

Abstract
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase that regulates processes including mRNA translation, proliferation, and survival. By assembling with different cofactors, mTOR forms two complexes with distinct biological functions. Raptor-bound mTOR (mTORC1) governs cap-dependent mRNA translation, whereas mTOR, rictor, and mSin1 (mTORC2) activate the survival and proliferative kinase Akt. How the balance between the competing needs for mTORC1 and -2 is controlled in normal cells and deregulated in disease is poorly understood. Here, we show that the ubiquitin hydrolase UCH-L1 regulates the balance of mTOR signaling by disrupting mTORC1. We find that UCH-L1 impairs mTORC1 activity toward S6 kinase and 4EBP1 while increasing mTORC2 activity toward Akt. These effects are directly attributable to a dramatic rearrangement in mTOR complex assembly. UCH-L1 disrupts a complex between the DDB1-CUL4 ubiquitin ligase complex and raptor and counteracts DDB1-CUL4-mediated raptor ubiquitination. These events lead to mTORC1 dissolution and a secondary increase in mTORC2. Experiments in Uchl1-deficient and transgenic mice suggest that the balance between these pathways is important for preventing neurodegeneration and the development of malignancy. These data establish UCH-L1 as a key regulator of the dichotomy between mTORC1 and mTORC2 signaling.
INTRODUCTION
As a master regulator of cellular metabolism, proliferation, and survival, mammalian target of rapamycin (mTOR) is of great and increasing interest to biologists and oncologists alike. mTOR is a serine/threonine kinase that assembles with at least five other proteins to form two different and functionally distinct multiprotein complexes, mTORC1 and mTORC2 (1). The mechanisms and molecules that dictate the absolute and relative amounts of mTORC1 and mTORC2 are largely unknown. We recently found that Akt phosphorylation was dramatically increased in transgenic mice overexpressing UCH-L1 (2). In that study, we found UCH-L1 to have no effect on the level of mTOR or any of its cofactors, though a suppressive effect on the antagonistic Akt phosphatase PHLPP1 was observed. The mechanism behind these findings was unclear.
Though UCH-L1 was the first deubiquitinating enzyme discovered, its functions remain poorly understood. In animals, UCH-L1 expression is restricted to the brain, peripheral nerves, endocrine tissues, and gonads of both sexes (3). Deletion of Uchl1 in mice leads to a fatal neurodegenerative disorder known as gracile axonal dystrophy (4), and mutations have been identified that impact the pathogenesis of Parkinson's disease (5, 6). Expression outside neuroendocrine tissues is found in various cancers, including B cell lymphoma (7), multiple myeloma (2, 8), and lung cancer (9). Using transgenic (Tg) mice, we demonstrated that UCH-L1 is in fact an oncogene that causes the same malignancies, most likely by boosting Akt signaling (2). Whether this function is relevant to the physiological role of UCH-L1 in the brain is not known. Here, we describe a previously unrecognized function of UCH-L1 as a powerful regulator of mTOR activities. Using a combination of animal and cell culture models, we find that UCH-L1 potently disrupts mTORC1 and promotes the assembly of mTORC2 under physiological and disease conditions. To accomplish this, UCH-L1 interferes with ubiquitination of raptor, which is catalyzed by the DDB1-CUL4 E3 ligase complex, leading to loss of mTORC1 integrity. Loss of this complex is accompanied by a concurrent increase in mTORC2, likely due to the increased availability of free mTOR. These data have important implications regarding the physiological role of UCH-L1 in the brain, as well as its oncogenic role in human cancer.
MATERIALS AND METHODS
Cell culture, lentiviral transduction, RNA interference, and reagents.
All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin, streptomycin, and 10% fetal calf serum (embryonic stem [ES] cell grade). Where indicated, cells were incubated for 48 h with 100 nM rapamycin (EMD-Millipore, Billerica, MA). UCH-L1 and N-terminally FLAG-tagged ubiquitin (FLAG-Ub) constructs were introduced to cells by lentivirus transduction using the TSiN vector (10). RNA interference was performed using pGIPZ (constitutive-expression) and pTRIPZ (doxycycline-inducible) short hairpin RNA (shRNA) obtained from Open Biosystems (Huntsville, AL). Doxycycline-inducible UCH-L1 shRNA was used as previously described (2). Relative cell size (forward scatter height [FSC-H]) was determined on ethanol-fixed cells by measuring forward scatter on a FACSCalibur (BD Bioscience) as described previously (11). Doxycycline-inducible hemagglutinin (HA)-tagged ubiquitin G76A (HAUbG76A) (a gift from Christian Schlieker, Yale University) was introduced into cells by transient transfection, along with a plasmid encoding the reverse tetracycline-controlled transactivator (rtTA) with Lipofectamine 2000 (Invitrogen, Carlsbad CA), and expression was induced by immediately culturing cells in the presence of doxycycline, as previously described (2).
