

Abstract
Antigen receptors activate pathways that control cell survival, proliferation, and differentiation. Two important targets of antigen receptors, NF-κB and Jun N-terminal kinase (JNK), are activated downstream of CARMA1, a scaffolding protein that nucleates a complex including BCL10, MALT1, and other IκB kinase (IKK)-signalosome components. Somatic mutations that constitutively activate CARMA1 occur frequently in diffuse large B cell lymphoma (DLBCL) and mediate essential survival signals. Mechanisms that downregulate this pathway might thus yield important therapeutic targets. Stimulation of antigen receptors induces not only BCL10 activation but also its degradation downstream of CARMA1, thereby ultimately limiting signals to its downstream targets. Here, using lymphocyte cell models, we identify a kinase-independent requirement for TAK1 and its adaptor, TAB1, in antigen receptor-induced BCL10 degradation. We show that TAK1 acts as an adaptor for E3 ubiquitin ligases that target BCL10 for degradation. Functionally, TAK1 overexpression restrains CARMA1-dependent activation of NF-κB by reducing BCL10 levels. TAK1 also promotes counterselection of NF-κB-addicted DLBCL lines by a dual mechanism involving kinase-independent degradation of BCL10 and kinase-dependent activation of JNK. Thus, by directly promoting BCL10 degradation, TAK1 counterbalances NF-κB and JNK signals essential for the activation and survival of lymphocytes and CARMA1-addicted lymphoma types.
INTRODUCTION
The transcription factor NF-κB has important roles in lymphocyte maturation, activation, and differentiation, usually by regulating the expression of cell survival genes (1). Mutations that result in the constitutive activation of NF-κB are essential to the survival of certain lymphocytic cancers, including both activated B cell (ABC)-diffuse large B cell lymphoma (DLBCL) and mucosa-associated lymphoid tissue (MALT) lymphoma (2). Defining how NF-κB is regulated in these cells is thus a fundamental requirement for developing therapeutic strategies aimed at disrupting its activity in these cancers.
The multiprotein molecular complex formed by the protein scaffolds CARMA1 and BCL10 and the protease MALT1, known as the CBM complex, represents the constriction point through which the antigen receptor (AR)-induced activation of the NF-κB and Jun N-terminal kinase (JNK) signaling pathways are controlled in lymphocytes (3–5). Downstream of the CBM complex, transforming growth factor β-activated kinase 1 (TAK1) participates in the phosphorylation of IκB kinase β (IKKβ) and a mitogen-activated protein kinase kinase (MAP2K) (6), which in turn activate the NF-κB and JNK signaling pathways, respectively (3, 7–10). Despite advances in our knowledge of the CARMA1 activation cascade, relatively little is known about how lymphocytes limit AR-driven NF-κB signals. Studies have indicated that activation of CARMA1 through the AR (or via phorbol ester stimulation mimicking the AR signal) induces K48-linked polyubiquitination (polyUb) of both CARMA1 (11) and BCL10 (12–15). This polyUb tag then targets these proteins for proteasomal, lysosomal, or autophagosomal degradation (14, 16), thereby downregulating the activity of the CBM complex.
The regulation of BCL10 has been extensively studied, predominantly in T cells. Although scaffolding by CARMA1 is essential for BCL10 activation, whether BCL10 requires additional signals for activation is unclear. AR stimulation induces BCL10 phosphorylation (17–20); however, conflicting evidence suggests that phosphorylation promotes BCL10 activity by scaffolding (20), signaling for K63-linked polyUb (21) or K48-linked polyUb targeting BCL10 for degradation (12–15) or, alternatively, for the induction of actin cytoskeletal rearrangements (22). Similarly, although the degradation of BCL10 after AR stimulation is a consistent finding, the proposed mechanisms and molecular players vary considerably. Some studies have described a negative feedback loop that controls BCL10 ubiquitination and degradation via IKKβ-dependent BCL10 phosphorylation at S81 and S85 or S134 and S138 (13, 15); however, other studies have indicated that BCL10 phosphorylation by the NF-κB essential modulator (NEMO)-IKK complex inactivates BCL10 independently of BCL10 degradation (14, 20). BCL10 polyUb has also been attributed to various E3 ligases (E3s) including βTrCP, ITCH, and NEDD4 or cIAP2 (12–15).
In this study, we used genetically deficient B cell lines to demonstrate that CARMA1 and TAK1, but not IKKβ, are necessary to induce AR- or phorbol ester-dependent degradation of BCL10 in DT40 B cells. Interestingly, our genetic and pharmacologic data suggest that TAK1 functions as an adaptor in lymphocytes. In support of these results, experiments in 293T cells demonstrate that TAK1 specifically interacted with multiple E3 ligases, including two members of the NEDD4 HECT E3 family: ITCH and, to a lesser extent, NEDD4. Consistently, AR-induced BCL10 degradation from ITCH−/− mice was significantly delayed in primary T cells but not in B cells, implying differential usage of E3s and/or redundancy in these cell types. Functionally, TAK1 overexpression limited CARMA1-dependent activation of NF-κB and reduced the BCL10 levels in 293T cells. In DLBCL lines, TAK1 promoted cell counterselection via kinase-independent BCL10 degradation and kinase-dependent JNK activation. Together, these findings imply that, besides its positive role in AR-dependent activation, TAK1 plays a crucial feedback role by promoting BCL10 degradation and limiting receptor-induced NF-κB activation.
MATERIALS AND METHODS
Mice, cell lines, and reagents.
ITCH−/− mice (Itcha-18H) (23) and conditional TAB1 mutant mice (24) in a C57BL/6J background were described previously. Control C57BL/6J mice were obtained from Jackson Laboratories. Ramos, EL4 T, and HEK 293T cells were obtained from the ATCC. The OCI-Ly7 and OCI-Ly10 DLBCL cell lines were kindly provided by Ari Melnick (Weill Cornell Medical College). PKCβ−/−, CARMA1−/−, TAK1−/−, and IKKβ−/− DT40 cell lines were kindly provided by T. Kurosaki (RIKEN, Japan). Cell lines were cultured as described previously (25, 26). 5Z-7-Oxozeaenol, phorbol 12-myristate 13-acetate (PMA), ionomycin, phosphatase inhibitor cocktails (set I and set II), JNK inhibitor II, and ALLN (calpain inhibitor 1) were obtained from EMD Biosciences; cycloheximide (CHX) (a drug that prevents de novo protein synthesis) and protease inhibitor cocktail (P8340) were from Sigma-Aldrich. BV6 cIAP1/2 antagonists were kindly provided by V. Dixit (Genentech). TAT-NLS-Cre recombinant protein was obtained from Excellgen. Human/mouse ITCH (catalog no. sc-40364) and human TAB1 (hTAB1) small interfering RNAs (siRNAs) (catalog no. sc-61851) were from Santa Cruz Biotechnology. Mouse NEDD4 ON-TARGETplus siRNA and hTAB1 ON-TARGETplus siRNA (catalog no. L-058562-00-0010 and L-004770-00-0005, respectively) were from Dharmacon RNA Technologies. Control siRNA was from Qiagen (catalog no. 1022076).
Antibodies.
The following antibodies were used: anti-BCL10 (331.3 and H-197), anti-phospho-extracellular signal-regulated kinase (anti-phospho-ERK) (Y204, E-4), anti-ERK (C-16), antiactin (C-2), antihemagglutinin (anti-HA) probe (Y-11), antiubiquitin (Fl-76), and anti-IκB-α (C-21) (Santa Cruz Biotechnology); anti-TAB1 (3225), anti-TAK1 (4505), anti-phospho-JNK (T183/Y185, 81E11), and anti-phospho-protein kinase D (anti-phospho-PKD) (S744/748, 2054) (Cell Signaling Technology); anti-myc epitope (9E10) and anti-Flag epitope (M2) (Sigma-Aldrich); pan anti-human/mouse cIAP1/2 (315301, R&D Systems); anti-ITCH (611198; BD Biosciences); anti-NEDD4 (07-049; Millipore); anti-chicken IgM (M4; Southern Biotech); anti-mouse IgM F(ab′)2 (Jackson ImmunoResearch); IRDye800-conjugated anti-rabbit IgG (Rockland Immunochemicals); and Alexa Fluor 680-conjugated anti-mouse IgG (Invitrogen).
Plasmid constructs.
