

Abstract
In the human-pathogenic bacterium Streptococcus pyogenes, the tagatose bisphosphate aldolase LacD.1 likely originated through a gene duplication event and was adapted to a role as a metabolic sensor for regulation of virulence gene transcription. Although LacD.1 retains enzymatic activity, its ancestral metabolic function resides in the LacD.2 aldolase, which is required for the catabolism of galactose. In this study, we compared these paralogous proteins to identify characteristics correlated with divergence and novel function. Surprisingly, despite the fact that these proteins have identical active sites and 82% similarity in amino acid sequence, LacD.1 was less efficient at cleaving both fructose and tagatose bisphosphates. Analysis of kinetic properties revealed that LacD.1's adaptation was associated with a decrease in kcat and an increase in Km. Construction and analysis of enzyme chimeras indicated that non-active-site residues previously associated with the variable activities of human aldolase isoenzymes modulated LacD.1's affinity for substrate. Mutant LacD.1 proteins engineered to have LacD.2-like levels of enzymatic efficiency lost the ability to function as regulators, suggesting that an alteration in efficiency was required for adaptation. In competition under growth conditions that mimic a deep-tissue environment, LacD.1 conferred a significant gain in fitness that was associated with its regulatory activity. Taken together, these data suggest that LacD.1's adaptation represents a form of neofunctionalization in which duplication facilitated the gain of regulatory function important for growth in tissue and pathogenesis.
INTRODUCTION
Adapted metabolic enzymes provide interesting models for examining the role of gene duplication in driving the process of protein evolution. Derived from metabolic enzymes, these proteins have been adapted by evolution to perform novel functions that are not directly associated with catalysis, often functioning as components of transcriptional regulatory complexes (1). Many of these enzymes are multifunctional, where the same protein entity participates in both catalysis and regulation (1, 2). However, others are divergent in function, participating in regulation while no longer retaining a clear metabolic role (3, 4). The members of the latter class are often the products of an apparent gene duplication event in which the ancestral protein is retained as a component of the original metabolic pathway. Since the duplicate is initially redundant, how it avoids the accumulation of deleterious mutations while undergoing adaptation is not well understood (5). However, the fact that adaptation often results in only minor alterations to the primary amino acid sequence and its three-dimensional structure suggests that careful comparison of the adapted enzyme and its ancestor can provide insight into the evolutionary forces driving adaptation. A challenge then becomes the identification of those specific amino acid residues whose divergence is associated with the acquisition of novel functionality.
In Streptococcus pyogenes (group A streptococcus), LacD.1 is an adapted metabolic enzyme implicated in the transcriptional regulation of several virulence factors, including the secreted SpeB cysteine protease (6). This Gram-positive bacterium is a human pathogen responsible for a large number of diseases ranging from largely superficial infection of the skin and mucous membranes (impetigo and pharyngitis) to highly invasive and life-threatening diseases (necrotizing fasciitis), as well as serious postinfection sequelae (rheumatic fever and glomerulonephritis) and immunopathological syndromes (pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection [PANDAS]) (reviewed in reference 7). Deletion of the gene encoding LacD.1 has been associated with derepression of transcription of virulence genes regulated by several different environmental cues, including pH, salt, and carbon source (8). Functionally, LacD.1 has the characteristics of a tagatose 1,6-bisphosphate (TBP) aldolase, a component of the tagatose 6-phosphate pathway used by Gram-positive bacteria to metabolize lactose and galactose (9). However, the ability to metabolize these carbohydrates in S. pyogenes resides not in the Lac.1 operon, of which LacD.1 is a member, but, rather, in the nearly identical Lac.2 operon (10). In fact, the Lac.1 operon is defective, lacking functional copies of the genes encoding several key enzymes, which instead reside in the operon as pseudogenes. While it cannot be ruled out that the Lac.2 operon was acquired following the adaptation of the LacD.1 gene, given the high degree of homology, and the highly conserved organization and genomic locations of these two operons, it is more likely that the Lac.1 operon arose as a duplication of the Lac.2 operon and, subsequently, the LacD.1 TPB aldolase was adapted for a regulatory role that specifically required the loss of function of other enzymes in the pathway. This hypothesis is supported by the fact that restoration of LacC.1 pseudogene function phenocopies deletion of LacD.1 and that this defective operon architecture is conserved in all S. pyogenes whole-genome sequences that are currently deposited in the GenBank database.
Interestingly, LacD.1 retains the enzymatic activity characteristic of an aldolase and has the ability to cleave TBP, and its isomer fructose 1,6-bisphosphate (FBP), into the glycolytic intermediates dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (GA3P) (6). However, its ability to participate in transcriptional regulation is independent of enzymatic activity, since mutation of a catalytic lysine residue (K204) abolishes the ability to cleave substrate but has no effect on its ability to regulate virulence factor expression. In contrast, mutation of key residues involved in binding substrate does lead to an abrogation of regulatory activity, suggesting that an ability to bind substrate is an essential component of LacD.1's adapted regulatory function (6).
Further support for the specific adaptation of LacD.1 to a regulatory role comes from the observation that LacD.2 cannot substitute for LacD.1 in regulation despite the fact the two aldolases are 82% similar. Comparison of all aldolases has revealed similarity in sequence, although they are distributed between 2 general classes that are distinguished by their mechanism of catalysis (class I and class II) (11). Unlike the metal-requiring class II aldolases found in most prokaryotes, LacD.1 and LacD.2 are class I enzymes that utilize a lysine to form a Schiff base intermediate. The crystal structures of LacD.2 in both its apo form and bound to DHAP and the apo form of LacD.1 have recently been determined, and they reveal strong similarity to the well-studied structure of rabbit muscle aldolase (RMA) (12, 13). All three enzymes share a conserved (α/β)8 fold, and the structures show that RMA and LacD.2 undergo similar conformational responses upon binding DHAP. In addition, the structures show that the LacD.1 and LacD.2 active sites share identical sequences. Taken together, these data suggest that LacD.1's adaptation to a regulatory function relied upon its original function as an aldolase and that adaptation involved alterations to residues outside the active site.
As a first step in understanding how these two similar proteins have been adapted to diverse functions, we sought to compare and contrast various properties of LacD.1 and LacD.2 in order to identify characteristics unique to each enzyme. We found that LacD.1's adaptation is associated with a measurable divergence in catalytic function and that this altered enzymatic activity is necessary for its adapted regulatory function. However, an altered enzymatic activity is not sufficient to account for LacD.1's divergence, as its regulatory function requires additional variant residues. LacD.1, but not LacD.2, was found to confer a significant competitive advantage under growth conditions that mimic a deep-tissue environment. Furthermore, this gain in fitness did not depend on its divergent catalytic properties but was specifically associated with its regulatory activity. Thus, these data provide insight into the molecular basis of the evolution of LacD.1's novel regulatory function.
MATERIALS AND METHODS
Strains, media, and growth conditions.
