with previous findings (15, 45, 47). Another possibility is that the increased concentration of DinB MPCs directly enhances recombination. It has been shown that DinB plays an active role in recombination by extending the 3′ OH of the invading strand; deletion of dinB in E. coli was shown to decrease the frequency of homologous recombination (46). Therefore, since DinB(C66A) seemingly results in increased ternary complex formation, this will ultimately result in an increase in the number of successful recombination events. The increase in homologous recombination detected for DinB(C66A) requires UmuD (Fig. 9), again suggesting an active role for the DinB(C66A) MPC in promoting homologous recombination.
Our model proposes that conservation of C66, and of other residues at this surface, is critical to maintain a balance between the activities of Pol IV and Pol V (Fig. 10). Both the DinB MPC and Pol V share a version of the accessory subunit, UmuD. While a dimer of the full-length form of UmuD is preferentially bound by the DinB MPC (22), a dimer of the processed form is bound by UmuC to form active Pol V complexes (3, 23, 57). UmuD, like the SOS gene network repressor LexA, undergoes autocleavage to remove the first 24 amino acids and form UmuD′ (1–3). Thus, if DinB(C66A) binds tightly to RecA and to full-length UmuD, then limited amounts of DNA Pol V would exist. Retention of a cysteine in this position of DinB is important to retain the functionality of both Pol IV and Pol V. This would ultimately control the level of mutagenesis in the cell, depending on the extent of DNA damage and the types of lesions present. Given the extensive conservation of DinB and the pivotal role Y-family polymerases play in a variety of processes, it is critical to understand the mechanisms by which cells regulate the potentially mutagenic activities of these enzymes.
Fig 10.
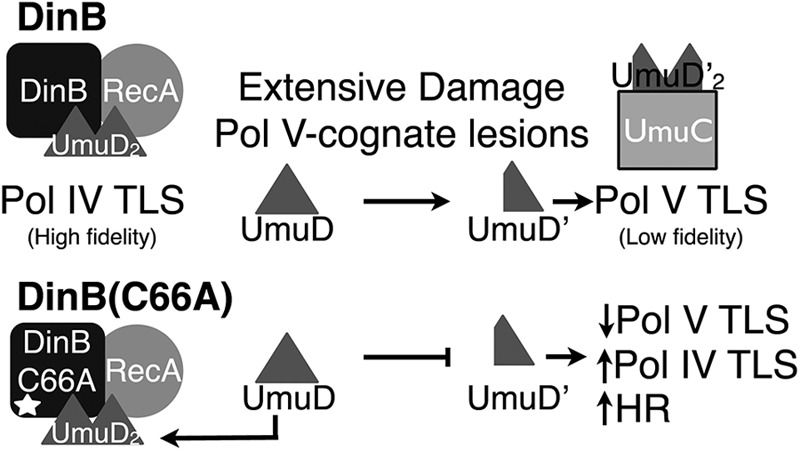
Model for DinB(C66A) activity in vivo. In dinB+ cells, Pol IV MPCs form relatively early in the SOS response or may be preexisting. However, as DNA damage becomes extensive or as Pol V cognate lesions are introduced, greater amounts of active Pol V are formed. The formation of Pol V requires the sequestration and cleavage of the accessory protein UmuD, forming UmuD′ and thereby reducing the activity of Pol IV complexes by direct competition for UmuD. In dinB(C66A) cells, Pol IV complexes form more frequently, as the MPC components exhibit an increased binding affinity for one another. The sequestration of UmuD to form Pol V mutasomes is consequently limited, as full-length UmuD remains bound to DinB(C66A) and RecA. This increases the activity of Pol IV, resulting in higher fidelity in the TLS of DinB cognate lesions, increased DinB-mediated extension to enhance HR, and limited TLS by Pol V complexes.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Woodgate for his gift of the parental and ΔumuDC strains, DE192 and RW86. Additionally, we thank A. E. MacGuire for careful reading of the manuscript.
This work was funded by the 1RO1GM088230-01A1 award from NIGMS.
We declare no conflict of interest.
Footnotes
Published ahead of print 4 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01349-12.