The following transient expression plasmids were used: pCMV 3×Flag-Cbl-b, -cIAP2, - ITCH, -NEDD4, and Flag-βTrCP (25); pmCARMA1-ΔPRD (26); pCMV Flag-IKKβ-WT and pCMV Flag-IKKβ-DA (Addgene plasmids 23298 and 11105) (27); pCR3-Flag-BCL10 (a gift from M. Thome) (28); pCMV5B-Flag-SMURF1 (Addgene plasmid 11752) (29); pCMV-SPORT6 muTAK1 (IMAGE clone 3499247; Open Biosystems); pcDNA3-Flag-PKD2 (a gift from S. Sidorenko) (30); pcDNA HA-TAK1 (created by ligating a double-stranded oligonucleotide encoding an HA tag into the 5′ end of the multiple-cloning site of pcDNA 3.1 [Invitrogen]; site-directed mutagenesis was then used [QuikChange II; Agilent] to generate restriction sites in pCMV-SPORT6 TAK1 to allow in-frame cloning of TAK1 downstream of the HA tag in the pcDNA backbone); pcDNA HA-TAK1-AAYA (to block binding of TAK1 to TAB1 or HECT E3 ligases, point mutations L122A, P123A, and Y125A were introduced into TAK1 using site-directed mutagenesis of parental construct pcDNA HA-TAK1); pcDNA HA-TAK1-ΔTAB2/3 (to remove residues binding TAB2 and TAB3 [31], amino acids 509 to 533 of TAK1 were deleted by site-directed mutagenesis of parental construct pcDNA HA-TAK1); pCMV-SPORT6 hTAB1 (IMAGE clone 6202841; Open Biosystems); pCMV 3×Flag TAB1 (site-directed mutagenesis was used to create restriction sites in pCMV-SPORT6 TAB1 that allowed direct cloning of the TAB1 coding sequence downstream of an N-terminal 3×Flag tag in vector p3×Flag-CMV-7.1 [Sigma-Aldrich]); His-Ub (a gift from K. Sabapathy, NCC, Singapore); pCS 8× HA-Ub (a gift from J. Roberts, FHCRC); Igκ2-IFN-luciferase and pmCARMA1 (gifts from J. Pomerantz) (32); pRL-TK (Promega); pmCARMA1-L232LI (for expression of the murine CARMA1 version of human CARMA1 L225LI [a constitutively active mutant identified in a primary human DLBCL] [33]; site-directed mutagenesis was performed on pmCARMA1 to introduce the L232LI mutation); pCMV C3×F-CARMA1-ΔPRD (site-directed mutagenesis was performed on plasmid pmCARMA1 to create restriction sites allowing in-frame cloning of the CARMA1 coding sequence upstream of a C-terminal 3×Flag epitope in plasmid p3×Flag-CMV-14 [Sigma-Aldrich]); pIRES Puro myc-BCL10 and pcDNA destabilized BCL10-6×myc (d-BCL10) (generated by amplifying a BCL10 coding sequence from pCR3 Flag-BCL10 using oligonucleotides with convenient restriction sites; amplicons were digested and then cloned in-frame to N-terminal myc or C-terminal 6×myc epitope tags in the specified plasmid backbones, and sequence analysis showed cloning artifacts in pcDNA d-BCL10 6×myc [P3H, E11G mutations of BCL10, and 35 random amino acids after the 6×myc tag] that decreased the stability of wild-type pcDNA BCL10 6×myc). The following plasmids were use to generate lentiviral vectors (LVs) for stable expression of the genes of interest: pRRL MND-HA-TAK1 2A-GFP and pRRL MND HA-TAK1-AAYA 2A-GFP (coding sequences of pcDNA HA-TAK1 and pcDNA HA-TAK1-AAYA were isolated by restriction digest and cloned into pRRL MND 2A-GFP as described previously [34]); pRRL MND-HA-TAK1-K63W 2A-GFP; pRRL MND GFP (34); pRRL MND-myc-BCL10 2A-mCherry; pRRL MND-myc-BCL10-S138A 2A-mCherry; pRRL MND-mCherry; pGIPZ shTAB1 V3LHS_350795; 5′-TGCTGTTGACAGTGAGCGAAACGGCTATGATGGCAACCGATAGTGAAGCCACAGATGTATCGGTTGCCATCATAGCCGTTGTGCCTACTGCCTCGGA-3′; pGIPZ shTAK1 V2LHS_153758; 5′-TGCTGTTGACAGTGAGCGCGCAGATGAGCCATTACAGTATTAGTGAAGCCACAGATGTAATACTGTAATGGCTCATCTGCTTGCCTACTGCCTCGGA-3′ (Open Biosystems); and pGIPZ shScrambled (Open Biosystems). The following plasmids were used to generate retrovirus for stable expression of the genes of interest: MSCV myc-CARMA1-ΔPRD IRES-GFP, MSCV myc-CARMA1-WT IRES-GFP (25), MSCV 3×Flag-TAK1 IRES-GFP, and MSCV 3×Flag-TAK1-K63W IRES-GFP (to create these constructs, site-directed mutagenesis of pCMV-SPORT6 TAK1 was performed to create restriction sites allowing cloning of TAK1 in-frame with the N-terminal 3×Flag epitope in plasmid 3×Flag-CMV-7.1; mutagenesis was also used to generate a K63W mutation within the kinase domain of TAK1, and subsequently, 3×Flag-TAK1 and 3× Flag-TAK1-K63W coding sequences were isolated by restriction digestion of the resulting plasmid and inserted into the MSCV-IRES-GFP backbone). All coding sequences and cloning junctions were sequenced to confirm sequence integrity.
Transient transfection and viral transduction.
CARMA1−/− DT40 cells (4 × 106 cells/condition) were transiently transfected with 5 μg of pCMV Flag-IKKβ-DA or empty vector plasmid DNA using an Amaxa Nucleofector and cell line Nucleofector kit T, program B-23 (Lonza AG). 293T cells (0.5 × 106/condition) were transfected with selected expression or empty vectors using FuGENE 6 (Roche) with a 1:3 DNA/FuGENE 6 ratio. All transfection experiments were carried out for 48 h before analysis. Retroviruses were generated for stable protein expression in DT40 or Ramos B cells; retrovirus production and target cell transduction and green fluorescent protein (GFP) selection were carried out as described previously (25) using the packaging plasmid 10A1 and the cDNA of interest subcloned into the retroviral MSCV-IRES-GFP plasmid. Recombinant LVs were used to stably express TAK1 proteins in human DLBCL lines or short hairpin RNAs (shRNAs) in 293T and Jurkat cells. LVs were generated as described previously (34, 35). Briefly, 60% confluent 293T cells were transfected with the LV constructs of interest, psPAX2 and MD2.G (4:2:1 psPAX2/MD2.G/DNA ratio), using polyethyleneimine (PEI) at 1 μg/ml or Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Seventy-two hours posttransfection, supernatants were harvested and cleared by centrifugation. Viral preparations were concentrated using PEG-it virus precipitation solution (System Biosciences). Cells (0.5 × 106/condition) were transduced using the following LVs: pRRL-MND-TAK1 2A-GFP, pRRL-TAK1-AAYA 2A-GFP, pRRL-TAK1-K63W 2A-GFP, or pRRL-MND-GFP. Counterselection and cell death were analyzed with a fluorescence-activated cell sorter (FACS) at different time points posttransduction by analysis of GFP expression or incorporation of 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich), respectively. For TAB1 knockdown, 293T cells were transduced for 72 h at 37°C using concentrated viral supernatants in growth medium with Polybrene (8 μg/ml). GFP-positive cells were purified using a FACSAria cell sorter and expanded. Knockdown efficiency was estimated by Western blotting. For TAK1 knockdown, Jurkat T cells were transduced in growth medium with Polybrene (8 μg/ml) using three additions of 100-fold-concentrated recombinant LVs expressing TAK1 shRNA over 48 h.
Cell activation and treatment with drugs.
All protein degradation experiments were carried out in the presence of 70 μM CHX (or 140 μM for DLBCL lines) to prevent de novo protein synthesis. For basal analysis of BCL10 degradation, Ramos (1 × 106/condition), DT40 (2 × 106/condition), or OCI-Ly7 (0.5 × 106/condition) cells were incubated with CHX for 0 to 7 h at 37°C in a CO2 incubator. For inducible BCL10 degradation, cells were serum starved in RPMI medium for 30 to 60 min before stimulation. DT40 cells (2 × 106/condition), Jurkat T cells (0.5 × 106/condition), OCI-Ly10 cells (0.5 × 106/condition), or primary murine splenocytes (4 × 106/condition) were preincubated with CHX for 30 min at room temperature and then stimulated with PMA (3.9 to 250 nM) and ionomycin (1 μg/ml) (P/I). For AR-stimulated DT40 cells, anti-chicken IgM (10 μg/ml) was added for 15 min at 4°C and washed, and cells were incubated at 37°C with anti-mouse IgM F(ab′)2 fragment (10 μg/ml) for various time points. For AR stimulation of Jurkat T cells, anti-hCD3e (OKT3) at 10 μg/ml and anti-hCD28 (catalog no. 555725; BD Pharmingen) at 5 μg/ml were added for 10 min on ice prior to the addition of anti-mouse IgG cross-linking antibody (Jackson ImmunoResearch) at 37°C for stimulation. For cells treated with inhibitors, cells were preincubated with either 5Z-7-oxozeaenol (0.1 to 20 μM), JNK inhibitor II (2.5 to 20 μM) for 30 min at room temperature or the cIAP1/2 antagonist BV6 (2.5 μM) for 1 h at 37°C before the addition of CHX, followed by stimulation with P/I. At each time point, cells were harvested and lysed with a highly stringent lysis buffer (radioimmunoprecipitation assay [RIPA], 250 mM NaCl, 20 mM Tris [pH 7.4], 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, with protease and phosphatase inhibitor cocktails).
Immunoprecipitation and immunoblotting.
Transfected 293T cells were preincubated with 25 μM ALLN for 1 h before harvesting in order to block proteasomal degradation. For analysis of protein ubiquitination, cells were lysed with RIPA buffer containing 1% SDS and then diluted 10-fold to allow immunoprecipitation (IP); for analysis of protein coimmunoprecipitation (co-IP), cells were lysed with RIPA buffer. IPs were carried out in postnuclear supernatants using a 25-μl bead volume of anti-Flag M2 affinity gel (Flag IP; Sigma-Aldrich). IPs were carried out for 2 h at 4°C; beads were washed 3 times with RIPA buffer without SDS and sodium deoxycholate and 2 times with TBS (50 mM Tris-HCl [pH 7.4] and 150 mM NaCl). Protein complexes were eluted with 150 ng/μl of 3×Flag peptide (Sigma-Aldrich), and eluates were treated with 6× Laemmli buffer. Proteins were resolved using 8 to 10% SDS-PAGE and transferred to PVDF-FL membranes (Millipore). Membranes were blocked with Odyssey blocking buffer (Li-Cor Biosciences). Primary antibodies were incubated overnight at 4°C with IRDye800- or Alexa Fluor 680-labeled secondary antibodies for 1.5 h at room temperature. Proteins were visualized and quantified using the Odyssey infrared imaging system from Li-Cor Biosciences.
siRNA transfection.
Murine EL4 T cells, Jurkat T cells, and A20 B cells were cultured at a density of 5 × 105 overnight 1 day before transfection. Transfection (2 × 106 cells/condition) was carried out using the Amaxa L kit (program C-009) for EL4 cells, the V kit (program L-13) for A20 cells, and the V kit (program X001) for Jurkat cells with 200 pmol of control, ITCH, and/or NEDD4 siRNA or two pooled TAB1 siRNAs (100 pmol each). Cells were incubated for 24 to 48 h before analysis.
NF-κB luciferase activity.
293T cells (5 × 105 per sample) were transfected for 48 h using FuGENE 6 (Roche Applied Science) with 1 μg of mammalian expression plasmids, 250 ng of Igκ2-IFN-luciferase (NF-κB reporter), and 50 ng of pRL-TK (transfection control). NF-κB and control activities were analyzed according to the dual-luciferase reporter assay protocol from Promega. Alternatively, DLBCL lines and Jurkat T cells were transduced to express a secreted Gaussia luciferase (GLuc) (36) under the control of 3×NF-κB response elements. Transduction efficiency was evaluated by the expression of the cis-linked marker mCherry under the control of an sEF1α promoter. NF-κB activity was analyzed in cells that were washed twice with PBS and resuspended in 200 μl of culture medium at a density of 2 × 105 cells/condition and plated in 96-well plates. Sample supernatants (20 μl/time point) were collected between 0 and 5 h without stimulation. For CD3/CD28-stimulated Jurkat T cells, plates were precoated with 5 μg/ml of anti-hCD3ε antibody and 1 μg/ml of anti-hCD28 for 0 to 5 h. GLuc activity was analyzed using the BioLux GLuc assay kit (New England BioLabs).
TAT-Cre treatment and stimulation of floxed-Tab1 splenic B cells.