Molecular cloning experiments utilized Escherichia coli TOP10 (Invitrogen), and BL21(DE3) was used for expression of recombinant proteins. Experiments with S. pyogenes used strains HSC5 (14) and JL151 (6), a ΔLacD.1 derivative of HSC5 with an in-frame deletion in the LacD.1 gene (SPy_1704). Culture for all assays of function was at 37°C in C medium (15). In selected experiments, C medium was buffered by the addition of 1 M HEPES (pH 7.5) to a final concentration of 0.1 M. Routine culture of S. pyogenes and E. coli utilized Todd-Hewitt yeast extract medium under anaerobic conditions and Luria-Bertani medium aerobically, respectively. When appropriate, antibiotics were added to media at the following concentrations: chloramphenicol, 15 μg/ml for E. coli and 3 μg/ml for S. pyogenes, and kanamycin, 50 μg/ml for E. coli and 500 μg/ml for S. pyogenes.
Purification of enzymes.
Proteins were expressed in BL21(DE3) cells as described previously (6). Following expression, cells were collected by centrifugation and disrupted by sonication (6). The six-His-tagged proteins were purified by chromatography over a cobalt affinity resin according to the recommendations of the manufacturer (Clontech). Eluted proteins were then dialyzed against a buffer consisting of 50 mM NaPO4 (pH 7.5), 300 mM NaCl, and 25 mM β-mercaptoethanol (βME) and further purified by gel filtration chromatography using Superdex 200 10/300 GL (GE Healthcare). Fractions containing purified protein were identified by SDS-PAGE, pooled, and then dialyzed against a buffer consisting of 50 mM NaPO4 (pH 7.5), 300 mM NaCl, and 1 mM dithiothreitol (DTT) in a final volume of 25% glycerol and stored at −20°C. Protein concentrations were determined by absorbance at 280 nm of aliquots diluted into 6 M guanidine hydrochloride, and final values were calculated from the averages of three different dilutions. Calculation of protein extinction coefficients utilized ProtParam (http://web.expasy.org/protparam/) (16).
DNA and computational techniques.
Plasmid purification and transformation of E. coli and S. pyogenes were performed as described previously (17, 18). The genes encoding various LacD chimeric enzymes were constructed using the PCR products generated using 5′ phosphorylated primers for three-way ligations, or through a process of overlap extension PCR (19). Restriction enzymes and additional reagents were used according to the manufacturer's recommendations (New England BioLabs). Site-directed mutagenesis was performed by PCR followed by DpnI digestion of template using a commercial kit (QuikChange site-directed mutagenesis kit; Stratagene) and the primers and templates described in Table S1 in the supplemental material. The fidelity of all constructs was confirmed by determination of DNA sequences using the services of a commercial vendor (SeqWright, Houston, TX). Coordinates for protein structures were obtained from the Protein Data Bank (http://www.pdb.org) and were compared using the PyMOL molecular graphics system (version 1.5.0.4; Schrödinger, LLC).
Aldolase cleavage assay and determination of steady-state kinetic constants.
Aldolase activity was measured using a standard enzyme-coupled assay that monitored oxidation of β-NADH by monitoring the decrease in absorbance at 340 nm in the presence of the coupling enzymes glycerol-3-phosphate dehydrogenase (GDH) and triosephosphate isomerase (TIM) (20). Enzyme was diluted to 100 to 170 nM into a reaction mixture consisting of 50 mM HEPES (pH 7.5), 100 mM NaCl, 2.5 U of glycerol-3-phosphate dehydrogenase, 20 U of triosephosphate isomerase, and 0.25 mM β-NADH. The initial velocity (V0) was plotted at various concentrations of substrate ([S]) and fit to a standard equation (V0 = Vmax[S]/Km + [S]) to determine the Michaelis-Menten constant (Km) and the maximum velocity (Vmax). The kcat was calculated by dividing the Vmax by the enzyme concentration. For cleavage of FBP at pH 7.5, a substrate range of 0.2 to 8 mM was used for LacD.2 and 0.2 to 40 mM was used for LacD.1. For cleavage of TBP at pH 7.5 (supplied by Wolf-Dieter Fessner of Technische Universität, Darmstadt, Germany), a substrate range of 0.1 to 2 mM was used for LacD.2 and 0.5 to 16 mM was used for LacD.1. For cleavage of FBP at pH 6.8, a substrate range of 0.2 to 8 mM was used for all enzymes. For cleavage of TBP at pH 6.8, a substrate range of 0.04 to 4 mM was used for all enzymes.
Influence of pH on aldolase activity.
The aldolase cleavage assay was performed as described above in a reaction mixture buffered by 50 mM bis-Tris (pH range, 6.0 to 6.5) or by 50 mM HEPES (pH range, 6.8 to 8.0). The kcat/Km ratio was calculated and fit to the equation log Y = log{C/(1+[H]/Ka + Kb/[H] + [H]2/KaKb)} using Graphpad Prism (version 5; GraphPad Software, San Diego, CA) (where [H] is the hydrogen ion concentration, C is the pH independent value of kcat, Ka is the acid dissociation constant, and Kb is the base dissociation constant).
Determination of molecular weight.
Purified protein and molecular weight standards (GE Healthcare) were either dialyzed against or resuspended in a buffer of 50 mM NaPO4, 300 mM NaCl, and 25 mM βME. Aliquots ranging from 100 to 200 μl were then developed over a Superdex 200 HR 10/300 GL column with a flow rate of 0.5 ml/min. Elution of protein was monitored by absorbance at 280 nm. The value of the partition coefficient (Kav) was calculated for each standard using the equation Kav = (Ve − Vo)/(Vt − Vo), where Ve is the elution volume, Vo is the void volume (8.107 ml), and Vt is the bed volume (24 ml). For each standard, the Kav was plotted against the log Mr (molecular weight). The calculated molecular weight of LacD.1 and LacD.2 represents the average of at least three separate experiments.
Influence of aldolase concentration on activity.
To determine the enzymatic activities of LacD.1 and LacD.2, an aliquot of 5 μl containing enzyme at concentrations ranging from 100 nM to 5 mM was added to 95 μl of a mixture containing 50 mM HEPES (pH 7.5), 100 mM NaCl, 15 U of GDH, 600 U of TIM, 22.5 mM FBP, and 250 or 500 μM β-NADH. The measured rate of decrease in absorbance at 340 nm was used to calculate the activity at each concentration of enzyme. The slope of enzyme activity plotted versus enzyme concentration is equal to the specific activity of each aldolase and was comparable to the specific activity calculated from the kcat of each enzyme determined from steady-state experiments.
SpeB protease activity.
Protease activity was measured as described previously by spotting a 5-μl aliquot of an overnight bacterial culture from each strain under analysis onto the surface of solidified unmodified C medium or modified C medium supplemented with 2% milk (15). Following overnight culture under anaerobic conditions, protease activity was apparent as a zone of clearing surrounding the bacterial growth. Quantitative analysis of protease activity following overnight culture in liquid medium was performed using a fluorescein isothiocyanate (FITC)-casein substrate as described previously (6).
Determination of fitness.