The antibodies used in this study include UCH-L1 (3524), phospho-mTORS2448 (5536), phospho-mTOR2481 (2974), raptor (2280), rictor (9476), mLST8 (3274), 4EBP1 (2845), p4EBP1T70 (5078), p4EBP1T37/46 (2855), p4EBP1S65 (9451), pS6K (9208), S6K (49D7), Akt (4691), pAktS473 (4060), TSC2 (4308), Sin1 (anti-DYKDDDDK Tag (8146), and CUL4A (2699) from Cell Signaling Inc. (Danvers, MA); CUL4B (GTX113875) from GeneTex (Irvine, CA); CUL4A/B (C19) from Santa Cruz Biotechnology (Santa Cruz, CA); mTOR (A301-143a) and Sin1 (A300-910A) from Bethyl Laboratories (Montgomery, TX); α-tubulin (T9026) from Sigma; HA (clone 3F10) from Roche Applied Sciences (Indianapolis, IN); and DDB1 from Abcam (ab7522) (Cambridge, MA) and Invitrogen (376200) (Carlsbad CA).
Immunoprecipitation.
Except where indicated, all immunoprecipitations were performed on extracts generated with 0.3% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} according to the method of Sarbassov et al. (12). Occasionally cells were treated with the cross-linking agent dithiobis [succinimidyl propionate] (DSP) prior to lysis, according to the manufacturers' instructions (Pierce, Rockford, IL). Immunoprecipitations involving HA-ubiquitin G76A were performed on extracts prepared in buffer containing 1% SDS. After clearing, these extracts were diluted to 0.1% SDS prior to adding antibodies. In vitro UCH-L1 reactions used His-tagged UCH-L1 purified from bacteria using standard procedures. The in vitro mTOR kinase assay was performed as described previously (13) using purified S6K (a generous gift from David Sabatini) as a substrate.
Mouse tissues.
All animal studies were reviewed and approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). Uchl1 transgenic mice were described previously (2). UCH-L1-null (Uchl1nm3419) animals were the generous gift of Scott Wilson, University of Alabama, Birmingham, AL (14). Tissues were disrupted using the gentleMACS tissue dissociator (Miltenyi, Auburn, CA) system prior to cell lysis. B and T lymphocytes were isolated using the untouched B or T cell magnetic isolation MACS kits (Miltenyi). Quantitative reverse transcription (RT)-PCR was performed on isolated B and T cells using TaqMan gene expression assays for UCH-L1 normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (all primer sets were from Applied Biosystems, Carlsbad, CA).
Immunofluorescence imaging.
For cells at steady state, indirect immunofluorescence assays were performed as described previously (15). HeLa cells transduced with either empty vector or UCH-L1-encoding lentivirus were seeded on 10-well glass slides (Fisher Scientific, Pittsburg, PA) 24 hours prior to fixation with paraformaldehyde (3% for 12 min; room temperature). For experiments involving amino acid-induced relocalization, starvation, restimulation, and immunostaining, procedures were performed as described previously (16). Images were obtained with a laser scanning microscope (LSM 510 v3.2SP2; Carl Zeiss, Inc.) with Axiovert 100 M (Carl Zeiss, Inc.), with a c-Apochromat 40× water objective used to analyze immunostained cells and to capture representative images.
RESULTS
UCH-L1 levels are inversely proportional to mTORC1 activity in vitro and in vivo.
Akt is activated through two essential phosphorylation events catalyzed by two distinct kinases. Downstream of phosphoinositide 3 (PI3) kinase, PDK1 acts to phosphorylate Thr308 of Akt (17). In order to achieve full activity, Akt must also be phosphorylated on Ser473, an essential function of mTORC2 (18). Despite the positive effect of UCH-L1 on Akt phosphorylation, it did not produce changes in the levels of PDK1, mTOR, or any mTORC1/2 components. We did find, however, that UCH-L1 suppressed the levels of the phosphatase PHLPP1, which antagonizes mTOR activity on Ser473 of Akt (2). The mechanism for this was unclear. Recently, PHLPP1 levels were shown to be highly dependent upon mTORC1-driven mRNA translation, suggesting that UCH-L1 may negatively regulate mTORC1 signaling (19). To test this, we transduced HeLa cells with a lentivirus encoding wild-type (WT) UCH-L1 (UCH-L1WTHA) or catalytic mutant UCH-L1 (UCH-L1C90AHA) or with a control empty vector and assayed the phosphorylation of the mTORC1 substrates 4EBP1 and S6K. UCH-L1 potently suppressed phosphorylation of 4EBP1 at Thr70, Thr37/46, and Ser65, as well as S6KT389, consistent with inhibition of mTORC1 activity (Fig. 1A). As we observed previously, UCH-L1 increased phosphorylation of Akt on both T308 and S473. As phosphorylation of AktT308 is inhibited by mTORC1 activity (via S6K phosphorylation of IRS-1 [20, 21]), these data are most consistent with UCH-L1 inhibiting mTORC1. This required catalytic activity, as expression of the catalytic mutant had no effect. As we observed previously, we found no change in the level of any mTOR binding protein. We also found similar results in splenocytes from Uchl1 transgenic (Uchl1Tg) mice (Fig. 1B). To determine if these changes affect cellular physiology, we studied the impact of UCH-L1 on cell size—a parameter that correlates with mTORC1 activity (22). HeLa cells expressing UCH-L1 are substantially smaller, as measured by flow cytometry, than those that do not (Fig. 1C). The magnitude of this change phenocopies that seen in cells treated with rapamycin, indicating that UCH-L1 profoundly inhibits the mTORC1 pathway. This effect is relevant in vivo, as we also found that UCH-L1 expression reduces the size of B cells, but not T cells, in Uchl1Tg mice (Fig. 1D and E). This may be due to relatively low transgene levels in transgenic T cells compared with transgenic B cells (Fig. 1F). Because endogenous UCH-L1 is nearly undetectable in lymphocytes, both T cells and B cells from transgenic mice have much higher levels of Uchl1 mRNA than wild-type animals, though transgenic T cells have only about 25% of the Uchl1 mRNA seen in transgenic B cells.