Splenic B cells were purified from flox/flox-Tab1, wild type (WT), and a flox/flox-GFP reporter mice. The B cells were washed 2 times with serum-free RPMI medium and resuspended at a density of 5 × 106 cells/ml. Cells were pretreated with 250 nM TAT-Cre for 1 h at 37°C, washed, resuspended in RPMI with 10% FCS at the same cell density, and cultured for 24 h at 37°C in 5% CO2 (37). Cells were harvested, serum starved in RPMI for 1 h at 37°C, and then stimulated with P/I (250 nM/1 μg/ml) for 0 to 3 h. Cells were lysed with 1× RIPA buffer, and 3 × 106 to 4 × 106 cell equivalents were loaded/lane for subsequent Western blotting. Of note, as Cre recombination deletes only the C-terminal TAK1 binding region of TAB1, TAB1 expression appeared to be equivalent in TAT-Cre-treated and nontreated cells, findings which are consistent with those in the original report (24).
Protein densitometry.
Signal intensities of immunoblots probed with fluorescently labeled secondary antibodies were measured using the Odyssey infrared imaging system (Li-Cor). In DT40 cells, Ramos B cells, Jurkat T cells, and primary splenocytes, endogenous BCL10 and myc-CARMA1 levels were determined relative to those of ERK, TAK1, or actin loading controls; for kinetic analyses, values were normalized relative to the zero time point, whose value was arbitrarily set as 1. For densitometry of protein knockdown, signal intensities were normalized relative to those of the loading controls, with the relative signal for cells transfected/transduced with control siRNA/shRNA set as 1. In 293T cells, endogenous BCL10 was quantified relative to the values of CARMA1. To quantify the levels of protein coimmunoprecipitated from 293T cell transfections, the BCL10/Flag, CARMA1/Flag, and TAK1/Flag ratios were obtained after dividing the densities of each protein in IPs by those of whole-cell lysates (WCLs).
RESULTS
In DT40 B cells, BCL10 degradation requires PKCβ, CARMA1, and TAK1.
Previous studies have demonstrated that CARMA1 expression is required for both BCL10 ubiquitination and degradation in T cell lines (13, 21). To determine whether CARMA1controls BCL10 degradation in B cells, CARMA1−/− DT40 B cells were stimulated with P/I in the presence of CHX, a drug that prevents de novo protein synthesis, and degradation of BCL10 was visualized by immunoblotting (Fig. 1A, middle). While BCL10 levels were reduced by 50% as early as 30 min post-P/I stimulation in WT cells, BCL10 was resistant to proteolysis in CARMA1−/− DT40 cells.
Fig 1.

Activation-induced BCL10 degradation in DT40 B cells requires PKCβ, CARMA1, and TAK1 but not IKKβ. (A) WT, PKCβ, CARMA1−/−, TAK1−/−, and IKKβ−/− DT40 cells were stimulated with P/I for 0 to 2 h. (B) BCL10 densitometry; graphs show the means ± standard errors of the mean (SEM) from six independent experiments. **, P < 0.01 using Student's unpaired t test compared to the same time points in WT and IKKβ−/− DT40 cells. (C) CARMA1−/− DT40 cells stably reconstituted with myc-CARMA1-ΔPRD were transfected with empty vector or Flag-IKKβ-DA for 48 h and incubated with CHX for 0 to 7 h. (D) CARMA1−/− and TAK1−/− DT40 cells stably reconstituted with myc-CARMA1-WT or myc-CARMA1-ΔPRD were stimulated with P/I for 0 to 120 min (left) or incubated with CHX (right) for 0 to 5 h. (E) WT, CARMA1−/−, or TAK1−/− DT40 cells were stimulated with anti-IgM plus an anti-mouse IgM (2nd) or with 2nd only as a control for 0 to 2 h. (F) Jurkat T cells transduced with TAK1 or control shRNAs were stimulated with CD3/CD28 or P/I for 0 to 3 h. (G) Human Ramos B cells stably reconstituted with myc-CARMA1 or myc-CARMA1-ΔPRD were incubated with CHX for 0 to 3 h. In each experiment, whole-cell lysates (WCLs) were immunoblotted with the indicated antibodies (left labels). The signal intensities of BCL10, myc-CARMA1, and pERK (E) are shown in a graph (B) or below the blots (A and C to G) and are calculated as described in Materials and Methods.
A primary downstream target of P/I in B cells is PKCβ, which phosphorylates and activates CARMA1 (3, 26). CARMA1 then assembles a BCL10-containing protein complex required for TAK1 activation and subsequent activation of JNK and IKKβ signaling pathways. To identify which proteins downstream of P/I signal BCL10 proteolysis besides CARMA1, we analyzed DT40 cell lines with targeted deletions of PKCβ, IKKβ, and TAK1 (9, 38). As expected, genetic deletion of PKCβ abolished P/I-induced BCL10 degradation, consistent with its function upstream of CARMA1 in this pathway (Fig. 1A, top). Surprisingly, although IKKβ controls induction-dependent degradation of BCL10 in T cells (13, 15), BCL10 degradation was normal in IKKβ−/− DT40 B cells (Fig. 1A, bottom, and B). Additionally, overexpression of a constitutively active IKKβ (f-IKKβ-DA) (Fig. 1C) (27), even in activated CARMA1−/− DT40 B cells (cells reconstituted with CARMA1-ΔPRD), did not further enhance BCL10 degradation, indicating that IKKβ does not synergize CARMA1-dependent BCL10 degradation in DT40 B cells. Interestingly, deletion of the distal signaling effector TAK1 abolished BCL10 proteolysis in P/I-activated cells (Fig. 1A, bottom), and while reexpression of WT CARMA1 (Fig. 1D, left) or a constitutively active CARMA1 molecule (myc-CARMA1-ΔPRD) (Fig. 1D, right) in CARMA1−/− cells reconstituted BCL10 degradation, deletion of TAK1 abolished BCL10 degradation in either case.
To confirm that P/I stimulation is a faithful surrogate for antigen receptor (AR) stimulation of BCL10 stability, TAK1−/− and CARMA1−/− DT40 cells or Jurkat T cells LV transduced with TAK1-specific shRNAs were stimulated using anti-IgM or anti-CD3/CD28, respectively. Similar to P/I stimulation, AR cross-linking required both CARMA1 and TAK1 in DT40 cells (Fig. 1E) and TAK1 in Jurkat T cells (Fig. 1F) to induce BCL10 degradation. Finally, we observed that BCL10 degradation constitutively occurred in a human B cell line (Ramos) stably expressing activated CARMA1 (Fig. 1G). Consistent with our previous report (25), myc-CARMA1-ΔPRD stability was also reduced, suggesting that CARMA1 and BCL10 undergo parallel protein degradation upon CARMA1 activation.
TAK1, but not its kinase activity, is required for BCL10 ubiquitination and degradation.
Studies have indicated that BCL10 turnover is signaled by IKKβ-dependent phosphorylation of BCL10 in T lymphocytes (13, 15). Because IKKβ was dispensable in DT40 B cells, we hypothesized that TAK1 instead might catalyze a signaling phosphorylation event in B cells. To test this hypothesis, BCL10 degradation was analyzed in TAK1−/− DT40 cells reconstituted with either WT or kinase dead (KD) TAK1 (K63W) (39) and stimulated with doses of PMA (Fig. 2A). Under these conditions, BCL10 degradation was equally rescued in both TAK1- and TAK1-KD-reconstituted cells. The absence of JNK phosphorylation confirmed the lack of TAK activity in cells reconstituted with TAK1-KD (Fig. 2B). To further confirm that TAK1 kinase activity is not required for BCL10 degradation in lymphocytes, WT DT40 B cells and murine EL4 T cells were treated with the specific TAK1 kinase inhibitor 5Z-7-oxozeaenol. Cells treated with the TAK1 inhibitor exhibited normal BCL10 degradation after P/I stimulation, although the inhibitor specifically blocked JNK phosphorylation (Fig. 2C and D). These data indicate that TAK1 is required for BCL10 degradation in lymphocytes and that TAK1 kinase activity is dispensable in this process.
Fig 2.

TAK1 promotes BCL10 degradation independent of its kinase activity in lymphocytes. TAK1−/− DT40 cells were retrovirally reconstituted with Flag-TAK1 or kinase-dead Flag-TAK1 (KD) and stimulated with 1 μg/ml ionomycin and increasing doses (3.9, 7.8, 31.2, and 250 nM) of PMA for 2 h (A) or 1 μg/ml ionomycin and 250 nM PMA for 0 to 20 min (B). WT and TAK1−/− DT40 B cells (C) or EL4 T cells (D) were pretreated with the indicated doses of the TAK1 inhibitor 5Z-7-oxozeaenol followed by CHX and then stimulated with P/I (250 nM/1 μg/ml) for 0 to 180 min. Cells were lysed with RIPA buffer and immunoblotted as indicated to the left of each blot. The signal intensities of BCL10 levels relative to those of ERK were measured as described in Materials and Methods and are shown below the BCL10 immunoblots.
We hypothesized that TAK1 might be an essential adaptor of a protein that either signals for or catalyzes the polyUb of BCL10. To test this idea, we first determined whether overexpressed TAK1 can promote the polyUb of BCL10. These experiments were carried out in 293T cells in order to consistently analyze BCL10 ubiquitination under coexpression of multiple proteins. 293T cells were transfected with combinations of TAK1, Flag-BCL10, HA-Ub, and activated CARMA1 (myc-CARMA1-ΔPRD), and BCL10 polyUb was visualized as a slower-migrating smear in Flag IPs using anti-HA or anti-Ub antibodies (Fig. 3A). Because BCL10 overexpression is sufficient for activating downstream signals (40, 41), it results in low constitutive polyUb. However, polyUb of BCL10 was significantly enhanced only when TAK1 and CARMA1-ΔPRD were coexpressed (Fig. 3A, lane 5), indicating that TAK1 acts in synergy with CARMA1 to promote BCL10 polyUb.
Fig 3.