Overnight cultures in C medium supplemented with chloramphenicol of bacterial strains were back-diluted 1:100 into fresh C medium with chloramphenicol and incubated for approximately 4 h. Culture densities were normalized by optical density at 600 nm (OD600), and cocultures were established by coinoculation of 50 μl of both test and competitor strains into 10 ml of C medium containing chloramphenicol. Following overnight incubation, each culture was back-diluted 1:1,000 in fresh medium, and this process was continued for 4 days. The competitor strain was ΔLacD.1 complemented with pLacD.1−kan, a plasmid identical to pLacD.1 except for removal of the kanamycin resistance cassette. The test strains were ΔLacD.1 complemented with the various plasmids indicated below. Cultures were grown in the presence of chloramphenicol, as both test and competitor plasmids carry identical chloramphenicol resistance cassettes. Growth rates were calculated for each individual strain in the absence of competition to confirm that these were identical. To enumerate CFU, serial dilutions from cultures were plated on Todd-Hewitt yeast extract media with and without kanamycin at the indicated time points. Data shown are the averages of at least 2 independent experiments.
RESULTS
LacD.1 and LacD.2 are structurally similar.
Recently, the crystal structures of LacD.1 and LacD.2 from S. pyogenes (PDB codes 3MYO and 3MHF, respectively) have both been solved (12, 13), but they have not been directly compared. To compare the tertiary structures of the aldolases, the algorithm FATCAT was utilized to allow for an alignment that takes flexibility into consideration (21). This comparison indicated that the two proteins were significantly similar (P < 0.001), with a root mean square deviation (RMSD) of 1.0 (Cα atoms) (Fig. 1A), and that the positions of active-site residues between the apo forms of the two enzymes are essentially identical (Fig. 1B).
Fig 1.
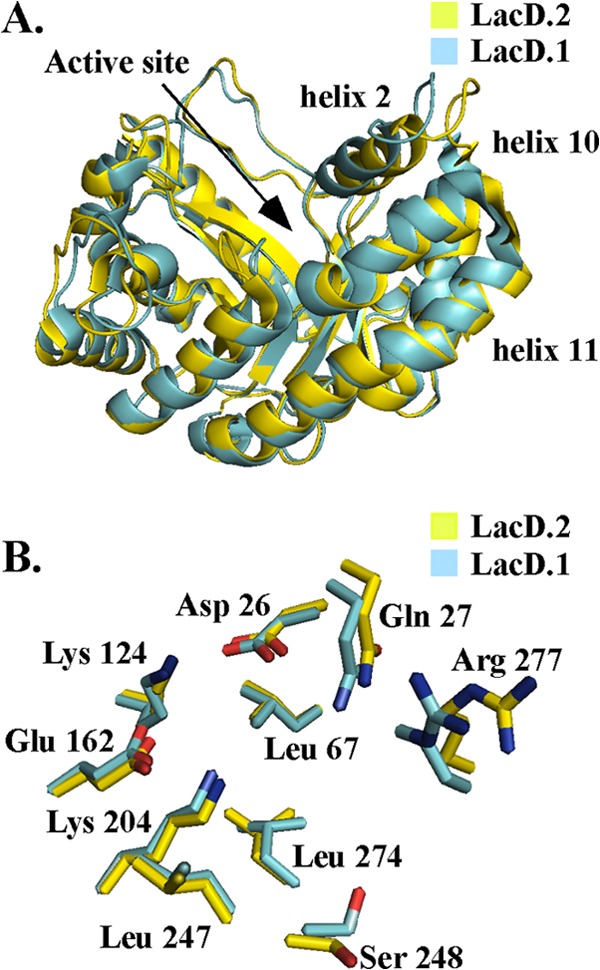
The crystal structures of LacD.1 (3MYO) and LacD.2 (3MHF) were compared. (A) Superposition of LacD.1's active site (cyan) and LacD.2's active site (yellow); active-site residues from LacD.1—D26, Q27, L67, K124, E162, K204, L247, S248, L274, and R277—are labeled. (B) Superposition of LacD.1 and LacD.2 crystal structures with thermal B-factors illustrated. The higher B-factors are associated with an increase in thickness.
LacD.1 has a higher Km for substrate than LacD.2.
Given that LacD.1 and LacD.2 share identical active-site residues and nearly identical three-dimensional structures, aldolase activities were directly compared to determine if LacD.1's divergent function was associated with any variation in enzyme efficiency. Activity was assessed using a standard enzyme-coupled assay linking cleavage of the phosphorylated sugar to oxidation of β-NADH by glycerol-3-phosphate dehydrogenase, which was measured spectroscopically. This analysis revealed that LacD.2 readily cleaved both TBP and FBP (Table 1) at rates similar to those reported previously (12) and similar to those with the class II TBP aldolase found in E. coli (22). When the S. pyogenes enzymes were compared, it was found that the kcat of LacD.1 was similar to that of LacD.2, although it was slightly lower for both substrates (approximately 1.5- to 2-fold) (Table 1). In contrast, Km for LacD.1 was approximately four times higher than for LacD.2, indicating a considerable decrease in affinity for both TBP and FBP (Table 1). Considering that the two enzymes share identical active sites, these data demonstrate that LacD.1's adaptation to a regulatory role is associated with a roughly 6- to 8-fold decrease in enzyme efficiency, as calculated by the kcat/Km ratio (Table 1). These data also suggest that variant non-active-site residues influence LacD.1's divergent properties.
Table 1.
Steady-state kinetic parameters of LacD chimeric enzymes for the cleavage reaction of TBP and FBPa
| Enzyme | FBP |
TBP |
||||
|---|---|---|---|---|---|---|
| kcat (s−1) | Km (μM) | kcat/Km (s−1 mM−1) | kcat (s−1) | Km(μM) | kcat/Km(s−1 mM−1) | |
| LacD.2 | 6.35 ± 0.19 | 498 ± 57 | 12.8 ± 0.1 | 24.3 ± 1.1 | 199 ± 28 | 122 ± 18 |
| LacD.1 | 4.41 ± 0.19 | 1,993 ± 144 | 2.1 ± 0.8 | 14.62 ± 1.3 | 920 ± 193 | 16 ± 3.6 |
| LacD.12A | 12.6 ± 0.6 | 293 ± 34 | 42.9 ± 5.4 | 18.3 ± 0.7 | 183 ± 19 | 100 ± 11 |
| LacD.12B | 5.20 ± 0.12 | 404 ± 39 | 12.8 ± 1.3 | 12.9 ± 0.38 | 298 ± 55 | 39 ± 7.6 |
| LacD.12C | 5.46 ± 0.11 | 684 ± 46 | 7.9 ± 0.8 | 15.1 ± 1.5 | 446 ± 112 | 33 ± 8.8 |
| LacD.12(L1) | 5.48 ± 0.18 | 864 ± 98 | 5.8 ± 0.4 | 10 ± 0.58 | 497 ± 88 | 20 ± 3.7 |
| LacD.12(T2) | 4.76 ± 0.19 | 907 ± 121 | 4.9 ± 0.3 | 12.6 ± 0.48 | 477 ± 55 | 26 ± 3.2 |
| LacD.12(L1+T2) | 5.56 ± 0.19 | 667 ± 84 | 8.4 ± 0.7 | 12.3 ± 0.39 | 343 ± 37 | 36 ± 4.0 |
| LacD.21G | 4.88 ± 0.11 | 538 ± 46 | 9.1 ± 0.8 | 18.5 ± 1.0 | 216 ± 31 | 86 ± 13 |
Kinetic parameters were determined using enzyme-coupled assay described in Materials and Methods. Enzyme assay solution conditions were 50 mM HEPES (pH 6.8) and 100 mM NaCl at 25°C. Values are means ± standard errors.