Fig 1.

UCH-L1 expression leads to reduced mTORC1 activity. (A) HeLa cells were stably transduced with either control (empty vector) or wild-type UCH-L1 (UCH-L1WTHA)- or catalytically inactive UCH-L1 (UCH-L1C90AHA)-encoding lentivirus, followed by immunoblot analysis of mTORC1 and mTORC2 substrates and binding proteins as indicated. (B) Spleens were harvested from age-matched mice of the indicated genotypes and immunoblotted for the indicated mTORC1 and mTORC2 substrates. (C) HeLa cells stably transduced or not with UCH-L1 were followed by flow cytometry to monitor cell size. For comparison, control HeLa cells were treated with rapamycin (100 nM) for 48 h. The green rapamycin tracing is largely hidden by that of UCH-L1. (D and E) B and T lymphocytes were isolated from the spleens of age-matched mice of the indicated genotypes by magnetic isolation. Cell size (FSC-H) was determined by flow cytometry. The graphs are representative of those seen in three independent experiments. (F) Relative expression of UCH-L1 in B or T cells from Uchl1Tg or wild-type mice was determined by quantitative real-time PCR using TaqMan gene expression assays. The graphs represent the means and standard errors for three different mice.
We next asked whether depletion or genetic loss of endogenous UCH-L1 would stimulate mTORC1 activity. Upon depletion of UCH-L1 in the multiple-myeloma cell line KMS-28, which expresses high levels of endogenous UCH-L1, we observed an increase in phosphorylation of 4EBP1 and S6K and increased cell size, indicating that UCH-L1 loss stimulates mTORC1 activity (Fig. 2A and B). This was confirmed with another shRNA construct and in two other cell lines (data not shown). Similarly, we found markedly increased phospho-4EBP1, phospho-S6K, and PHLPP1, and decreased phospho-AktS473 levels in the brains (the tissue where UCH-L1 is expressed) of Uchl1-null mice compared with wild-type animals (Fig. 2C). Taking the data together, we conclude that UCH-L1 dictates the balance of mTOR activity in physiological and pathophysiological states.
Fig 2.

UCH-L1 depletion leads to increased mTORC1 activity. (A) KMS-28doxsh cells were incubated with or without doxycycline (Dox) for 5 days and were then analyzed by immunoblotting using the indicated antibodies. (B) Sizes (FSC-H) of KMS-28doxsh cells from panel A as measured by flow cytometry. (C) Brain tissue was harvested from age-matched mice of the indicated genotypes, followed by immunoblotting using the indicated antibodies. (D) HeLa cells with or without stable expression of UCH-L1 were grown under standard conditions and analyzed by confocal immunofluorescence microscopy using the indicated antibodies. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). (E) HeLa cells with or without stable expression of UCH-L1 were starved of amino acids (−AA) for 50 min and restimulated with amino acids as detailed in Materials and Methods. The cells were fixed and stained for mTOR as indicated. Bars = 5 μm.
UCH-L1 catalytic activity promotes the disassembly of mTORC1.
We next asked whether UCH-L1 impacted the membrane localization of mTOR, which is required for its nutrient-dependent activation (16). Regardless of UCH-L1 expression, at steady state, we observed mTOR in punctate structures that encircle the nucleus, suggestive of a membrane localization pattern (Fig. 2D). While UCH-L1 is localized more diffusely, we observed substantial colocalization and enrichment in these perinuclear regions. Recently, mTOR has been found to undergo dramatic relocalization from the cytoplasm to lysosomal surfaces when stimulated with amino acids after a period of starvation (16, 23). To determine if UCH-L1 impacts this process, we incubated cells transduced with empty vector or UCH-L1-encoding lentivirus and cultured them for 50 min in the absence of amino acids. In this setting, mTOR localized diffusely throughout the cytoplasm regardless of UCH-L1 expression (Fig. 2E). Following the reintroduction of amino acids, there was an accumulation of mTOR into larger structures in the perinuclear region consistent with its described clustering on the surfaces of lysosomes (16). In contrast, this pattern of mTOR recruitment was substantially blunted in cells transduced with UCH-L1-encoding lentivirus, suggesting that the UCH-L1-induced changes in lysosome recruitment may underlie the reduced mTORC1 activity in these cells.