TAK1 interaction with TAB1 controls CARMA1-dependent ubiquitination and degradation of BCL10. (A to D) 293T cells were cotransfected for 48 h with combinations of expression vectors for the proteins indicated at the top of each blot. WCLs were prepared for Flag IPs and Western blots as described in Materials and Methods. BCL10 polyUb (A, C, and D) or TAK1-TAB1 interaction (B, middle) in Flag IPs (A to D, top panels) or WCLs (A to D, bottom panels) was detected using the indicated antibodies (left of the blots). (B) The top diagram shows TAK1 mutations that disrupt interaction with TAB2/3 (TAK1-ΔTAB2/3) or TAB1 (TAK1-AAYA). Middle, TAK1 blots with levels of TAK1 in the Flag IPs are normalized to those in WCLs (to compensate for TAK1-WT protein stabilization induced by TAB1 (50, 51). Bottom, NF-κB activity in 293T cells cotransfected with Igκ2-IFN-luciferase (NF-κB reporter), pRL-TK (transfection control), and combinations of the indicated plasmids. The graph shows means ± SEM from four independent experiments normalized as the percentage of the highest value in each experiment. (C) Immunoblot analysis of BCL10 polyUb by the CARMA1 and TAK1 mutants described for panel B. (D) Transfections were performed in cells transduced with LVs expressing control or TAB1-specific shRNAs. (E) Jurkat T cells were transfected with control or TAB1 siRNAs, stimulated with anti-CD3/CD28, lysed, and blotted with the antibodies indicated on the left. (F) Jurkat T cells, transduced to express a secreted NF-κB-controlled GLuc, were transfected with control or TAB1 siRNAs and stimulated with anti-CD3/CD28 for 0 to 5 h. GLuc activity was analyzed as described in Materials and Methods, normalized to that of unstimulated cells, and plotted as the percentage of the highest GLuc value in each experiment. (G) Splenic B cells purified from flox/flox-TAB1, WT, and flox/flox-GFP reporter mice were treated with TAT-Cre and stimulated as described in Materials and Methods. Left, GFP induction in untreated (−) or TAT-Cre-treated flox/flox-GFP reporter B cells. Middle, cells were lysed and blotted with the antibodies indicated on the left. The signal intensities of BCL10 and pJNK levels relative to that of actin are displayed below the immunoblots and graphically for BCL10 (right). (E and G) The graphs show the means ± SEM from three independent experiments. BCL10 levels were normalized relative to that of actin. *, P > 0.05, unpaired Student's t test.
The activation and substrate specificity of TAK1 are mediated through its interactions with TAB1 and TAB2 and with TAB3 adaptors, respectively (42–44). To determine whether these adaptors have a role in the polyUb of BCL10, we generated TAK1 mutants with an impaired ability to interact with TAB proteins (Fig. 3B, top). TAB2/3 interactions are eliminated by the deletion of amino acids 509 to 533 of TAK1 (31) (TAK1-Δταβ2/3), and for TAB1 binding, the crystal structure of a TAK1-TAB1 chimeric protein suggested tyrosine 125 (Y125) of TAK1 as one of the important interaction residues (45). Y125 is also part of the LPYY motif (residues 122 to 125), a canonical binding site for the NEDD4 family of E3s (46, 47). Therefore, we mutated this LPYY motif to AAYA (48, 49) in order to determine whether this mutation would delete TAB1 binding and/or eliminate any potential NEDD4-E3 binding partners. Co-IP of TAB1 with TAK1-AAYA revealed 10-fold less interaction than with WT TAK1 (Fig. 3B, middle). Although the overexpression of TAB1 enhances spontaneous TAK1-dependent NF-κB activity in 293T cells (43, 50, 51), coexpressed TAK1-AAYA and TAB1 failed to enhance NF-κB activity (Fig. 3B, bottom). We next evaluated the effect of TAB binding-deficient TAK1 mutants on polyUb of BCL10 (Fig. 3C). WT and mutant TAK1 constructs were cotransfected with Flag-BCL10, CARMA1-ΔPRD, and His-Ub in 293T cells, and BCL10 polyUb was detected in Flag IPs. Only the TAK1-AAYA mutation interfered with polyUb of BCL10 (Fig. 3C, lane 3), suggesting that TAK1 interaction with TAB1, but not TAB2/3, might be required to promote BCL10 polyUb. Although reduced expression of TAK1-ΔTAB2/3 was observed, it did not interfere with BCL10 polyUb.
Genetic analyses of the role for TAB1 in activation-induced BCL10 polyUb and degradation included shRNA and siRNA knockdown in 293T and Jurkat T cells, respectively (Fig. 3D to F). TAB1 knockdown reduced CARMA1/TAK1-codependent polyUb of BCL10 in 293T cells (Fig. 3D) and delayed CD3/CD28-induced BCL10 degradation in Jurkat cells (despite a knockdown efficiency of only ∼30%) (Fig. 3E). We next tested the requirement for TAB1 in primary splenic B cells purified from conditional-TAB1 mutant mice (24) (Fig. 3G). After treatment with TAT-Cre ex vivo (excising the TAK1 binding site from TAB1), we found that P/I-induced BCL10 degradation was markedly reduced (Fig. 3G), indicating a requirement for TAK1-TAB1 interaction for BCL10 degradation in primary B cells. Similar to studies suggesting that enhanced BCL10 stability in Jurkat T cells results in increased NF-κB activity and interleukin 2 (IL-2) production (13, 15), we observed that TAB1 knockdown resulted in increased NF-κB GLuc activity (∼30 to 40%) in Jurkat T cells relative to that in controls (Fig. 3F). We also observed higher JNK phosphorylation in TAT-Cre-treated floxed-TAB1 B cells (Fig. 3G), consistent with a role for TAB1 in downmodulating CARMA1-triggered signals. Overall, these results indicate a nonenzymatic requirement for TAK1 and its adaptor TAB1 for activation-induced BCL10 polyUb and degradation as a mechanism for downregulating CARMA1-dependent signals in T and B lymphocytes.
Preferential interaction of the NEDD4 family of HECT ubiquitin ligases with TAK1.
Multiple E3s have been reported to target BCL10 for polyUb and turnover in lymphocytes (12–14). Among these candidates, cIAP2 and ITCH have been shown to co-IP with BCL10 under mild solubilization conditions (12). Because protein overexpression might result in an overestimation of protein interactions under such conditions, we tested the potential interactions of BCL10 with these and other E3s using relatively stringent lysis conditions (RIPA buffer) (Fig. 4A). The positive-control co-IP of BCL10 with CARMA1-ΔPRD was clearly preserved under these conditions (Fig. 4A, lane 8). Surprisingly, none of the E3s coimmunoprecipitated with BCL10 (Fig. 4A, top); nonetheless, a high-molecular-weight smear of BCL10 (indicative of polyUb) was observed in WCLs when coexpressed with either ITCH or NEDD4 (Fig. 4A, bottom). Because TAK1, CARMA1, and BCL10 have been shown to coimmunoprecipitate from activated lymphocytes (7, 9), and because BCL10 degradation requires a kinase-independent TAK1 function, we tested the ability of TAK1 to coimmunoprecipitate with candidate E3s under identical lysis conditions (Fig. 4B, top). Quantification of E3 and TAK1 levels in Flag IPs revealed that TAK1 preferentially interacted with members of the NEDD4 family of HECT E3s, ITCH, and, to a less significant extent, NEDD4 (Fig. 4B, bottom). Similar to BCL10, CARMA1 interacted less efficiently with the selected E3s than TAK1 (Fig. 4C).
Fig 4.

TAK1, but not BCL10 or CARMA1, preferentially interacts with ITCH E3 ligase. 293T cells were cotransfected for 48 h with combinations of the indicated Flag-tagged E3s and BCL10 (A), TAK1 or TAK1-AAYA (B to D), or CARMA1 (C). WCLs were prepared using RIPA buffer, and proteins were detected by immunoblot (left of blots) on Flag IPs (top blots) and WCLs (bottom blots). (A, bottom) Overexposed BCL10 blot in WCLs. (B) The graph shows means ± SEM from four independent experiments; NEDD4 interaction levels with TAK1 varied between experiments and did not reach statistical significance. Blots show representative data. (B and C) Quantification of immunoprecipitated TAK1 or CARMA1 by E3s in Flag IPs relative to that of WCLs. The highest value in each experiment was set as 1. *, P > 0.05; ns, not significant, unpaired Student's t test comparing TAK1 interaction with ITCH or NEDD versus that each of the other non-NEDD4 family E3s.
Although we demonstrated that TAB1 interaction is diminished by the TAK1-AAYA mutation (Fig. 3B), it remained possible that this mutation also eliminates a putative binding site for NEDD4 family E3s (46, 47). Thus, we next compared the interaction of TAK1 or TAK1-AAYA with Flag-tagged ITCH (Fig. 4D). Equivalent levels of both proteins coimmunoprecipitated with ITCH, indicating that deficient BCL10 ubiquitination by the TAK1-AAYA mutation was mainly due to defective interactions between TAK1 and TAB1. In conclusion, these combined data support the hypothesis that TAK1 functions as an adaptor of NEDD4 family HECT E3s in order to promote BCL10 ubiquitination and degradation.
Although ITCH and NEDD4 increased polyUb of BCL10, a reduction of BCL10 protein levels was not readily apparent (Fig. 4A). To define which of these E3s preferentially targeted BCL10 for degradation in 293T cells, we utilized a destabilized BCL10 protein (d-BCL10). Previous studies have suggested that BCL10 turnover preferentially targets phosphorylated BCL10 (15). Coexpression of d-BCL10 with E3s revealed that levels of the phosphorylated forms p1 and p2 of BCL10 were markedly reduced by ITCH (Fig. 5A). To determine whether reduced p1 and/or p2 levels in cells expressing ITCH result from enhanced degradation rather than defective phosphorylation of BCL10, we evaluated the stability of WT-BCL10 and d-BCL10 in the presence of ITCH and CHX (Fig. 5B). While the phosphorylated and nonphosphorylated forms of WT-BCL10 were stable under these conditions, robust degradation of both species was observed with d-BCL10 (Fig. 5B), indicating that reduced p1 and/or p2 levels of d-BCL10 result mainly from enhanced degradation. As nonphosphorylated d-BCL10 also rapidly degraded (Fig. 5B), and the association of BCL10 phosphorylation with its degradation is still controversial (15, 20, 22), we reevaluated BCL10 degradation using a mutant in which serine 138 was replaced with alanine (S138A) (Fig. 5C). As expected, S138A-BCL10 was expressed mostly as p0 (Fig. 5C, left), even after stimulation (17, 20, 22). In agreement with previous studies (20, 22), we observed that activation-induced degradation of BCL10 was identical in the WT and the S138A mutant in Jurkat cells (Fig. 5C, right and bottom), indicating that both the phosphorylated and the nonphosphorylated BCL10 isoforms can be targeted for degradation in T lymphocytes.
Fig 5.