REFERENCES
- 1. Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis, 2nd ed ASM Press, Washington, DC [Google Scholar]
- 2. Sutton MD, Smith BT, Godoy VG, Walker GC. 2000. The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 34:479–497 [DOI] [PubMed] [Google Scholar]
- 3. Goodman MF. 2002. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 71:17–50 [DOI] [PubMed] [Google Scholar]
- 4. Jarosz DF, Cohen SE, Delaney JC, Essigmann JM, Walker GC. 2009. A DinB variant reveals diverse physiological consequences of incomplete TLS extension by a Y-family DNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 106:21137–21142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang W. 2003. Damage repair DNA polymerases Y. Curr. Opin. Struct. Biol. 13:23–30 [DOI] [PubMed] [Google Scholar]
- 6. Qiu Z, Goodman MF. 1997. The Escherichia coli polB locus is identical to dinA, the structural gene for DNA polymerase II. Characterization of Pol II purified from a polB mutant. J. Biol. Chem. 272:8611–8617 [DOI] [PubMed] [Google Scholar]
- 7. Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. 2001. The Y-family of DNA polymerases. Mol. Cell 8:7–8 [DOI] [PubMed] [Google Scholar]
- 8. Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. 2001. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol. Genet. Genomics 266:207–215 [DOI] [PubMed] [Google Scholar]
- 9. Benson RW, Norton MD, Lin I, Du Comb WS, Godoy VG. 2011. An active site aromatic triad in Escherichia coli DNA Pol IV coordinates cell survival and mutagenesis in different DNA damaging agents. PLoS One 6:e19944 doi:10.1371/journal.pone.0019944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. 2006. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 439:225–228 [DOI] [PubMed] [Google Scholar]
- 11. Jarosz DF, Godoy VG, Walker GC. 2007. Proficient and accurate bypass of persistent DNA lesions by DinB DNA polymerases. Cell Cycle 6:817–822 [DOI] [PubMed] [Google Scholar]
- 12. Yuan B, Cao H, Jiang Y, Hong H, Wang Y. 2008. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 105:8679–8684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bjedov I, Dasgupta CN, Slade D, Le Blastier S, Selva M, Matic I. 2007. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics 176:1431–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, Woodgate R. 2008. Eukaryotic Y-family polymerases bypass a 3-methyl-2′-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 36:2152–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sommer S, Bailone A, Devoret R. 1993. The appearance of the UmuD′C protein complex in Escherichia coli switches repair from homologous recombination to SOS mutagenesis. Mol. Microbiol. 10:963–971 [DOI] [PubMed] [Google Scholar]
- 16. Woodgate R, Ennis DG. 1991. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol. Gen. Genet. 229:10–16 [DOI] [PubMed] [Google Scholar]
- 17. Jarosz DF, Beuning PJ, Cohen SE, Walker GC. 2007. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 15:70–77 [DOI] [PubMed] [Google Scholar]
- 18. Tang M, Shen X, Frank EG, O'Donnell M, Woodgate R, Goodman MF. 1999. UmuD′(2)C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl. Acad. Sci. U. S. A. 96:8919–8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rajagopalan M, Lu C, Woodgate R, O'Donnell M, Goodman MF, Echols H. 1992. Activity of the purified mutagenesis proteins UmuC, UmuD′, and RecA in replicative bypass of an abasic DNA lesion by DNA polymerase III. Proc. Natl. Acad. Sci. U. S. A. 89:10777–10781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reuven NB, Arad G, Maor-Shoshani A, Livneh Z. 1999. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 274:31763–31766 [DOI] [PubMed] [Google Scholar]
- 21. Woodgate R, Rajagopalan M, Lu C, Echols H. 1989. UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc. Natl. Acad. Sci. U. S. A. 86:7301–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Godoy VG, Jarosz DF, Simon SM, Abyzov A, Ilyin V, Walker GC. 2007. UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol. Cell 28:1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schlacher K, Leslie K, Wyman C, Woodgate R, Cox MM, Goodman MF. 2005. DNA polymerase V and RecA protein, a minimal mutasome. Mol. Cell 17:561–572 [DOI] [PubMed] [Google Scholar]
- 24. Kohanski MA, DePristo MA, Collins JJ. 2010. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 37:311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pillaire MJ, Selves J, Gordien K, Gourraud PA, Gentil C, Danjoux M, Do C, Negre V, Bieth A, Guimbaud R, Trouche D, Pasero P, Mechali M, Hoffmann JS, Cazaux C. 2010. A ‘DNA replication’ signature of progression and negative outcome in colorectal cancer. Oncogene 29:876–887 [DOI] [PubMed] [Google Scholar]
- 26. Wang H, Wu W, Wang HW, Wang S, Chen Y, Zhang X, Yang J, Zhao S, Ding HF, Lu D. 2010. Analysis of specialized DNA polymerases expression in human gliomas: association with prognostic significance. Neuro Oncol. 12:679–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou J, Zhang S, Xie L, Liu P, Xie F, Wu J, Cao J, Ding WQ. 2012. Overexpression of DNA polymerase iota (Poliota) in esophageal squamous cell carcinoma. Cancer Sci. 103:1574–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim SR, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, Nohmi T, Ohmori H. 1997. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc. Natl. Acad. Sci. U. S. A. 94:13792–13797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RP, Nohmi T. 1999. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell 4:281–286 [DOI] [PubMed] [Google Scholar]