The amino acid-dependent recruitment of mTORC1 to lysosomal membranes requires the association of raptor with the two small GTPases RagB and RagD, which in turn complex with the trimeric “ragulator” complex, consisting of p14, p18, and MB1 (16). Accordingly, amino acid-induced relocalization of mTOR to the lysosomal surface is impaired in raptor-depleted cells (16). This, together with our observation that UCH-L1 inhibits mTORC1 activity without affecting the level of any tested mTOR pathway component, led us to question the impact of UCH-L1 on the mTOR-raptor complex. We therefore depleted UCH-L1 in KMS-28 myeloma cells stably transduced with a doxycycline-inducible shRNA targeting UCH-L1 (KMS-28doxsh cells) and analyzed mTOR complex levels by immunoprecipitation and immunoblotting. We found a dramatic shift in mTOR complexes, so that UCH-L1 knockdown increased the association of mTOR with raptor, with a concomitant loss of rictor (Fig. 3A). We obtained similar results from brain extracts from wild-type and Uchl1-null mice (Fig. 3B). To test whether UCH-L1 overexpression impairs mTORC1 stability while increasing mTORC2 and to establish the role of UCH-L1 catalytic activity, we overexpressed UCH-L1WTHA or the catalytic mutant UCH-L1C90AHA and again performed mTOR immunoprecipitation. Due to inconsistent efficiency of mTOR immunoprecipitation from many cell types, we used 293T cells for this analysis. Compared with the control, there was a dramatic reduction in the recovery of raptor with mTOR in cells expressing UCH-L1WTHA, with a corresponding increase in the precipitation of rictor (Fig. 3C). Similarly, UCH-L1 reduced the recovery of mTOR from raptor immunoprecipitates and increased its recovery with rictor compared with the control. This effect is dependent on UCH-L1 catalytic activity, as expression of UCH-L1C90AHA did not lead to a similar change. Although reduced phosphorylation of mTORC1 targets may be the result of the loss of mTORC1, the data do not exclude a direct effect on mTOR kinase activity within mTORC1. We found no effect of UCH-L1 on the autophosphorylation status of mTOR in extracts (data not shown) or in immunoprecipitates (Fig. 3D). To further investigate this, we performed an in vitro kinase assay in which we immunoprecipitated mTORC1 (using raptor antibodies) and tested its ability to phosphorylate purified S6K (13). As expected, we found reduced phosphorylation of S6K in reaction mixtures containing raptor precipitates from cells expressing UCH-L1 (Fig. 3E). However, despite similar amounts of raptor, these reaction mixtures contained less mTOR kinase, possibly explaining the reduced activity. When we normalized mTOR levels with those of the control, we found that S6K phosphorylation was restored. To determine if UCH-L1 affects mTORC1 assembly or stability, we examined the effect of bacterially purified UCH-L1 on mTORC1 in vitro. Strikingly, the addition of active, but not catalytically inactive, UCH-L1 to mTOR immunoprecipitates resulted in loss of raptor from mTOR (Fig. 3F). In this in vitro setting, the amount of rictor bound to mTOR remained unchanged, presumably due to the lack of free protein under these conditions. These data suggest that UCH-L1 acts catalytically toward one or more members of mTORC1, resulting in the displacement of raptor from mTOR, and that the increase in mTORC2 depends on subsequent binding of rictor with free mTOR. We conclude that UCH-L1 does not directly affect mTOR kinase activity, but rather, leads to mTORC1 instability, with a secondary effect of increased mTORC2 assembly, leading to increased Akt phosphorylation.
Fig 3.

UCH-L1 reorganizes the assembly of competing mTOR complexes. (A) KMS-28doxsh cells were incubated with or without doxycycline for 5 days to deplete UCH-L1 and were then subjected to immunoprecipitation (IP) with the indicated antibodies. Ratios were determined using ImageJ. (B) Brain tissue was harvested from age-matched mice of the indicated genotypes and subjected to immunoprecipitation using the indicated antibodies. The precipitates were then immunoblotted using the indicated antibodies. Ratios were determined using ImageJ. (C and D) HEK-293T cells, stably transduced with the indicated constructs, were subjected to immunoprecipitation using the indicated antibodies. The precipitates were then immunoblotted using the indicated antibodies. (E) mTORC1 was purified from HEK-293T cells by immunoprecipitation using raptor or control IgG antibodies as indicated. Purified S6K was added to the resulting precipitates, and after further incubation, the reactions were stopped by adding 2× SDS sample buffer. The reaction mixtures were subjected to immunoblotting with the indicated antibodies. Where indicated, ([2×]), the amount of mTORC1 was increased 2-fold to equalize the amount of mTOR in the reaction mixture. (F) mTOR complexes were immunoprecipitated from 293T cells, washed extensively, and incubated with purified UCH-L1. Where indicated, N-ethylmaleimide (NEM) was included to inactivate catalytic activity. After extensive washing, the remaining bound proteins were analyzed by immunoblotting using the indicated antibodies.