ITCH overexpression promotes BCL10 turnover. (A and B) 293T cells were cotransfected with candidate Flag-tagged E3s and a modified 6×myc-BCL10 construct (A and B) (d-BCL10; see Materials and Methods for details) or WT-BCL10 (B). WCLs were prepared and blotted for BCL10 and E3 expression (anti-Flag) (left of blots). Unphosphorylated (p0) and phosphorylated BCL10 bands (p1 and p2) are indicated by arrowheads. (A) Right, densitometry of phosphorylated (p1 + p2) relative to unphosphorylated (p0) BCL10 levels. **, P > 0.01, unpaired Student's t test comparing p-BCL10 levels in ITCH-expressing cells to those in vector-, cIAP2-, NEDD4-, Cbl-b-, and βTrCP-expressing cells. (B) Cells were incubated with CHX for 0 to 6 h and analyzed for BCL10 and Flag-ITCH expression. Quantification of p0 or p1 levels were normalized relative to that of Flag (shown below the BCL10 blots, zero time point = 1). (C) Left, levels of BCL10 and ERK in 293T cells transfected with myc-tagged WT or serine 138 to alanine (S138A)-mutated BCL10. Right, Jurkat T cells stably expressing myc-BCL10 or myc-BCL10-S138A were stimulated with CD3/CD28 for 0 to 3 h and analyzed for the indicated proteins (left of blots). Bottom, BCL10 levels relative to that of actin (means ± SEM from three independent experiments, zero time point = 1).
Delayed BCL10 degradation in ITCH-deficient T lymphocytes.
Because ITCH preferentially increased BCL10 polyUb and degradation and interacted with TAK1 in 293T cells, we next tested the requirement for ITCH in BCL10 turnover in total splenocytes or purified T and B cells derived from WT versus ITCH−/− mice (Fig. 6A). Consistent with our results in 293T cells (Fig. 5), we observed increased phospho-BCL10 levels in ITCH−/− primary lymphocytes (Fig. 6A, overexposed blots). Quantification of BCL10 protein in stimulated WT versus ITCH−/− lymphocytes revealed that BCL10 degradation was significantly delayed in ITCH−/− total splenocytes and in splenic T cells but not in B cells (Fig. 6A, bottom). The partial delay in BCL10 degradation did not appreciably impact downstream signaling pathways, as IκBα degradation remained intact, suggesting that other E3s, perhaps including NEDD4, might have redundant roles in BCL10 turnover. To test this idea, endogenous ITCH, NEDD4, or both were knocked down using siRNAs in EL4 T cells (Fig. 6B). Following P/I stimulation, siRNAs for ITCH and NEDD4 alone or together delayed BCL10 degradation. Quantification of BCL10 from multiple experiments showed that ITCH deletion delayed BCL10 degradation more efficiently than NEDD4 deletion at 30 to 60 min poststimulation (Fig. 6B, bottom left). In agreement with our data from 293T cells (Fig. 4 and 5), the deletion of both E3s did not further delay the process, indicating a less predominant role for NEDD4 in this T cell line (Fig. 6B, bottom left). Similar to the results in ITCH−/− B cells (Fig. 6A), ITCH and NEDD4 knockdown in the murine A20 B cell line showed no impact on BCL10 degradation (Fig. 6C). We conclude that ITCH and, to a lesser extent, NEDD4 have roles in BCL10 degradation in T cells but not B cells and that it is likely that additional E3s signal the turnover of BCL10 in lymphocytes.
Fig 6.

Itch regulates BCL10 degradation in primary T but not B cells. (A) Splenocytes or purified splenic B and T cells derived from WT or ITCH−/− mice were stimulated with P/I for 0 to 180 min and immunoblotted with the antibodies indicated on the left. Bottom, levels of BCL10 (combined signal intensities of unmodified and phosphorylated BCL10/ERK) (zero time points = 1). *, P > 0.05, unpaired Student's t test comparing ITCH−/− versus WT cells. Inset blot, expression of ITCH and actin in splenocytes of WT and ITCH−/− mice. Murine EL4 T cells (B) and A20 B cells (C) were transfected with the indicated siRNAs for 24 h, stimulated with P/I, and analyzed for the proteins indicated on the left. (B) Left bottom, the relative levels of BCL10 (zero time point = 1). **, P > 0.01, and *, P > 0.05, unpaired Student's t test comparing time points in ITCH or NEDD4 siRNA-transfected cells versus those transfected with control siRNA. (B and C) siRNA knockdown of ITCH and NEDD4 protein intensities are shown beneath the blots and were determined relative to that of ERK.
cIAPs are not required for BCL10 degradation in B cells.
Previous reports indicated that ubiquitination and degradation of BCL10 were partially controlled by the RING family E3 ubiquitin ligase cIAP2 in T cells (12, 52); however, its role in B cells was unknown. Because ITCH and NEDD4 did not participate in BCL10 degradation in B cells, we evaluated the role of cIAP1/2 in BCL10 degradation in B cell lines using a specific cIAP1/2 antagonist, BV6 (53), that depletes the pool of endogenous cIAP1/2 (Fig. 7). BCL10 degradation was unchanged by cIAP1/2 depletion in either DT40 B cells stimulated with P/I (Fig. 7A) or CHX-treated Ramos B cells expressing CARMA1-ΔPRD (Fig. 7B), indicating that cIAP1 and cIAP2 are also dispensable for BCL10 degradation in B cell lines.
Fig 7.
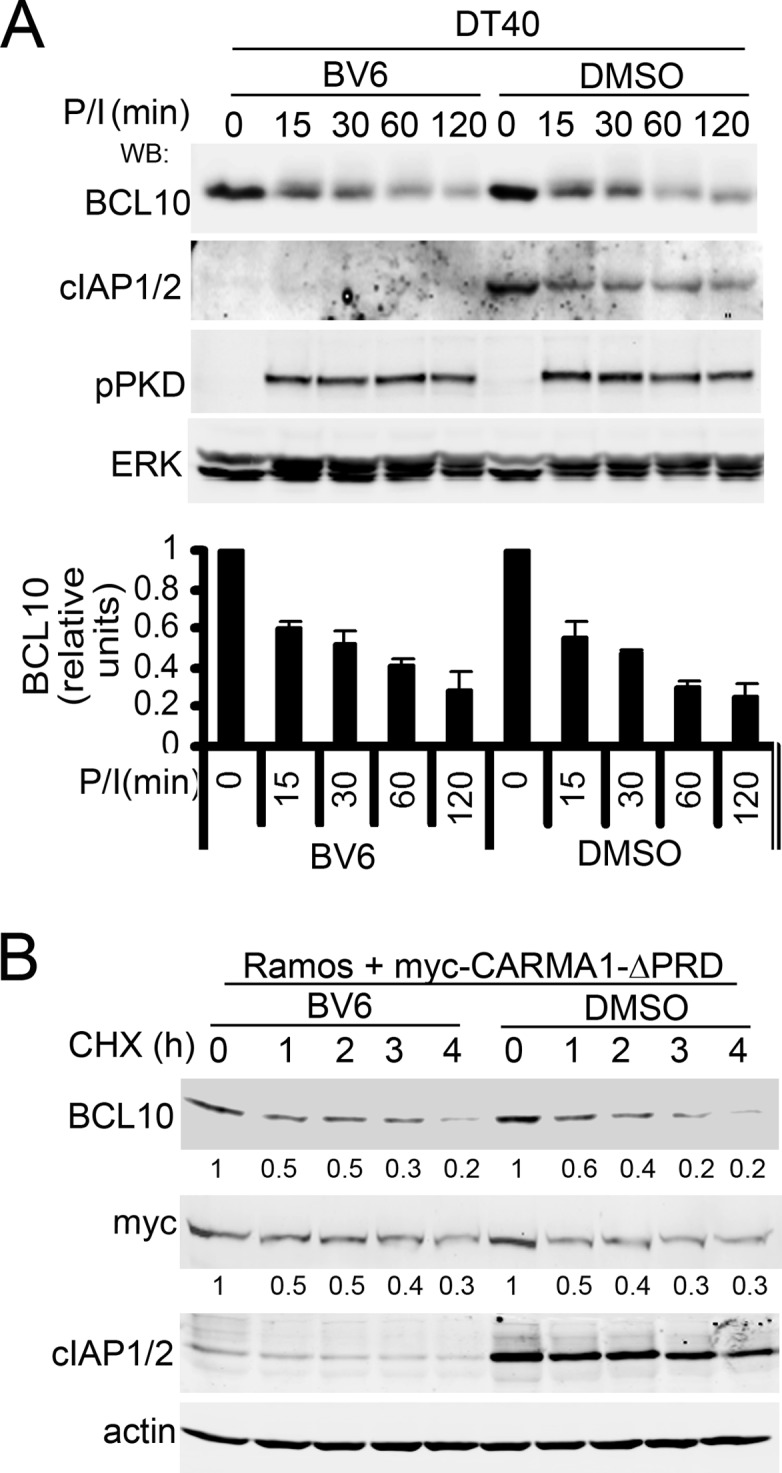
CARMA1-dependent BCL10 degradation in B cells does not require cIAP1/2 expression. WT DT40 cells (A) or Ramos cells expressing myc-CARMA1-ΔPRD (B) were pretreated with the cIAP1/2 antagonist BV6 or vehicle (DMSO) for 1 h at 37°C and were incubated with P/I and CHX (A) or just CHX (B) for the indicated times. WCLs were immunoblotted with the antibodies indicated on the left. Signal intensities of BCL10 or CARMA1 relative to ERK (A) or actin (B) are shown beneath the blots (zero time points = 1).
Overexpression of TAK1 negatively impacts CARMA1-dependent NF-κB activation and reduces BCL10 expression in 293T cells.
Our data indicate that TAK1 promotes BCL10 polyUb and degradation downstream of CARMA1 and suggest that it provides a negative feedback loop during NF-κB activation. To test this hypothesis, we evaluated the effect of TAK1 overexpression on NF-κB-dependent reporter gene assays induced by the constitutively active CARMA1-ΔPRD in 293T cells. In parallel, we compared TAK1 with other serine/threonine kinases, including PKD2 and IKKβ (Fig. 8A). Notably, TAK1 coexpression with CARMA1-ΔPRD or CARMA1-L232LI (a murine version of the L225LI mutation identified in an ABC-DLBCL) (33), resulted in ∼3-fold inhibition of CARMA1-dependent NF-κB activation. Altered CARMA1 expression or enhanced NF-κB consumption was unlikely to account for this change; CARMA1 protein levels were comparable with or without TAK1 coexpression (Fig. 8A and B, immunoblots), and coexpression of PKD2 or IKKβ did not diminish NF-κB activation (Fig. 8A).
Fig 8.