Reaction pH has similar influences on both LacD.1 and LacD.2.
The observation that pH is one signal that modulates LacD.1-dependent regulation (6) suggested that adaptation may have involved a modification to a pH-sensitive step associated with LacD.1's differential catalytic activity. To test this, steady-state kinetic parameters for FBP cleavage were determined over a range of pH values that reflect those encountered by S. pyogenes during infection. Comparison of enzyme efficiencies revealed a decrease for LacD.1 at every pH value tested (Fig. 2). However, the profiles for LacD.1 and LacD.2 resembled similar bell-shaped curves, with optimum enzyme efficiency occurring at pH 6.8 and efficiency decreasing at both more acidic and basic solution conditions (Fig. 2). Although two different buffer systems were required for the full range of pH values analyzed, measurement of activity of each buffer at their overlap (pH 6.8) produced similar results for both enzymes, indicating that the buffer composition had no measurable influence on catalysis (data not shown). Comparison of the apparent pKas and pKbs of the two enzymes revealed essentially identical values (Fig. 2). Thus, pH has similar influences on catalysis for both LacD.1 and LacD.2, suggesting that adaptation did not require a change in LacD.1's catalytic mode.
Fig 2.

The influence of pH on the enzyme efficiencies of both LacD.1 and LacD.2 was determined by measuring the Michaelis-Menten constants for cleavage of FBP at several different pHs in the range of 6.0 to 8.0. See Materials and Methods for details.
Catalysis is not influenced by changes in enzyme concentration.
Class I aldolases in higher eukaryotes typically function as homotetramers with an extremely low oligomerization constant (23), with quaternary structure linked to enzyme thermostability (24) and noncatalytic aldolase function (25). However, to determine if a change to oligomeric state is associated with altered enzymatic activity, LacD.1 and LacD.2 quaternary structures were compared. Analysis using size exclusion chromatography revealed that both proteins eluted as a single peak, with nearly identical elution volumes (Fig. 3). The calculated apparent molecular masses for LacD.1 and LacD.2 were 67 ± 3 and 71 ± 3 kDa, close to the expected molecular mass for a dimer (75 kDa for LacD.1 and 76 kDa for LacD.2) and consistent with the crystallization of each as homodimers (12, 13). To investigate if enzyme concentration influences catalysis, the FBP aldolase activities of both LacD.1 and LacD.2 were measured at six different concentrations ranging between 100 nM to 5 mM. Plots of enzyme activity as a function of enzyme concentration revealed a linear relationship for both LacD.1 and LacD.2 (Fig. 4), suggesting that the differential activity of LacD.1 is not the result of a concentration-dependent change in activity. Taken together, these data suggest that adaptation did not involve major changes to the quaternary structure of LacD.1.
Fig 3.

Gel filtration chromatography of LacD.1 and LacD.2. Purified LacD.1 and LacD.2 were subjected to gel filtration chromatography over Superdex 200 10/200 GL. Based on the elution of the standards (thyroglobin [669 kDa] [A], aldolase [158 kDa] [B], conalbumin [75 kDa] [C], ovalbumin [43 kDa] [D], and RNase A [13.7 kDa] [E]), the molecular masses of LacD.1 and LacD.2 were calculated to be 67 ± 3 and 71 ± 3 kDa, respectively.
Fig 4.

Influence of enzyme concentration on enzyme activity. The FBP aldolase activities of LacD.1 and LacD.2 were measured at the enzyme concentrations 0.1, 0.325, 0.625, 1.25, 2.5, and 5 mM. Plotting of activity versus enzyme concentration revealed a linear relationship, with an R2 of 0.99 for both LacD.1 and LacD.2. See Materials and Methods for details.
The C terminus of LacD.1 contributes to its differential enzymatic activity.
To determine the amino acid residues involved in LacD.1's differential catalytic activity, the divergent LacD.1 residues were mapped onto the structure of LacD.2. This comparison revealed that these residues were located predominantly on the surface of the enzyme (Fig. 5). However, they were not noticeably clustered in any specific region but, rather, were dispersed throughout the protein (Fig. 6). Thus, in the absence of any obvious candidate residues involved in the differential activity, several chimeric proteins were created that swapped extended contiguous regions of LacD.1 with the corresponding LacD.2 region (Fig. 6), followed by analysis of their steady-state kinetic properties (Table 1). Initially, LacD.1 was bisected, replacing its C-terminal half with 162 amino acid residues from the C terminus of LacD.2 (LacD.12A) (Fig. 6). This enzyme behaved very similarly to LacD.2 in the cleavage of TBP but was over 3 times more efficient than LacD.2 at cleaving FBP, as measured by the quotient for kcat/Km (Table 1). The increase in enzyme activity by LacD.12A compared to LacD.1 suggested that the residues in the C terminus contributed to the decreased activity displayed by LacD.1. To help further identify the specific residues involved in modulating LacD.1's enzymatic activity, two smaller swaps were made, LacD.12B and LacD.12C (Fig. 6), resulting in the exchange of 19 and 13 polymorphic residues, respectively. Comparison of LacD.12B to LacD.2 revealed that the enzyme was similar in activity in cleaving FBP but less efficient at cleaving TBP due to a lower kcat (Table 1). LacD.12C exhibited a decrease in the Km for both TBP and FBP compared to LacD.1 (Table 1). However, compared to LacD.12B or LacD.2, LacD.12C was less efficient at cleaving both FBP and TBP due to a relative increase in Km, suggesting that the 13 polymorphic residues mutated in LacD.12C, as well as the 6 additional polymorphic residues mutated in LacD.12B, contribute to the enzymatic efficiency of LacD.1. Taken together, comparisons of the chimeric enzymes illustrate that polymorphic residues found throughout the C-terminal half of LacD.1 contribute to its decrease in enzymatic activity, primarily by influencing LacD.1's Km for both TBP and FBP and,to a lesser extent, the turnover number or kcat for both substrates.
Fig 5.

Identification of polymorphic amino acid residues in LacD.1. (A) The polymorphic residues of LacD.1 (cyan) are mapped on the crystal structure of LacD.2. Catalytic residues identified above are highlighted in red. The locations of the loop 1 and turn 2 regions are labeled with arrows. (B) Identification of LacD.1 regions swapped with LacD.2; regions swapped are highlighted. LacD.12A includes red, green, and purple residues; LacD.12B includes green and purple residues; and LacD.12C includes purple residues.
Fig 6.

Construction of LacD.1 chimeras. The amino acid sequence of LacD.2 is illustrated at the top, with black boxes representing the polymorphic residues between LacD.2 and LacD.1. The catalytic residues from LacD.1 are labeled. The LacD.1 sequence is indicated by the white box, and for each chimera, the black and white boxes represent the sequences derived from LacD.2 and LacD.1, respectively. The loop 1 and turn 2 sequences are highlighted below the boxes of selected chimeras, with the specific mutations introduced into the chimera underlined as represented by the sequence shown on the lower line. The name of each chimera is shown at the right.