UCH-L1 disruption of the DDB1-raptor complex destabilizes mTORC1.
Because of the dependence on catalytic activity for the effect of UCH-L1 on mTOR complex assembly, we suspected that it was impacting a ubiquitin-dependent activity associated with mTOR. Recently, a novel complex between a Ub-ligase complex containing DDB1, CUL4, and ROC1 and raptor was described (24). This ligase complex is best known for its role in the recognition of DNA lesions induced by UV light (25), though by binding to different adaptor proteins known as DDB1- and CUL4-associated factors (DCAFs), the complex participates in a broad range of cellular activities (26). We therefore explored the impact of UCH-L1 on the association of DDB1 with raptor. In 293T cells, we readily confirmed the precipitation of DDB1, CUL4A, and CUL4B from raptor immunoprecipitates, but not from those of rictor (Fig. 4A and C). Similarly, we recovered mTOR and raptor, but not rictor, from DDB1 immunoprecipitates (Fig. 4C). Strikingly, ectopic UCH-L1 reduced the recovery of DDB1 with raptor (Fig. 4B and C). The effect was somewhat less apparent when we examined the coprecipitation of DDB1 with mTOR itself (Fig. 4C), likely reflective of the described association with the mTOR complex subunit mLST8 that is present in both mTORC1 and mTORC2 (24). Again, the effect required catalytic activity, as UCH-L1C90AHA had no effect on the association of DDB1 with raptor. Consistent with UCH-L1 regulating this association, there was a substantial increase in the association of DDB1 with mTOR in KMS-28doxsh cells upon depletion of UCH-L1 (Fig. 4D). To further understand the nature of UCH-L1-driven DDB1 loss from raptor, we again performed in vitro reconstitution assays in which we immunopurified mTOR complexes and added recombinant active or inactive UCH-L1. When we performed this analysis on mTOR complexes retrieved with an anti-mTOR antibody, we observed a substantial loss of raptor upon the addition of catalytically active UCH-L1 (Fig. 4E). Displacement of DDB1 in this setting was again less obvious, likely due to its independent association with mTOR via mLST8 (24). In comparison, when we added UCH-L1 to mTORC1 precipitated with an antiraptor antibody, there was a dramatic loss of both mTOR and DDB1 from the complex. We conclude that UCH-L1 drives the dissolution of the mTOR-raptor-DDB1 complex through a mechanism that requires catalytic activity.
Fig 4.

UCH-L1 disrupts the DDB1-raptor complex that is required for mTORC1 stability. (A to C) HEK-293T cells, stably transduced with the indicated constructs, were subjected to immunoprecipitation using the indicated antibodies. The precipitates were then immunoblotted using the indicated antibodies. (D) KMS-28doxsh cells were incubated with or without doxycycline for 5 days to deplete UCH-L1 and were then subjected to immunoprecipitation with the indicated antibodies. The precipitates were then immunoblotted using the indicated antibodies. (E and F) mTOR complexes were immunoprecipitated from 293T cells, washed extensively, and incubated with purified UCH-L1. Where indicated, NEM was included to inactivate catalytic activity. After extensive washing, the remaining bound proteins were analyzed by immunoblotting using the indicated antibodies.
To determine its role in mTORC1 signaling, we depleted the DDB1-CUL4 complex and examined substrate phosphorylation and mTOR complex association. Strikingly, depletion of DDB1 or codepletion of CUL4A and CUL4B led to reduced phosphorylation of S6K and increased phosphorylation of Akt (Fig. 5A and D). Depletion of CUL4A or CUL4B individually had no effect on mTOR signaling (data not shown). These changes were also accompanied by loss of mTORC1 and increased levels of mTORC2, as determined by coimmunoprecipitation (Fig. 5B). These results indicate that DDB1 is essential for mTORC1 stability. Further demonstrating the importance of DDB1 for mTORC1 activity, we found that depletion of DDB1 entirely prevented the increase in mTORC1 activity seen upon UCH-L1 knockdown (Fig. 5C). UCH-L1 expression in cells depleted of DDB1 or CUL4A/CUL4B did not further increase phosphorylation of the mTORC2 substrate Akt, though a modest additional effect of UCH-L1 expression on suppressing phosphorylation of the mTORC1 substrates S6K and 4EBP1 was seen in DDB1-depleted cells. This suggests that even at reduced levels, DDB1-CUL4 associates with mTORC1 in a UCH-L1-sensitive manner. As we observed with UCH-L1 expression, depletion of the DDB1-CUL4 complex did not change the level of mTOR, the core mTOR complex subunits, the mTOR inhibitor TSC2, or Akt, indicating that the regulation of the pathway by DDB1-CUL4 is likely nondegradative. Interestingly, overexpression of DDB1 did not further increase mTORC1 activity (data not shown), suggesting either that in the absence of UCH-L1, DDB1 is not rate limiting in the formation of the complex with raptor or that a regulatory step governs its abundance. Taken together, these data demonstrate that the DDB1-CUL4 complex is an essential component of mTORC1 that regulates its stability through a nondegradative mechanism.
Fig 5.