TAK1 overexpression downregulates CARMA1-dependent NF-κB activity and reduces BCL10 levels. (A) 293T cells were transfected for 48 h with Igκ2-IFN-luciferase (NF-κB reporter) and pRL-TK (transfection control) and combinations of empty vector (−) or the indicated expression vectors (labels at the bottom of the graphs). Luciferase activity was determined as described in Materials and Methods. Bottom, WCLs were analyzed by immunoblotting for the proteins indicated to the left or right of the blots. (B) 293T cells were cotransfected for 48 h with empty vector (−) or the indicated expression vectors (top of blots) and were immunoblotted for BCL10, CARMA1, and TAK1 (left). Quantifications, carried out as described in Materials and Methods, are shown below the BCL10 blots. **, P > 0.01; *, P > 0.05; ns, not significant, unpaired Student's t test.
To determine whether TAK1 induction of BCL10 polyUb was responsible for this reduction in NF-κB activation, CARMA1-dependent NF-κB activation was assessed in the presence of the WT or AAYA forms of TAK1 (Fig. 8A, right). Cells expressing WT TAK1 inhibited NF-κB activity twice as efficiently as TAK1-AAYA (Fig. 8A, compare bars 3 and 5); TAK1-AAYA had a nonsignificant tendency to inhibit NF-κB activation relative to cells expressing CARMA1-ΔPRD alone. These data are consistent with a model of TAK1 that, although essential for NF-κB activation downstream of AR activation, also drives activation-induced BCL10 polyUb and proteolysis to downmodulate NF-κB activity. Indeed, analysis of BCL10 expression showed that CARMA1-ΔPRD coexpression with TAK1, but not TAK1-AAYA, reduced BCL10 levels ∼2- to 3-fold relative to cells expressing CARMA1-ΔPRD alone (Fig. 8B).
TAK1 induces counterselection of DLBCL lines by promoting both BCL10 degradation and JNK activation.
The survival of ABC-DLBCLs requires constitutive NF-κB activation, while the germ center B (GCB)-DLBCL subtype is less dependent on this pathway for survival (54). Based on our findings that TAK1 expression downmodulates NF-κB activity downstream of CARMA1, we hypothesized that TAK1 overexpression would impact the survival of NF-κB-addicted ABC-DLBCL subtypes. Quantification of NF-κB GLuc activity in ABC-DLBCL (OCI-Ly10) and GCB-DLBCL (OCI-Ly7) lines (Fig. 9A) confirmed the predominant activity of NF-κB in OCI-Ly10 relative to OCI-Ly7 cells (∼75% higher). We then compared the effect of TAK1 overexpression in these cell lines by stable transduction with LVs expressing TAK1-WT or TAK1-AAYA, monitored by bicistronic expression of GFP. As a negative control, cells were also transduced with LVs to express GFP alone (vector) (Fig. 9B and C). We found that cells transduced with TAK1 lost GFP expression over time in culture, likely due to counterselection against TAK1 expression (Fig. 9B). In order to quantify this effect, we evaluated the frequencies of TAK1-expressing cells (using GFP as a surrogate, GFP+) and cell death by DAPI incorporation in transduced cells using flow cytometry (Fig. 9B and C). We observed that while WT TAK1 did not affect the frequency of GFP+ OCI-Ly7 cells, it greatly reduced GFP-expressing OCI-Ly10 cells over a 9-day time course (Fig. 9B). To determine whether this counterselection was due to cell death, we analyzed DAPI incorporation in GFP-expressing cells at early (day 3) and late (day 9) time points posttransduction (Fig. 9C). While TAK1 promoted robust cell death in OCI-Ly10 cells, OCI-Ly7 cells were ∼4-fold less affected at day 3. By day 9, a small but significant fraction of TAK1-expressing OCI-Ly10 cells still exhibited higher levels of cell death (3-fold over vector). TAK1-AAYA-expressing cells paralleled the vector-only control (Fig. 9B and C).
Fig 9.

TAK1 overexpression promotes cell death and counterselection of DLBCL lines. (A) The ABC-DLBCL line OCI-Ly10 and the GCB-DLBCL line OCI-Ly7 were transduced to express the secreted NF-κB-dependent GLuc reporter. Cells were analyzed for GLuc activity in culture supernatants after 0 and 4 h. −, media only. (B and C) OCI-Ly10 and OCI-Ly7 cells were transduced with LVs (MOI, 27) expressing either HA-TAK1 2A-GFP, HA-TAK1-AAYA 2A-GFP, or GFP only (vector). (B) To assess for counterselection, the GFP+ fraction at each time point is compared to GFP marking at day 1 (set as 100%). (C) Cell death was analyzed at days 3 and 9 posttransduction by DAPI incorporation into GFP+ cells and represented as fold difference over vector (set as 1). Graphs show means ± SEM from six (A) or three (B and C) independent experiments. **, P > 0.01; *, P > 0.05; ns, not significant, unpaired Student's t test. (D) OCI-Ly7 cells were transduced with LVs expressing TAK1 2A-GFP, TAK1-AAYA 2A-GFP, or GFP only (vector). GFP+ cells were sorted at 48 h. Left, GFP expression in untransduced cells or cells expressing TAK1, TAK1-AAYA, or GFP only (vector). Right, cells were incubated with 140 μM CHX for 0 to 6 h, and BCL10, TAK1, and actin levels were analyzed by immunoblotting. BCL10 levels, relative to that of actin, are shown below the blots.
To test whether these TAK1 effects were associated with enhanced BCL10 turnover, OCI-Ly7 cells were transduced with LVs expressing TAK1-WT or TAK1-AAYA and cis-linked GFP or GFP alone for 48 h, sorted for GFP expression, and incubated with CHX for 0 to 6 h (Fig. 9D). Note that this experiment could not be carried out using OCI-Ly10 cells, which were rapidly counterselected as early as 24 h posttransduction. OCI-Ly7 cells expressing TAK1-WT showed increased degradation of BCL10 relative to cells expressing either TAK1-AAYA or GFP alone (Fig. 9D, right), implying that TAK1 (interacting with TAB1) induces degradation of BCL10.
Because BCL10 turnover occurs independently of TAK1 kinase activity in B and T cell lines (Fig. 2), we predicted that TAK1 would mediate kinase-independent BCL10 degradation in DLBCLs (Fig. 10). Consistent with this prediction, equivalent P/I-induced BCL10 degradation occurred with and without blockade of TAK1 activity (Fig. 10A). Interestingly, and contrary to our initial prediction, overexpression of the kinase-inactive TAK1 did not promote cell death or counterselection of OCI-Ly10 cells (data not shown), suggesting that a TAK1-kinase dependent pathway contributes to efficient counterselection in this ABC-DLBCL line. Because AR-stimulated TAK1 also mediates JNK signaling, and JNK activation has been linked to cell death of AR-stimulated B-lymphoma lines (55, 56), we hypothesized that enhanced JNK signaling downstream of overexpressed TAK1 might help to explain our observations. Indeed, transduction with TAK1-WT, but not TAK1-AAYA or vector alone, resulted in substantial JNK phosphorylation in OCI-Ly10 cells (Fig. 10B). To test the role for JNK in counterselection against TAK1 overexpression, OCI-Ly10 cells transduced with LVs expressing TAK1 or GFP alone were treated with a JNK inhibitor or a vehicle (dimethyl sulfoxide [DMSO]) (Fig. 10C). While JNK inhibition did not impact the proportion of GFP+ vector-transduced cells, the inhibitor increased the proportion of GFP+ TAK1-expressing cells (∼2-fold), implying that, in the absence of pharmacologic blockade, TAK1-driven JNK activation promotes counterselection.
Fig 10.

The kinase activity of TAK1 is dispensable for BCL10 degradation but required for counterselection of OCI-Ly10 cells. (A) OCI-Ly10 cells were pretreated with TAK1 inhibitor (20 μM) and then stimulated with P/I in the presence of CHX for the indicated times. Cells were lysed, and BCL10, pJNK, actin, and ERK were analyzed by immunoblotting. (B) Cells were transduced with LVs expressing TAK1 2A-GFP, TAK1-AAYA 2A-GFP, or GFP alone (vector). GFP+ cells were purified and immunoblotted for pJNK, HA-tagged TAK1, and actin. (C) Cells were preincubated for 30 min with the indicated doses of JNK inhibitor or DMSO and transduced with LVs expressing TAK1 2A-GFP or GFP only (vector). The proportion of GFP+ cells at day 2 posttransduction is displayed as the fold difference relative to that of DMSO-treated cells. Data are means ± SEM from 4 independent experiments. *, P > 0.05. (D) Cells were transduced with LV-expressing BCL10 2A-mCherry or mCherry (vector) alone, and mCherry+ cells were sorted. Left, mCherry levels in vector only (gray), myc-BCL10 (black), and untransduced (white) cells; immunoblots show myc-BCL10 and ERK expression. mCherry+ cells were treated with JNK inhibitor (at the indicated doses) or DMSO and concurrently transduced with LVs expressing TAK1 2A-GFP. The proportion of GFP+ cells at day 2 is displayed as described for panel C.
We then sought to determine if the counterselection against TAK1-expressing OCI-Ly10 cells was due to downstream activation of JNK alone or if a TAK1 influence on BCL10 protein expression also played a role. To do so, we evaluated whether TAK1-mediated positive selection in the presence of JNK inhibition was potentiated by increasing the expression of BCL10 (Fig. 10D). After OCI-Ly10 cells overexpressing BCL10 or vector alone (vector-mCherry) had been generated, cells were transduced with LVs expressing TAK1-WT 2A-GFP, and the proportions of GFP+ cells in the presence or absence of JNK inhibition were evaluated (Fig. 10D). Cells coexpressing BCL10 and TAK1 increased nearly 3-fold in the setting of JNK inhibition compared with an ∼1.9-fold increase in the absence of BCL10 coexpression (mCherry only; an impact equivalent to the TAK1 expression on non-mCherry-transduced OCI-Ly10) (Fig. 10C). These results imply that counterselection against ABC-DLBCL cells expressing TAK1 occurs via both enhanced BCL10 turnover and JNK activation.
DISCUSSION
TAK1 has an essential role in AR-dependent activation of the NF-κB and JNK signaling pathways in B and T cells (7, 9, 57, 58). However, in this study, we provide evidence for an additional role for TAK1 in this pathway as a regulator of its feedback inhibition. We utilized genetic knockout in DT40 B cell lines to demonstrate that CARMA1 and PKCβ (upstream proteins proximal to BCL10) were both required for BCL10 degradation, suggesting that a downstream signaling component mediates this process. Interestingly, deletion of TAK1 (a kinase that phosphorylates IKKβ downstream of BCL10) but not of IKKβ abolished BCL10 degradation in DT40 B cells (Fig. 1). Although this finding agrees with studies showing that IKKβ inhibition in T cells (20) or deletion in B cells (14) does not impact BCL10 stability, it contrasts with other studies in Jurkat T cells showing that IKKβ phosphorylates BCL10, marking it for subsequent ubiquitination and degradation (13, 15). TAK1 is, thus far, the most distal protein in the AR/NF-κB signaling cascade involved in this particular feedback inhibition mechanism in B cells.