Examination of the polymorphic residues swapped on the crystal structures of LacD.1 and LacD.2 revealed that the identified residues primarily clustered into two structural regions. The first region, loop 1, connects the 9th alpha helix to the 8th beta sheet and contains 4 polymorphic residues. The second region, turn 2, connects the 10th helix to the 11th helix (Fig. 5) and contains 6 polymorphic residues, including a proline that changes position and likely influences the flexibility of the protein (Fig. 6).
Loop 1 and turn 2 influence the differential enzymatic activity of LacD.1.
To determine the role of these two structural regions in the enzymatic activity of LacD.1, the amino acid sequence of the LacD.1 loop 1 or the turn 2 region was swapped with the corresponding LacD.2 sequence. The two enzymes' catalytic activities were then characterized and compared to those of LacD.1 and LacD.2. Mutation of loop 1 to create LacD.12(L1) (Fig. 6) resulted in an enzyme with a considerably lower Km for both FBP and TBP than that of LacD.1, along with an increase in kcat for FBP (Table 1). However, the loop 1 chimera remained less efficient than LacD.2 in cleavage of both phosphorylated sugars. The turn 2 chimera, LacD.12(T2) (Fig. 6), also resulted in an enzyme that was more efficient for cleavage of both substrates than LacD.1, although not to the same extent as LacD.2 (Table 1). To examine the contribution of these two structural regions together, an additional chimera was constructed to introduce both LacD.2 loop 1 and turn 2 into LacD.1 [LacD.12(L1+T2)] (Fig. 6). The swap of both regions generated an enzyme with activity comparable to that of LacD.2 for FBP (Table 1), but the enzyme was less efficient at cleaving TBP due to a lower kcat and a slightly higher Km (Table 1). Chimeras with the reciprocal swaps to exchange LacD.2 loop 1 and turn 2 into LacD.1 were constructed; however, expression of these in E. coli resulted in low yields and proteins that were insoluble [LacD.21(L1+T2)] (Fig. 6; see also Fig. S1 in the supplemental material). This protein instability was further seen with the chimeric enzyme where LacD.2's 36 C-terminal residues were swapped with the corresponding LacD.1 residues (LacD.21F) (Fig. 6; see also Fig. S1). Additionally, a LacD.2 chimera was created that replaced the LacD.2 residues 165 through 260 with the LacD.1 sequence, leaving intact both LacD.2's loop 1 and turn 2 sequences (LacD.21G) (see Fig. 8 below). Kinetic analysis of this aldolase revealed that for both FBP and TBP, this enzyme behaved similarly to LacD.2 (Table 1). Taken together, these data indicate (i) that the residues responsible for divergent activity reside in the C-terminal half of LacD.1, (ii) that the polymorphisms in loop 1 and turn 2 make a major contribution to differential catalysis of TBP by LacD.1 due to an increase in the enzyme's Km and a decrease in the kcat, and (iii) that LacD.2 loop 1 and turn 2 do not completely restore LacD.1's catalysis of TBP, with enzyme efficiency remaining approximately 4-fold lower than for LacD.2.
Fig 8.

Identification of regulatory regions of LacD.1. (A) Several additional LacD.1 and LacD.2 chimeras are shown and are represented as described for Fig. 6. (B) Protease activity of the indicated strains determined by a spotting assay on protease indicator media. The clear zone around the bacterial growth indicates production of proteolytic activity. (C) Quantitative determination of proteolytic activity in cell-free supernatant fluid of the indicated strains. Data represent the means of at least three independent experiments, with samples analyzed in triplicate.
The polymorphic residues responsible for differential enzyme activity contribute to regulatory function.
The adaptation of LacD.1 to a regulator resulted in an aldolase with decreased enzyme efficiency. However, it is unclear if this decrease in aldolase activity was necessary for adaptation or coincidental. To begin to answer this question, the ability of the chimeric enzymes to regulate a virulence factor, the secreted cysteine protease encoded by speB, was examined. Expression of SpeB is routinely monitored on protease indicator plates, where the diameter of zone of clearing around bacterial growth in milk-containing media results from the proteolytic cleavage of casein reflecting the amount of SpeB produced (Fig. 7A, WT, Unmod.). However, buffering the medium to pH 7.5 results in repression of speB transcription (8) and a loss of the halo (Fig. 7A, HSC5 Empty Vector, pH 7.5). In contrast, speB expression is derepressed in a LacD.1 null mutant under these conditions (Fig. 7A, ΔLacD.1 Empty Vector, pH 7.5), and the wild-type phenotype can be complemented by introduction of a plasmid expressing LacD.1 but not LacD.2 (Fig. 7A; compare pLacD.1 to pLacD.2, pH 7.5). To determine the influence of polymorphic residues on this regulatory activity, plasmids encoding various chimeric enzymes were tested for the ability to complement a LacD.1-deficient mutant. Expression of the bisected chimera (LacD.12A), which has LacD.2-like enzymatic properties, revealed a regulatory phenotype similar to that of LacD.2, as it was unable to repress expression of SpeB (Fig. 7). In contrast, the chimera with only the 67 C-terminal residues of LacD.2 (LacD.12B) and reduced enzyme efficiency was fully functional in regulation, with a phenotype similar to that of LacD.1 (Fig. 7). Swaps of the 40 C-terminal residues (LacD.12C) or the individual regions of loop 1, turn 2, or loop 1 plus turn 2 did not alter LacD.1's ability to regulate SpeB expression, as similar to the case with the wild type or pLacD.1, no halo formed around growth at pH 7.5 on the indicator plates (Fig. 7A). Measurement utilizing a more quantitative FITC-casein cleavage assay to measure SpeB activity following growth in liquid media revealed that chimeras containing LacD.2 turn 2 [LacD.12C and LacD.12(T2)] were even more effective repressors than LacD.1, as they exhibited a superrepressor phenotype by producing significantly less SpeB activity (Fig. 7B). Interestingly, superrepression was abrogated when LacD.2 turn 2 was combined with the cognate LacD.2 loop 1 residues, as chimeras with matched domains had regulatory activity identical to that of LacD.1 [LacD.12(L1+T2)] (Fig. 7). This context-dependent superrepressor phenotype along with the influence of these domains on aldolase activity (see above) suggests that the polymorphic residues in loop 1 and turn 2 are functionally coupled. While there were some differences in expression of these chimeric enzymes, these differences did not correlate with the observed regulatory phenotypes of each chimeric enzyme (see Fig. S2 in the supplemental material). Taken together, these data indicate that chimeras with LacD.2 levels of catalytic efficiency lacked the ability to function as regulators. The latter property was associated only with those chimeras that have a 4-fold or greater decrease in efficiency for cleavage of TBP.
Fig 7.