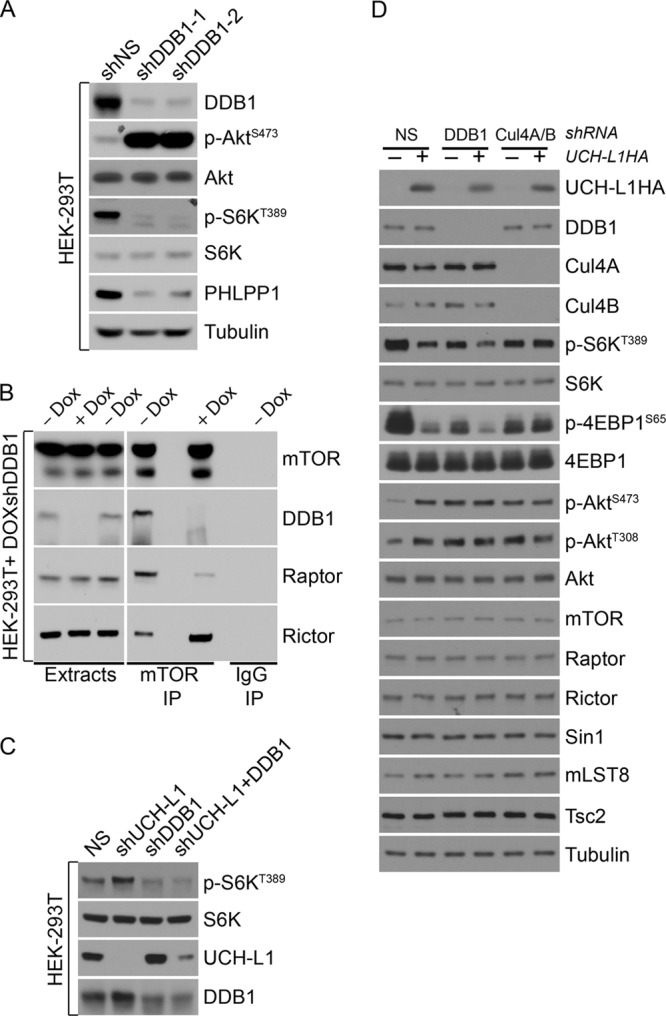
Depletion of the DDB1-CUL4 complex results in a phenocopy of UCH-L1 expression. (A and C) HEK-293T cells were transduced with lentiviruses carrying the indicated shRNAs for 5 days, and extracts were immunoblotted using the indicated antibodies. (B) HEK-293T transduced to carry a doxycycline-regulated shRNA targeting DDB1 were incubated with or without doxycycline for 5 days, followed by immunoprecipitation and immunoblotting with the indicated antibodies. (D) HEK-293T cells were transduced with the indicated constructs, followed by immunoblotting for the indicated antibodies. shNS and NS, nonsilencing shRNA.
To further understand the mechanism of UCH-L1 in regulating mTORC1 stability, we asked whether UCH-L1 associates with components of mTORC1 or mTORC2. We performed a series of immunoprecipitations targeting endogenous subunits of mTORC1/2 and associated proteins under detergent conditions that dissociate mTORC complexes themselves and probed for UCH-L1WTHA. Consistent with its effect on the two proteins, we found UCH-L1 to coprecipitate with DDB1 and raptor (Fig. 6A). Surprisingly, a similar fraction of DDB1 was recovered with UCH-L1C90AHA, indicating that the catalytic mutant can associate with DDB1 but cannot disrupt its complex with raptor (Fig. 6B). The coprecipitation of endogenous UCH-L1 and DDB1 was also seen in wild-type mouse brain extracts, indicating that the mechanism is physiologically relevant (Fig. 6C). Based on the importance of the DDB1-CUL4 complex for mTORC1 stability and the requirement for UCH-L1 catalytic activity in antagonizing this role, we hypothesized that UCH-L1 interferes with the ubiquitination of mTORC1 subunits. To test this, we made use of the ubiquitin mutant UbG76A, which is efficiently conjugated to target proteins but is resistant to deubiquitinating enzyme removal, thus increasing the sensitivity of detecting Ub modifications (27). We transfected cells with HAUbG76A and immunoprecipitated endogenous mTORC components, followed by HA immunoblotting. To enhance our ability to detect covalently attached ubiquitin, extracts were prepared with 1% SDS, denaturing conditions that minimize the coprecipitation of proteins associated through noncovalent means. Under these conditions, there was a robust and specific coprecipitation of HAUbG76A from raptor immunoprecipitates (Fig. 7A). To examine the importance of DDB1-CUL4 and UCH-L1 for this modification, we depleted DDB1 or expressed UCH-L1 in cells expressing N-terminally FLAG-tagged ubiquitin (FLAG-Ub), and then performed raptor immunoprecipitations under conditions preserving mTOR complex associations and probed for FLAG-Ub. UCH-L1 expression or DDB1 depletion had no effect on the global pattern of FLAG-Ub conjugates (Fig. 7B). However, there was a profound suppressive effect of UCH-L1 on reducing raptor ubiquitination that required catalytic activity. Similarly, depletion of DDB1 dramatically reduced raptor ubiquitination. We hypothesize that UCH-L1 may affect raptor ubiquitination through at least two mechanisms: direct deubiquitination and/or prevention of ubiquitination by disrupting the DDB1-raptor complex. To determine what role the disruption of raptor-DB1 binding plays in this process, we performed raptor immunoprecipitation from cells expressing HAUbG76A with or without UCH-L1. Despite the nonhydrolyzable nature of UbG76A, we observed a significant reduction in HAUbG76A modification of raptor in cells expressing UCH-L1, demonstrating that UCH-L1 affects raptor ubiquitination at least in part through preventing its modification (Fig. 7C). We conclude that DDB1-CUL4 maintains mTORC1 stability by ubiquitinating raptor and that UCH-L1 destabilizes mTORC1 by counteracting this process.