Surprisingly, we found that the role for TAK1 in BCL10 degradation was independent of its kinase activity (Fig. 2). This finding led us to investigate the hypothesis that TAK1 is an adaptor for other protein(s) that mediate BCL10 degradation. We first tested whether TAK1 is an adaptor for any of the E3s with previously reported roles in BCL10 ubiquitination (12–14) (Fig. 4). Using high-stringency cell lysis conditions in 293T cells, we found that TAK1 (but not BCL10 or CARMA1) preferentially interacted with ITCH and less significantly with NEDD4 (Fig. 4A to C). This NEDD4 family of HECT E3 ligases was previously shown to induce BCL10 polyUb and degradation in 293T and Jurkat T cells (14).
Genetic analysis of BCL10 degradation using ITCH−/− splenic lymphocytes or siRNA knockdown of E3s revealed a partial requirement for ITCH and NEDD4 in primary T cells but not in B cells (Fig. 6). In both T lymphocytes and the EL4 T cell line, a loss of ITCH increased the stability of BCL10 (Fig. 6A and B). NEDD4 depletion using siRNA also enhanced the stability of BCL10 in EL4 T cells, although it did so less efficiently than ITCH. However, combined depletion of ITCH and NEDD4 at levels achieved using siRNA did not further enhance BCL10 stability in these cells (Fig. 6B). Furthermore, our observation that NF-κB activation, measured as IκBα degradation, was comparable in WT and ITCH−/− lymphocytes suggests that additional E3s must participate to control BCL10 degradation and repress NF-κB activation in primary T lymphocytes. Interestingly, B lymphocytes were insensitive to ITCH, NEDD4, or cIAP1/2 depletion, indicating that as-yet-unidentified E3s regulate BCL10 degradation in B cells (Fig. 6 and 7).
Earlier work has implicated BCL10 site-specific phosphorylation by various enzymes as a key degradation-inducing trigger (13, 15, 17). Other work has challenged some of these conclusions (20, 22). We report here that in B cells, neither TAK1 nor IKKβ kinase activities are required for BCL10 turnover (Fig. 1 and 2). Notably, we observed that coexpression of ITCH depleted the phospho-BCL10 forms (Fig. 5 and 6) previously suggested to comprise its main degradation targets in 293T cells (12). However, we also found that nonphosphorylated BCL10 was normally degraded in CHX-treated 293T cells and AR-stimulated Jurkat cells (Fig. 5), suggesting that BCL10 turnover is independent of its phosphorylation status. This is consistent with reports showing that BCL10 turnover does not require phosphorylation (20, 22). Thus, while distinct phosphorylation sites and/or kinases might redundantly and/or additively participate in this process in other cell types, our findings in 293T and T cells show that TAK1 efficiently targets ITCH or another E3(s) to both phospho-modified and unphosphorylated BCL10, thereby orchestrating its turnover.
Both TAB1 and TAB2/3 are interacting partners of TAK1, and blocking TAB1/TAK1 interaction blocks spontaneous NF-κB activation in 293T cells (Fig. 3B). However, previous work has suggested that TAB1 is not required by TAK1 in AR-driven activation of NF-κB in B cells (8). Instead, our results imply that TAB1 has a role downmodulating this pathway (Fig. 3F and G). TAB1 has been postulated to affect the conformation of TAK1 (45, 59); thus, we hypothesize that TAB1 might control BCL10 polyUb by bringing into proximity its TAK1 binding partner and a TAK1-associated E3 complex. This hypothesis is supported by our results showing that TAB1 knockdown delayed polyUb and degradation of BCL10 and increased AR-dependent NF-κB activation in fibroblasts and Jurkat T cells (Fig. 3D and F) and by our finding that primary B cells expressing a mutant form of TAB1, incapable of interacting with TAK1, exhibited delayed P/I-dependent BCL10 turnover and increased P/I-dependent JNK activation (Fig. 3G). These combined results are also consistent with a previous study (60) implying a negative regulatory role for this adaptor in lymphocytes.
In concert with these ideas, we provide data that implicate TAK1 in negative modulation of NF-κB signals downstream of the AR and CARMA1. Perturbation of the balance between NF-κB activation and inhibition was also achieved by TAK1 overexpression, which resulted in a reduction of CARMA1-dependent NF-κB activation, in parallel with enhanced CARMA1-dependent induction of BCL10 turnover in 293T cells (Fig. 8). These effects required an intact TAB1 binding motif, as the TAK1-AAYA mutant did not reduce NF-κB activation or enhance BCL10 turnover. Similarly, we demonstrated that increased expression of TAK1 but not the TAB1 binding-deficient mutant TAK-AAYA reduced the survival of the ABC and GCB subtypes of DLBCLs and enhanced BCL10 degradation in GCB-DLBCLs (an experiment that could not be performed using the ABC subtype cells due to their rapid selection against TAK1 expression), implying that increased expression of TAK1 can promote BCL10 degradation and cell death (Fig. 9) (61). The limited effect of TAK1 observed in the GCB-DLBCL subtype (OCI-Ly7) is likely due to the ability of these cells to survive independently of NF-κB signals (54, 62). In contrast, the ABC-DLBCL cells (OCI-Ly10), which depend on NF-κB for survival, are susceptible to increased dosages of TAK1 (Fig. 9).
Interestingly, our data indicate that the counterselection against TAK1-expressing OCI-Ly10 cells occurs via two downstream signals of TAK1: increased JNK activation and BCL10 turnover. While the kinase activity of TAK1 was not required to promote BCL10 degradation (Fig. 10A), its enzymatic function was important for robust negative selection. TAK1 overexpression promoted JNK phosphorylation and pharmacologic inhibition of JNK-reversed counterselection in this DLBCL model (Fig. 10). Indeed, counterselection of overexpressed TAK1 was converted to positive selection in the setting of JNK inhibition, and this process was further augmented by increased BCL10 expression (Fig. 10B to D). These observations suggest that while basal JNK signals might be important for growth of B cell lymphomas, including the ABC- and GCB-DLBCL lines (63), increased activation of JNK, induced by TAK1 overexpression, achieves a threshold that promotes proapoptotic signals. Taken together, our data suggest that altering TAK1 dosage modulates survival of ABC-DLBCLs by dual proapoptotic signals, including kinase-dependent JNK activation and kinase-independent degradation of BCL10.
In summary, we propose a working model for the role of TAK1 in controlling the CBM signaling cascade (Fig. 11). AR-dependent proximal signals lead to the activation of PKC isoforms, which leads to direct phosphorylation of CARMA1 (26, 64), and this drives conformational changes required for the recruitment of BCL10 and MALT1 (3, 5). The CBM complex then recruits and activates the TAK1/TAB2/TAB3 complex, which, in turn, phosphorylates both IKKβ and MKK family members (likely MKK4/7) to initiate NF-κB and AP1 transcriptional activation, respectively. In parallel, TAK1 (specifically in association with TAB1) modulates the extent of cell activation by recruiting E3 ligases (including ITCH in T cells), shuttling them into close proximity for BCL10 ubiquitination, a process not restricted to but likely facilitated by site-specific BCL10 phosphorylation. As a whole, our data show for the first time that TAK1 exerts negative regulatory feedback in lymphocyte signaling as it adapts the machinery necessary to polyUb and degrade BCL10, downregulating CBM activity.
Fig 11.

Working model of BCL10 ubiquitination and degradation by TAK1. (A) AR or PMA signals result in activation of PKC isoforms (β in B cells or θ in T cells). PKCs then phosphorylate the PKC-regulated domain (PRD) of CARMA1, causing conformational changes that allow its interaction with BCL10/MALT1, forming the CBM complex. The CBM complex recruits several intermediate molecules, including TRAF2/6, and through a cascade of K63-polyUb and phosphorylation, the TAK1/TAB2/3 complex is recruited and activated. TAK1 phosphorylates IKKβ and MKKs leading to the activation of NF-κB and JNK, respectively. (B) Concurrently, TAK1 also downmodulates CBM activity by interacting specifically with TAB1 and recruiting E3 ubiquitin ligases that function to ubiquitinate and target BCL10 for degradation (right).
ACKNOWLEDGMENTS
We thank Marc Schwartz for technical assistance with flow cytometry studies, Socheath Khim for assistance with animal studies, and the other members of the Rawlings lab for helpful discussions.
We have no conflicting financial interest.
Support for this work has included NIH grants HD037091 and AI071163 (to D.J.R.).