- 30. Ennis DG, Fisher B, Edmiston S, Mount DW. 1985. Dual role for Escherichia coli RecA protein in SOS mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 82:3325–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woodgate R. 1992. Construction of a umuDC operon substitution mutation in Escherichia coli. Mutat. Res. 281:221–225 [DOI] [PubMed] [Google Scholar]
- 32. Benson RW, Cafarelli TM, Godoy VG. 2011. SOE-LRed: a simple and time-efficient method to localize genes with point mutations onto the Escherichia coli chromosome. J. Microbiol. Methods 84:479–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nguyen LH, Jensen DB, Burgess RR. 1993. Overproduction and purification of sigma 32, the Escherichia coli heat shock transcription factor. Protein Expr. Purif. 4:425–433 [DOI] [PubMed] [Google Scholar]
- 34. Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41:207–234 [DOI] [PubMed] [Google Scholar]
- 35. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 2001. Current protocols in molecular biology. J. Wiley, New York, NY [Google Scholar]
- 36. Tang WJ. 1993. Blot-affinity purification of antibodies. Methods Cell Biol. 37:95–104 [DOI] [PubMed] [Google Scholar]
- 37. Sreerama N, Woody RW. 2004. Computation and analysis of protein circular dichroism spectra. Methods Enzymol. 383:318–351 [DOI] [PubMed] [Google Scholar]
- 38. Lenski RE. 2010, posting date TA agar plates. Michigan State University, East Lansing, MI: http://myxo.css.msu.edu/ecoli/taagar.html [Google Scholar]
- 39. Maloy SR, Cronan JE, Freifelder D. 1994. Microbial genetics. Jones and Bartlett, Boston, MA [Google Scholar]
- 40. Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 41. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. doi:10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol. Rev. 53:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Niebuhr K, Wehland J. 1997. Screening of antibody epitopes and regions of protein-protein interaction sites using SPOT peptides, p 797–800 In Lefkovits I. (ed), Immunology methods manual: the comprehensive sourcebook of techniques. Academic Press, San Diego, CA [Google Scholar]
- 44. Kato T, Shinoura Y. 1977. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol. Gen. Genet. 156:121–131 [DOI] [PubMed] [Google Scholar]
- 45. Boudsocq F, Campbell M, Devoret R, Bailone A. 1997. Quantitation of the inhibition of Hfr x F- recombination by the mutagenesis complex UmuD′C. J. Mol. Biol. 270:201–211 [DOI] [PubMed] [Google Scholar]
- 46. Lovett ST. 2006. Replication arrest-stimulated recombination: dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair 5:1421–1427 [DOI] [PubMed] [Google Scholar]
- 47. Hawver LA, Gillooly CA, Beuning PJ. 2011. Characterization of Escherichia coli UmuC active-site loops identifies variants that confer UV hypersensitivity. J. Bacteriol. 193:5400–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lloyd RG, Buckman C. 1995. Conjugational recombination in Escherichia coli: genetic analysis of recombinant formation in Hfr x F- crosses. Genetics 139:1123–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith GR, Amundsen SK, Dabert P, Taylor AF. 1995. The initiation and control of homologous recombination in Escherichia coli. Philos. Trans. R. Soc. Lond. B Biol. Sci. 347:13–20 [DOI] [PubMed] [Google Scholar]
- 50. Jonczyk P, Nowicka A. 1996. Specific in vivo protein-protein interactions between Escherichia coli SOS mutagenesis proteins. J. Bacteriol. 178:2580–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koch WH, Ennis DG, Levine AS, Woodgate R. 1992. Escherichia coli umuDC mutants: DNA sequence alterations and UmuD cleavage. Mol. Gen. Genet. 233:443–448 [DOI] [PubMed] [Google Scholar]
- 52. Bi X, Barkley LR, Slater DM, Tateishi S, Yamaizumi M, Ohmori H, Vaziri C. 2006. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell. Biol. 26:3527–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Uchida K, Furukohri A, Shinozaki Y, Mori T, Ogawara D, Kanaya S, Nohmi T, Maki H, Akiyama M. 2008. Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol. Microbiol. 70:608–622 [DOI] [PubMed] [Google Scholar]
- 54. Manning M, Colon W. 2004. Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. Biochemistry 43:11248–11254 [DOI] [PubMed] [Google Scholar]
- 55. Pham P, Bertram JG, O'Donnell M, Woodgate R, Goodman MF. 2001. A model for SOS-lesion-targeted mutations in Escherichia coli. Nature 409:366–370 [DOI] [PubMed] [Google Scholar]
- 56. Rehrauer WM, Bruck I, Woodgate R, Goodman MF, Kowalczykowski SC. 1998. Modulation of RecA nucleoprotein function by the mutagenic UmuD′C protein complex. J. Biol. Chem. 273:32384–32387 [DOI] [PubMed] [Google Scholar]
- 57. Jiang Q, Karata K, Woodgate R, Cox MM, Goodman MF. 2009. The active form of DNA polymerase V is UmuD′(2)C-RecA-ATP. Nature 460:359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.