Fig 6.

UCH-L1 associates with DDB1 and raptor. (A and B) HEK-293T cells, stably transduced with the indicated constructs, were subjected to immunoprecipitation and immunoblotting using the indicated antibodies. Extr., extracts; *, UCH-L1; **, monoubiquitinated UCH-L1. (C) Extracts from wild-type mouse brain were subjected to immunoprecipitation and immunoblotting with the indicated antibodies.
Fig 7.

UCH-L1 reduces ubiquitination of raptor. (A and C) HEK-293T cells were transiently transfected (+) or not transfected (−) with HAUbG76A, and extracts (prepared with 1% SDS) were subjected to immunoprecipitation and immunoblotting with the indicated antibodies. Lanes separated by dashed lines were from different immunoblots. (B) HEK-293T cells were transduced with the indicated constructs, and extracts (prepared with CHAPS) were subjected to immunoprecipitation and immunoblotting (IB) with the indicated antibodies.
DISCUSSION
We previously observed that the ubiquitin hydrolase UCH-L1 is a potent oncogene that is frequently overexpressed in B cell non-Hodgkin's lymphoma and that transgenic expression of the enzyme is sufficient to drive the development of lymphoma and lung tumors in mice (2). At the signaling level, we observed a dramatic effect of UCH-L1 on Akt phosphorylation, but the underlying mechanism was unclear. In our investigations into this mechanism, we uncovered a novel antagonistic relationship between the DDB1-CUL4 ubiquitin ligase and UCH-L1 in regulating the ubiquitination status of raptor, thereby dictating the stability of mTORC1 through a nondegradative mechanism. Based on our findings, we propose a model in which UCH-L1 forms a complex involving DDB1 and raptor within mTORC1 that facilitates raptor deubiquitination. This leads to a conformational change in raptor that reduces its affinity for mTOR and DDB1, leading to mTORC1 instability. The reduced affinity between DDB1 and raptor further reduces raptor ubiquitination. The resulting increased availability of free mTOR leads to a secondary increase in the level of mTORC2, the principal kinase that targets AktS473. Consistent with our results, lower mTORC1 activity has been shown to impair translation of the phosphatase PHLPP that dephosphorylates AktS473. It is therefore likely that the net effect of UCH-L1 on Akt phosphorylation reflects the combined impacts of increased kinase activity and reduced phosphatase activity. These data provide the first evidence that targeted ubiquitination plays a role in regulating mTOR complex stability and provide important new insights into potential therapeutic approaches to modulate these signaling activities.
To better understand the biochemical functions of UCH-L1, we sought an approach to introduce UCH-L1 into cells where it is not normally expressed and to deplete it in cells where it is expressed endogenously at high levels. We therefore used HeLa and HEK293T cells (with little or no endogenous UCH-L1) to examine the impact of gain of UCH-L1 and the KMS-28 myeloma cell line (with high endogenous UCH-L1) to examine the impact of losing UCH-L1 in an in vitro setting. By comparing the results obtained from both experimental settings, we largely avoid artifacts that may result from either design. To allow better extrapolation of the data beyond cancer cell lines, we also examined the loss of endogenous UCH-L1 from its normal tissue (brain), as well as stable long-term expression of UCH-L1 in lymphocytes from Uchl1 transgenic mice. As our observations were consistent across these tissues and cell lines, under conditions of overexpression and depletion, the data strongly support the idea that UCH-L1 regulates mTOR activities in tissues where it is normally expressed (brain) and in tissues where cancers arise (lymphocytes).
The mTOR signaling network is of great importance in the understanding of basic cellular metabolism and is of interest to neurobiologists and cancer biologists alike. These data have implications for the understanding of the use of mTOR inhibitors in neurodegenerative disorders and B cell lymphoma. Furthermore, as deubiquitinating enzymes are increasingly considered potential drug targets (28–31), these data provide important insight into the mechanisms by which UCH-L1 inhibition may affect cellular physiology.