Footnotes
Published ahead of print 7 January 2013
REFERENCES
- 1. Gerondakis S, Siebenlist U. 2010. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2: a000182 doi:10.1101/cshperspect.a000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Staudt LM. 2010. Oncogenic activation of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2: a000109 doi:10.1101/cshperspect.a000109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rawlings DJ, Sommer K, Moreno-Garcia ME. 2006. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat. Rev. Immunol. 6: 799–812 [DOI] [PubMed] [Google Scholar]
- 4. Schaefer BC, Kappler JW, Kupfer A, Marrack P. 2004. Complex and dynamic redistribution of NF-kappaB signaling intermediates in response to T cell receptor stimulation. Proc. Natl. Acad. Sci. U. S. A. 101: 1004–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thome M. 2004. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat. Rev. Immunol. 4: 348–359 [DOI] [PubMed] [Google Scholar]
- 6. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412: 346–351 [DOI] [PubMed] [Google Scholar]
- 7. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. 2005. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 6: 1087–1095 [DOI] [PubMed] [Google Scholar]
- 8. Shinohara H, Kurosaki T. 2009. Comprehending the complex connection between PKCbeta, TAK1, and IKK in BCR signaling. Immunol. Rev. 232: 300–318 [DOI] [PubMed] [Google Scholar]
- 9. Shinohara H, Yasuda T, Aiba Y, Sanjo H, Hamadate M, Watarai H, Sakurai H, Kurosaki T. 2005. PKC beta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J. Exp. Med. 202: 1423–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. 2004. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14: 289–301 [DOI] [PubMed] [Google Scholar]
- 11. Moreno-Garcia ME, Sommer K, Haftmann C, Sontheimer C, Andrews SF, Rawlings DJ. 2009. Serine 649 phosphorylation within the protein kinase C-regulated domain down-regulates CARMA1 activity in lymphocytes. J. Immunol. 183: 7362–7370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu S, Du MQ, Park SM, Alcivar A, Qu L, Gupta S, Tang J, Baens M, Ye H, Lee TH, Marynen P, Riley JL, Yang X. 2006. cIAP2 is a ubiquitin protein ligase for BCL10 and is dysregulated in mucosa-associated lymphoid tissue lymphomas. J. Clin. Invest. 116: 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lobry C, Lopez T, Israel A, Weil R. 2007. Negative feedback loop in T cell activation through IkappaB kinase-induced phosphorylation and degradation of Bcl10. Proc. Natl. Acad. Sci. U. S. A. 104: 908–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scharschmidt E, Wegener E, Heissmeyer V, Rao A, Krappmann D. 2004. Degradation of Bcl10 induced by T-cell activation negatively regulates NF-kappa B signaling. Mol. Cell. Biol. 24: 3860–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeng H, Di L, Fu G, Chen Y, Gao X, Xu L, Lin X, Wen R. 2007. Phosphorylation of Bcl10 negatively regulates T-cell receptor-mediated NF-kappaB activation. Mol. Cell. Biol. 27: 5235–5245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. 2012. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity 36: 947–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishiguro K, Ando T, Goto H, Xavier R. 2007. Bcl10 is phosphorylated on Ser138 by Ca2+/calmodulin-dependent protein kinase II. Mol. Immunol. 44: 2095–2100 [DOI] [PubMed] [Google Scholar]
- 18. Oruganti SR, Edin S, Grundstrom C, Grundstrom T. 2010. CaMKII targets Bcl10 in T-cell receptor induced activation of NF-kappaB. Mol. Immunol. 48: 1448–1460 [DOI] [PubMed] [Google Scholar]
- 19. Ruefli-Brasse AA, Lee WP, Hurst S, Dixit VM. 2004. Rip2 participates in Bcl10 signaling and T-cell receptor-mediated NF-kappaB activation. J. Biol. Chem. 279: 1570–1574 [DOI] [PubMed] [Google Scholar]
- 20. Wegener E, Oeckinghaus A, Papadopoulou N, Lavitas L, Schmidt-Supprian M, Ferch U, Mak TW, Ruland J, Heissmeyer V, Krappmann D. 2006. Essential role for IkappaB kinase beta in remodeling Carma1-Bcl10-Malt1 complexes upon T cell activation. Mol. Cell 23: 13–23 [DOI] [PubMed] [Google Scholar]
- 21. Wu CJ, Ashwell JD. 2008. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. U. S. A. 105: 3023–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rueda D, Gaide O, Ho L, Lewkowicz E, Niedergang F, Hailfinger S, Rebeaud F, Guzzardi M, Conne B, Thelen M, Delon J, Ferch U, Mak TW, Ruland J, Schwaller J, Thome M. 2007. Bcl10 controls TCR- and FcgammaR-induced actin polymerization. J. Immunol. 178: 4373–4384 [DOI] [PubMed] [Google Scholar]
- 23. Matesic LE, Haines DC, Copeland NG, Jenkins NA. 2006. Itch genetically interacts with Notch1 in a mouse autoimmune disease model. Hum. Mol. Genet. 15: 3485–3497 [DOI] [PubMed] [Google Scholar]
- 24. Inagaki M, Komatsu Y, Scott G, Yamada G, Ray M, Ninomiya-Tsuji J, Mishina Y. 2008. Generation of a conditional mutant allele for Tab1 in mouse. Genesis 46: 431–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moreno-Garcia ME, Sommer K, Shinohara H, Bandaranayake AD, Kurosaki T, Rawlings DJ. 2010. MAGUK-controlled ubiquitination of CARMA1 modulates lymphocyte NF-kappaB activity. Mol. Cell. Biol. 30: 922–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, Rawlings DJ. 2005. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 23: 561–574 [DOI] [PubMed] [Google Scholar]
- 27. Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. 1997. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278: 860–866 [DOI] [PubMed] [Google Scholar]
- 28. Gaide O, Favier B, Legler DF, Bonnet D, Brissoni B, Valitutti S, Bron C, Tschopp J, Thome M. 2002. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat. Immunol. 3: 836–843 [DOI] [PubMed] [Google Scholar]
- 29. Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. 1999. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400: 687–693 [DOI] [PubMed] [Google Scholar]
- 30. Kovalevska LM, Yurchenko OV, Shlapatska LM, Berdova GG, Mikhalap SV, Van Lint J, Sidorenko SP. 2006. Immunohistochemical studies of protein kinase D (PKD) 2 expression in malignant human lymphomas. Exp. Oncol. 28: 225–230 [PubMed] [Google Scholar]
- 31. Besse A, Lamothe B, Campos AD, Webster WK, Maddineni U, Lin SC, Wu H, Darnay BG. 2007. TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J. Biol. Chem. 282: 3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pomerantz JL, Denny EM, Baltimore D. 2002. CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. EMBO J. 21: 5184–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Chan WC, Staudt LM. 2008. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319: 1676–1679 [DOI] [PubMed] [Google Scholar]
- 34. Sather BD, Ryu BY, Stirling BV, Garibov M, Kerns HM, Humblet-Baron S, Astrakhan A, Rawlings DJ. 2011. Development of B-lineage predominant lentiviral vectors for use in genetic therapies for B cell disorders. Mol. Ther. 19: 515–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kappes JC, Wu X, Wakefield JK. 2003. Production of trans-lentiviral vector with predictable safety. Methods Mol. Med. 76: 449–465 [DOI] [PubMed] [Google Scholar]
- 36. Tannous BA. 2009. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc. 4: 582–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F. 2002. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc. Natl. Acad. Sci. U. S. A. 99: 4489–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shinohara H, Maeda S, Watarai H, Kurosaki T. 2007. IkappaB kinase beta-induced phosphorylation of CARMA1 contributes to CARMA1 Bcl10 MALT1 complex formation in B cells. J. Exp. Med. 204: 3285–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. 1995. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270: 2008–2011 [DOI] [PubMed] [Google Scholar]
- 40. Costanzo A, Guiet C, Vito P. 1999. c-E10 is a caspase-recruiting domain-containing protein that interacts with components of death receptors signaling pathway and activates nuclear factor-kappaB. J. Biol. Chem. 274: 20127–20132 [DOI] [PubMed] [Google Scholar]
- 41. Koseki T, Inohara N, Chen S, Carrio R, Merino J, Hottiger MO, Nabel GJ, Nunez G. 1999. CIPER, a novel NF kappaB-activating protein containing a caspase recruitment domain with homology to Herpesvirus-2 protein E10. J. Biol. Chem. 274: 9955–9961 [DOI] [PubMed] [Google Scholar]
- 42. Cheung PC, Nebreda AR, Cohen P. 2004. TAB3, a new binding partner of the protein kinase TAK1. Biochem. J. 378: 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. 1996. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 272: 1179–1182 [DOI] [PubMed] [Google Scholar]
- 44. Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. 2000. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell 5: 649–658 [DOI] [PubMed] [Google Scholar]
- 45. Brown K, Vial SC, Dedi N, Long JM, Dunster NJ, Cheetham GM. 2005. Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. J. Mol. Biol. 354: 1013–1020 [DOI] [PubMed] [Google Scholar]
- 46. Bernassola F, Karin M, Ciechanover A, Melino G. 2008. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14: 10–21 [DOI] [PubMed] [Google Scholar]
- 47. Rotin D, Kumar S. 2009. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 10: 398–409 [DOI] [PubMed] [Google Scholar]
- 48. Dhananjayan SC, Ramamoorthy S, Khan OY, Ismail A, Sun J, Slingerland J, O'Malley BW, Nawaz Z. 2006. WW domain binding protein-2, an E6-associated protein interacting protein, acts as a coactivator of estrogen and progesterone receptors. Mol. Endocrinol. 20: 2343–2354 [DOI] [PubMed] [Google Scholar]
- 49. Nabhan JF, Pan H, Lu Q. 2010. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the beta2-adrenergic receptor. EMBO Rep. 11: 605–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sakurai H, Miyoshi H, Mizukami J, Sugita T. 2000. Phosphorylation-dependent activation of TAK1 mitogen-activated protein kinase kinase kinase by TAB1. FEBS Lett. 474: 141–145 [DOI] [PubMed] [Google Scholar]
- 51. Sakurai H, Miyoshi H, Toriumi W, Sugita T. 1999. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. J. Biol. Chem. 274: 10641–10648 [DOI] [PubMed] [Google Scholar]
- 52. Hu S, Alcivar A, Qu L, Tang J, Yang X. 2006. CIAP2 inhibits anigen receptor signaling by targeting Bcl10 for degredation. Cell Cycle 5: 1438–1442 [DOI] [PubMed] [Google Scholar]
- 53. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. 2007. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 131: 669–681 [DOI] [PubMed] [Google Scholar]
- 54. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. 2006. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441: 106–110 [DOI] [PubMed] [Google Scholar]
- 55. Takada E, Hata K, Mizuguchi J. 2006. Requirement for JNK-dependent upregulation of BimL in anti-IgM-induced apoptosis in murine B lymphoma cell lines WEHI-231 and CH31. Exp. Cell Res. 312: 3728–3738 [DOI] [PubMed] [Google Scholar]
- 56. Takada E, Toyota H, Suzuki J, Mizuguchi J. 2001. Prevention of anti-IgM-induced apoptosis accompanying G1 arrest in B lymphoma cells overexpressing dominant-negative mutant form of c-Jun N-terminal kinase 1. J. Immunol. 166: 1641–1649 [DOI] [PubMed] [Google Scholar]
- 57. Liu HH, Xie M, Schneider MD, Chen ZJ. 2006. Essential role of TAK1 in thymocyte development and activation. Proc. Natl. Acad. Sci. U. S. A. 103: 11677–11682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. 2006. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 7: 851–858 [DOI] [PubMed] [Google Scholar]
- 59. Conner SH, Kular G, Peggie M, Shepherd S, Schuttelkopf AW, Cohen P, Van Aalten DM. 2006. TAK1-binding protein 1 is a pseudophosphatase. Biochem. J. 399: 427–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ohkusu-Tsukada K, Tominaga N, Udono H, Yui K. 2004. Regulation of the maintenance of peripheral T-cell anergy by TAB1-mediated p38 alpha activation. Mol. Cell. Biol. 24: 6957–6966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. 2000. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403: 503–511 [DOI] [PubMed] [Google Scholar]
- 62. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, Xu W, Shaffer AL, Wright G, Xiao W, Powell J, Jiang JK, Thomas CJ, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Johnson NA, Rimsza LM, Campo E, Jaffe ES, Wilson WH, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pierce SK, Staudt LM. 2010. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463: 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gururajan M, Chui R, Karuppannan AK, Ke J, Jennings CD, Bondada S. 2005. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells. Blood 106: 1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Lin X. 2005. Phosphorylation of CARMA1 plays a critical role in T cell receptor-mediated NF-kappaB activation. Immunity 23: 575–585 [DOI] [PubMed] [Google Scholar]