Regulation of speB by LacD chimeras. (A) Spotting of overnight cultures on protease indicator media. Protease activity is apparent as a zone of clearing around the colony. The medium was unmodified (Unmod.) or buffered to a pH of 7.5 with the addition of 100 mM HEPES (pH 7.5). The HSC5 and LacD.1− strains contain empty vectors, while the other strains in a LacD.1− background contain either wild-type (WT) LacD.1, LacD.2, or the indicated chimeras described in Fig. 6. (B) Secreted-protease assay from supernatant fluid of the indicated strains described in Fig. 6. Buffered media were prepared as described in Materials and Methods. Activity is reported as a percentage of that of LacD.1−. Data represent the means of at least three independent experiments, with samples analyzed in triplicate. An asterisk denotes a significant difference from LacD.1− pLacD.1/LacD.1− complemented with LacD chimeric enzymes in the same medium (P < 0.05).
Extensive regions of LacD.1 are required for regulatory activity.
The ability of LacD.12B to function as a regulator and the inability of LacD.12A to complement a LacD.1 mutant suggested that variable residues within the 94-amino-acid sequence located in the center of LacD.1 are necessary for its regulatory function. To determine if these residues are sufficient, an additional chimera was constructed to swap the LacD.1 region into LacD.2 (LacD.21G, Fig. 8A). However, when examined on indicator plates and in liquid culture, this chimera was unable to repress expression of SpeB (Fig. 8B and C), suggesting that additional variable residues within the N terminus are required. Three additional chimeric enzymes were then constructed to swap larger regions of LacD.1 into the LacD.2 sequence (Fig. 8A). The introduction of neither 126 or 173 LacD.1 residues was able to generate a chimera with regulatory activity (LacD.21H and LacD.21I) (Fig. 8B and C). Furthermore, a final chimera which swapped the 87 N-terminal residues of LacD.1 along with the central 94-amino-acid region into LacD.2 also did not gain regulatory activity (LacD.21J) (Fig. 8B and C). These data suggest that multiple polymorphic residues dispersed throughout LacD.1 are required for its function as a regulator.
The adaptation of LacD.1 results in increased fitness.
In order to determine if LacD.1's adaptation is associated with any selective advantage, competition analyses were performed using culture conditions formulated to mimic growth in a deep-tissue environment (8). This analysis was conducted by complementing ΔLacD.1 with plasmids that expressed LacD.2 or selected enzyme chimeras. These plasmids were identical in all other respects, including the ectopic promoter driving expression of the chimeric gene and the kanamycin and chloramphenicol resistance determinants. An exception was the plasmid for the competitor strain, which expressed LacD.1 as described above but lacked the kanamycin resistance determinant (pLacD.1−kan). A coculture was then initiated between the competitor strain (ΔLacD.1 with pLacD−kan) and a test strain expressing one of the chimeric enzymes. The number of CFU for each strain was then determined following serial passage of the culture over a period of several days. As measured by the competitive index, the LacD.1 test strain (pLacD.1) had fitness equivalent to that of the competitor (pLacD.1−kan) over a 4-day period (Fig. 9). In contrast, the LacD.2 test strain (pLacD.2) had a loss of fitness that was apparent by day 2 and that continued to decline precipitously, achieving a net decrease of 4 logs by day 4 (Fig. 9). Test strains expressing LacD.1 chimeras with increased catalytic properties but intact regulatory activity (pLacD.12B and pLacD.12C) demonstrated no loss of fitness over the period of observation (Fig. 9). However, when the chimera tested also lacked regulatory activity (pLacD.12A), it also lost fitness in a pattern identical to that of the LacD.2 test strain (Fig. 9). These data demonstrate that LacD.1 makes an important contribution to fitness and that mutations in the C terminus linked to increased enzymatic activity did not influence this fitness under the conditions tested. Taken together, these results implicate its regulatory activity in LacD.1's contribution to fitness.
Fig 9.

The regulatory activity of LacD.1 confers increased fitness. Competition during growth in vitro was determined in coculture with a competitor strain (ΔLacD.1 complemented with pLacD.1−kan) and a test strain (ΔLacD.1 complemented with one of the various plasmids indicated). Cultures were back-diluted into fresh medium following 24 h of growth, and CFU were enumerated by plating on media with and without kanamycin for calculation of the competitive index. Data are the means of at least two independent experiments.
DISCUSSION
Comparison of protein paralogs with divergent functions can provide detailed information regarding the structure/function relationship of each protein as well as insight into the evolutionary mechanism that resulted in novel protein function. In this study, comparison of the paralogs LacD.1 and LacD.2 has revealed that while these enzymes share extensive sequence identity and structures, they are divergent with regard to their enzymatic properties and overall functions. The polymorphic residues associated with divergent catalytic activity were primarily confined to two discrete segments. However, the residues responsible for LacD.1's novel regulatory function were more widely dispersed. Furthermore, LacD.1's less efficient catalytic activity was associated with its ability to function as a regulator. Together, these data provide insight into the specific functions of LacD.1 and LacD.2 and into the evolutionary process that fostered their divergence.
One interesting insight came from the observation that the loop 1 and turn 2 features were functionally coupled. This is consistent with studies showing that protein evolution can be driven by networks of statistically coupled amino acids, termed sectors, that coevolve throughout protein families (26, 27). Sectors may be linked to conservation of specific function, including enzymatic reactions or substrate-binding motifs, and thus constitute fundamental units of evolution for repurposing to novel function. Examples of specific sectors include conserved networks for allosteric regulation of enzymes that link residues on the surface of the protein to the active site (28). While the residues within the sector can continue to change, their coevolution allows allosteric regulation to remain intact. Similar to what was observed for LacD.1 and LacD.2, cognate sectors can be adapted in order to modulate an enzymes activity (28).
This mechanism of variation has also been reported for other families of aldolase enzymes, suggesting that a common mechanism underlies the evolution of the S. pyogenes enzymes. Similar to the streptococcal enzymes, the aldolases found in higher eukaryotes are 70% identical and have identical active sites, yet they can have divergent activity with respect to their substrates. Mutational studies have shown that the residues responsible for differential activity are clustered in two specific patches, the terminal surface patch (TSP) and the distal surface patch (DSP), that are distal to their active sites (29–32). Examination of cysteine reactivity and the analysis of crystal structure B-factors suggest that these two patches influence flexibility to coordinate motion of the enzyme (33–35). Comparison of these aldolase isozymes to the S. pyogenes enzymes reveals that the polymorphic residues of turn 2 adjacent to helix 10 and helix 11 and across from helix 2 correspond to a region within the TSP. Thus, turn 2 could have a similar influence on movement. An increase in motion could explain the increase in the Km as a result of the higher entropic penalty accompanying binding. Additionally, a disruption in the coordination of the protein motions that are required for substrate binding could also be a contributing factor. Taken together, these similarities suggest that loop 1 and turn 2 are sectors that are constituents of an allosteric network in type 1 aldolases.
Conservation of an allosteric network in type I aldolases further supports the hypothesis that these enzymes evolved from a common ancestor. Structural comparison of LacD.2 and rabbit muscle aldolase revealed a strong conservation of active-site and catalytic residues (12), with the absence of the flexible C-terminal region found on class I FBP aldolases being the largest deviation in structure. This C-terminal region aids in product release and likely contributes to LacD.2's decrease in FBP cleavage rate compared to those of other FBP aldolases. Interestingly, the LacD.12A chimera showed an increase in cleavage of FBP compared to that of LacD.2, suggesting that polymorphic residues found between positions 163 and 258 have an influence on catalysis. However, swapping this region in LacD.2 to create LacD.21G did not drastically influence this enzyme's activity toward TBP or FBP. The epistatic nature of these residues suggests that additional amino acids found in the N terminus of LacD.1 are functionally linked to this region and the enzymatic activity of the aldolase. Furthermore, given that the residues in this region are required for the regulatory function of LacD.1, this further suggests that the adaptation of LacD.1 into a metabolic sensor required the coevolution of multiple amino acid residues.
Often, the best option for informing the cell of its metabolic state are enzymes that have evolved to be sensitive to the concentration of a specific metabolite (36–38). Thus, the manipulation of allostery may serve as a general mechanism to repurpose enzymes to more diverse function. Support for this concept comes from several examples where the presence or absence of substrate alters the regulatory output of a repurposed enzyme (32, 39–41). The data presented in this report are consistent with this model, suggesting that adaptation of LacD.1 acted at the level of allostery. However, for LacD.1 and for many of these other enzymes, adaptation that acts to harness an enzyme's ability to sense a metabolite to a transcriptional regulatory output occurred subsequent to a gene duplication event. Since the most common fate of the initially redundant gene is that it accumulates deleterious mutations leading to gene death (39), how these duplicated genes avoid negative selection is not clear.
Retention of the duplicate requires that it be adapted to provide a selective advantage for the organism. In this regard, LacD.1's adaptation may share some features in common with the well-characterized GAL network in the yeast Saccharomyces cerevisiae, in which an adapted galactose kinase functions as the sensory component of a transcriptional regulatory complex. This adapted enzyme is apparently the product of a gene duplication event, and comparisons to the ancestral enzyme found in related yeasts have suggested that duplication was necessary in order resolve an adaptive conflict that arose when positive selection for regulatory activity proved deleterious to enzymatic function. The ancestral enzyme has dual functionality, with a limited ability to act both as a transcriptional regulator and as a galactose kinase. However, in S. cerevisiae, duplication promoted a process of subfunctionalization that partitioned regulatory and enzymatic activities between the two genes. This “escape from conflict” then allowed positive selection to optimize each of these individual functions. In the streptococci, it is not yet known whether regulatory function is unique to LacD.1 of S. pyogenes or whether it was derived from an ancestor with dual function. However, since one of LacD.1's regulatory targets, the SpeB protease, is unique to S. pyogenes, it is possible that LacD.1's regulatory activity is also novel, and thus, its adaptation may be distinct from that of the yeast enzyme. In this scenario, instead of partitioning and perfecting existing functions, the gene would gain a novel function.
Another distinction is that while the subfunctionalization of the yeast regulatory paralog resulted in the loss of its galactose kinase activity (4), LacD.1 retains an aldolase activity. Interestingly, an increase in aldolase efficiency to levels comparable to those of LacD.2 resulted in a loss of activity. Currently, it is unclear whether these residues have a direct role in regulation or if an increase in activity indirectly influences LacD.1's regulatory function; it is possible that the reduced efficiency of LacD.1's catalytic activity represents subfunctionalization of catabolic activity between LacD.1 and LacD.2. For the yeast regulatory paralog, it has been shown that both the partitioning of regulatory activity and the loss of enzymatic activity each contributes to significant gains in fitness (3, 40). However, the gain of fitness provided by LacD.1 appears to be associated with a gain in regulatory activity acquired concomitantly with a decrease in enzymatic activity. Thus, while LacD.1's decrease in enzymatic activity toward TBP represents subfunctionalization, the polymorphisms that occur in loop 1 and turn 2 of LacD.1, and their associated role in enzymatic activity, are not clear. In the present study, we examined fitness under conditions that mimicked growth in soft tissue, which is relatively poor in its content of carbohydrates that can be metabolized by S. pyogenes. This is likely not the case at other sites that more commonly support streptococcal colonization. Thus, an alternative is that rather than being required for LacD.1's function during growth in deeper tissue, polymorphisms in loop 1 and turn 2 may be required to maintain fitness under carbohydrate-rich conditions where the Lac.2 operon is highly expressed. An inappropriate aggregate level of aldolase activity could alter the flow of carbon through the glycolytic pathway or alter the fine balance of regulation of the Lac.2 operon relative to other carbohydrate processing pathways. It is likely that the deep-tissue environment modeled here has less influence on long-term evolution because transmission from this environment is a rare occurrence. Thus, whether these conditions also mimic aspects of an environment that has a greater impact on long-term fitness, or whether LacD.1 will confer a similar advantage in other tissue compartments, awaits further analysis.
LacD.1's regulatory function has been demonstrated to include genes expressed both in exponential and in early stationary phases (41). This study represents the initial stage of an investigation into the complex evolutionary process that resulted in LacD.1's novel protein function. Taken together, the data suggest that LacD.1's adaptation represents a form of neofunctionalization where duplication facilitated the gain of regulatory function important to growth in tissue and pathogenesis. However, confirmation of this idea will require further investigation into a number of different areas, including an understanding of the molecular details of how LacD.1 acts to control transcription and its partners in the regulatory pathway. This information will prove invaluable for identification of residues that are specifically required for LacD.1's regulatory function and the role of selection in divergence at these positions relative to LacD.2. The rapid accumulation of sequences from additional S. pyogenes strains and from other streptococcal species will allow a higher-resolution analysis of selective pressure on individual residues and should provide insight into the relative contributions of subfunctionalization versus neofunctionalization in the evolution of virulence in S. pyogenes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Wolf-Dieter Fessner for his generous gift of tagatose bisphosphate. We also thank Jurgen Sygusch for his insight and conversations regarding the structures of LacD.2 and LacD.1.
This study was supported by Public Health Service grant NIH 5 R01 AI070759 from the National Institutes of Health.
Footnotes
Published ahead of print 11 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01997-12.
REFERENCES
- 1. Kim JW, Dang CV. 2005. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 30:142–150 [DOI] [PubMed] [Google Scholar]
- 2. Raffaelli N, Lorenzi T, Mariani PL, Emanuelli M, Amici A, Ruggieri S, Magni G. 1999. The Escherichia coli NadR regulator is endowed with nicotinamide mononucleotide adenylyltransferase activity. J. Bacteriol. 181:5509–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lohr D, Venkov P, Zlatanova J. 1995. Transcriptional regulation in the yeast GAL gene family: a complex genetic network. FASEB J. 9:777–787 [DOI] [PubMed] [Google Scholar]
- 4. Platt A, Ross HC, Hankin S, Reece RJ. 2000. The insertion of two amino acids into a transcriptional inducer converts it into a galactokinase. Proc. Natl. Acad. Sci. U. S. A. 97:3154–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hahn MW. 2009. Distinguishing among evolutionary models for the maintenance of gene duplicates. J. Hered. 100:605–617 [DOI] [PubMed] [Google Scholar]
- 6. Loughman JA, Caparon MG. 2006. A novel adaptation of aldolase regulates virulence in Streptococcus pyogenes. EMBO J. 25:5414–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13:470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loughman JA, Caparon M. 2006. Regulation of SpeB in Streptococcus pyogenes by pH and NaCl: a model for in vivo gene expression. J. Bacteriol. 188:399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosey EL, Oskouian B, Stewart GC. 1991. Lactose metabolism by Staphylococcus aureus: characterization of lacABCD, the structural genes of the tagatose 6-phosphate pathway. J. Bacteriol. 173:5992–5998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loughman JA, Caparon MG. 2007. Comparative functional analysis of the lac operons in Streptococcus pyogenes. Mol. Microbiol. 64:269–280 [DOI] [PubMed] [Google Scholar]
- 11. Marsh JJ, Lebherz HG. 1992. Fructose-bisphosphate aldolases: an evolutionary history. Trends Biochem. Sci. 17:110–113 [DOI] [PubMed] [Google Scholar]
- 12. LowKam C, Liotard B, Sygusch J. 2010. Structure of a class I tagatose-1,6-bisphosphate aldolase: investigation into an apparent loss of stereospecificity. J. Biol. Chem. 285:21143–21152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee SJ, Kim HS, JKim do, Yoon HJ, Kim KH, Yoon JY, Suh SW. 2011. Crystal structures of LacD from Staphylococcus aureus and LacD.1 from Streptococcus pyogenes: insights into substrate specificity and virulence gene regulation. FEBS Lett. 585:307–312 [DOI] [PubMed] [Google Scholar]
- 14. Hanski E, Horwitz PA, Caparon MG. 1992. Expression of protein F, the fibronectin-binding protein of Streptococcus pyogenes JRS4, in heterologous streptococcal and enterococcal strains promotes their adherence to respiratory epithelial cells. Infect. Immun. 60:5119–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lyon WR, Gibson CM, Caparon MG. 1998. A role for trigger factor and an rgg-like regulator in the transcription, secretion and processing of the cysteine proteinase of Streptococcus pyogenes. EMBO J. 17:6263–6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31:3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kushner SH. 1978. An improved method for transformation of Escherichia coli with ColE1-derived plasmids. Elsevier/North Holland Biomedical Press, New York, NY [Google Scholar]
- 18. Caparon MG, Scott JR. 1991. Genetic manipulation of pathogenic streptococci. Methods Enzymol. 204:556–586 [DOI] [PubMed] [Google Scholar]
- 19. Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 20. Blostein R, Rutter WJ. 1963. Comparative studies of liver and muscle aldolase. II. Immunochemical and chromatographic differentiation. J. Biol. Chem. 238:3280–3285 [PubMed] [Google Scholar]
- 21. Ye Y, Godzik A. 2004. FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 32:W582–W585 doi:10.1093/nar/gkh430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Williams GJ, Domann S, Nelson A, Berry A. 2003. Modifying the stereochemistry of an enzyme-catalyzed reaction by directed evolution. Proc. Natl. Acad. Sci. U. S. A. 100:3143–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tolan DR, Schuler B, Beernink PT, Jaenicke R. 2003. Thermodynamic analysis of the dissociation of the aldolase tetramer substituted at one or both of the subunit interfaces. Biol. Chem. 384:1463–1471 [DOI] [PubMed] [Google Scholar]
- 24. Beernink PT, Tolan DR. 1994. Subunit interface mutants of rabbit muscle aldolase form active dimers. Protein Sci. 3:1383–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sherawat M, Tolan DR, Allen KN. 2008. Structure of a rabbit muscle fructose-1,6-bisphosphate aldolase A dimer variant. Acta Crystallogr. D Biol. Crystallogr. 64:543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Halabi N, Rivoire O, Leibler S, Ranganathan R. 2009. Protein sectors: evolutionary units of three-dimensional structure. Cell 138:774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Süel GM, Lockless SW, Wall MA, Ranganathan R. 2003. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 10:59–69 [DOI] [PubMed] [Google Scholar]
- 28. Reynolds KA, McLaughlin RN, Ranganathan R. 2011. Hot spots for allosteric regulation on protein surfaces. Cell 147:1564–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arakaki TL, Pezza JA, Cronin MA, Hopkins CE, Zimmer DB, Tolan DR, Allen KN. 2004. Structure of human brain fructose 1,6-(bis)phosphate aldolase: linking isozyme structure with function. Protein Sci. 13:3077–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Motoki K, Kitajima Y, Hori K. 1993. Isozyme-specific modules on human aldolase A molecule. Isozyme group-specific sequences 1 and 4 are required for showing characteristics as aldolase A. J. Biol. Chem. 268:1677–1683 [PubMed] [Google Scholar]
- 31. Kusakabe T, Motoki K, Sugimoto Y, Takasaki Y, Hori K. 1994. Human aldolase B: liver-specific properties of the isozyme depend on type B isozyme group-specific sequences. Protein Eng. 7:1387–1393 [DOI] [PubMed] [Google Scholar]
- 32. Kusakabe T, Motoki K, Sugimoto Y, Hori K. 1998. Role of isozyme group-specific sequence 4 in the isozyme-specific properties of human aldolase C. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120:665–673 [DOI] [PubMed] [Google Scholar]
- 33. Pezza JA, Choi KH, Berardini TZ, Beernink PT, Allen KN, Tolan DR. 2003. Spatial clustering of isozyme-specific residues reveals unlikely determinants of isozyme specificity in fructose-1,6-bisphosphate aldolase. J. Biol. Chem. 278:17307–17313 [DOI] [PubMed] [Google Scholar]
- 34. Pezza JA, Stopa JD, Brunyak EM, Allen KN, Tolan DR. 2007. Thermodynamic analysis shows conformational coupling and dynamics confer substrate specificity in fructose-1,6-bisphosphate aldolase. Biochemistry 46:13010–13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heyduk T, Michalczyk R, Kochman M. 1991. Long-range effects and conformational flexibility of aldolase. J. Biol. Chem. 266:15650–15655 [PubMed] [Google Scholar]
- 36. Henderson B, Martin A. 2011. Bacterial virulence in the moonlight: multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect. Immun. 79:3476–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Commichau FM, Stulke J. 2008. Trigger enzymes: bifunctional proteins active in metabolism and in controlling gene expression. Mol. Microbiol. 67:692–702 [DOI] [PubMed] [Google Scholar]
- 38. Shi Y. 2004. Metabolic enzymes and coenzymes in transcription—a direct link between metabolism and transcription? Trends Genet. 20:445–452 [DOI] [PubMed] [Google Scholar]
- 39. Conant GC, Wolfe KH. 2008. Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 9:938–950 [DOI] [PubMed] [Google Scholar]
- 40. Hittinger CT, Carroll SB. 2007. Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449:677–681 [DOI] [PubMed] [Google Scholar]
- 41. Anbalagan S, Dmitriev A, McShan WM, Dunman PM, Chaussee MS. 2012. Growth phase-dependent modulation of Rgg binding specificity in Streptococcus pyogenes. J. Bacteriol. 194:3961–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.