Our data strongly suggest that the DDB1-CUL4 ligase complex is an essential component of mTORC1. With our data, it is not possible to accurately determine the proportion of mTORC1 that contains DDB1-CUL4. While it is clear that displacement of DDB1-CUL4 by UCH-L1 or depletion of DDB1 or CUL4 results in reduced mTORC1, it is not clear whether the DDB1-CUL4 complex must remain bound to mTORC1 continuously or whether transient association is sufficient to maintain its stability. However, our in vitro data, in which we observe destabilization of the DDB1-raptor complex and mTORC1 upon the addition of recombinant UCH-L1, can be interpreted to indicate that continuous association is required. The requirement for DDB1 and CUL4 in maintaining mTORC1 signaling was also observed by Ghosh et al. (24), though the mechanism behind the observation remained unclear. We found that both wild-type and catalytic mutant UCH-L1 coprecipitate with DDB1 but that UCH-L1 catalytic activity drives the dissociation of the mTOR-raptor-DDB1 complex. The continued association of catalytically dead UCH-L1 with DDB1 without disrupting DDB1-raptor binding or mTORC1 integrity indicates that UCH-L1 does not compete for binding sites on these proteins. It also suggests that UCH-L1 first associates with mTORC1-DDB1 and then destabilizes the complex, rather than UCH-L1 interfering with the formation of the DDB1-raptor complex. Preventing the formation of this complex through DDB1 depletion, however, is sufficient to destabilize mTORC1. Consistent with its effect on mTORC1 integrity, we found that UCH-L1 interferes with the amino acid-dependent relocalization of mTOR to lysosomal surfaces. We did not, however, detect an appreciable change in mTOR localization at steady state in cells expressing UCH-L1. It seems likely that under static conditions there is sufficient remaining mTORC1 to retain the partial membrane localization, but in the context of the massive shift that occurs upon reintroducing amino acids to starved cells, this remaining amount is insufficient to match the demand. Our results are also consistent with those recently showing that targeting of cullin-containing ubiquitin ligases with a small-molecule neddylation inhibitor (MLN4924) inhibits mTORC1 activity (32). Our results differ somewhat in that UCH-L1 overexpression and DDB1 depletion led to an increase in mTORC2 activity that was not observed in cells exposed to MLN4924, suggesting that this inhibitor may have other effects that prevent the increase in mTORC2 activity that results from DDB1 depletion.
Our results demonstrate that ubiquitination of raptor is driven by DDB1 and antagonized by UCH-L1. Raptor ubiquitination was also observed in high-throughput ubiquitin proteomic studies (33). UCH-L1 may have a dual impact on this modification—a direct deubiquitination step and an indirect effect on preventing the ongoing raptor modification by DDB1. The fact that we do not see any changes in the absolute levels of any mTOR complex subunit further indicates that the role of Ub in the process is nondegradative. Though raptor ubiquitination was reduced to very low levels, neither depletion of DDB1 nor overexpression of UCH-L1 entirely eliminated it in our experiments. Further, UCH-L1 could not further reduce raptor ubiquitination beyond the level seen with DDB1 depletion. It is possible that there is a small pool of mTORC1 that is relatively inaccessible to UCH-L1 deubiquitination. It is also possible that UCH-L1 exhibits chain specificity with the remaining ubiquitin molecules in alternative linkages, possibly installed by other Ub ligases. Our in vitro data in which we found that purified UCH-L1 can disrupt the mTOR-raptor complex without increasing mTOR-rictor (mTORC2) indicate that rictor itself is not directly involved in the destabilization of mTORC1 and suggest that increased mTORC2 is likely a secondary effect of increasing the availability of free mTOR that passively assembles with rictor. Competition of raptor and rictor for mTOR binding has been suggested by the work of Sarbassov et al., in which either overexpression of raptor or depletion of rictor leads to increased mTORC1 (18). Others have seen that knockdown of raptor in Drosophila cells led to increased phosphorylation of Akt, a finding consistent with increased mTORC2 (34). Despite the dramatic changes in association of raptor and rictor with mTOR, we saw no changes in the autophosphorylation status of mTOR. Phosphorylation of mTORS2481 was reported to be an indicator of mTORC2 levels, with phospho-mTORS2448 specific for mTORC1 (35). Other reports have called this finding into question (36). It is likely that differences in the experimental approaches underlie these contradictory findings, as the association between the mTOR phosphoforms was primarily observed after serum starvation and insulin restimulation. It is possible that these forms do not accurately reflect the levels of these complexes under our steady-state conditions.
As UCH-L1 is present only in neuroendocrine tissues and in certain cancers, most tissues may regulate the balance of mTOR activities through a different mechanism. The mTORC1-inhibitory complex comprised of TSC1/2 is expressed in a broad tissue distribution (37). The TSC1/2 complex regulates mTORC1 activity without altering complex stability through a mechanism in which it controls the required mTOR activator, Rheb, on lysosomal membranes (1). We propose that UCH-L1 may therefore be representative of a new class of regulators that affect the relative balance of mTOR complex abundance, with other DUBs potentially performing a similar function in other tissues. The identification of UCH-L1 as a negative regulator of mTORC1 makes the Uchl1-null mouse, to our knowledge, the second genetic model characterized by mTORC1 overactivity in the brain, after tuberous sclerosis (38). It is noteworthy that Uchl1-null mice, like those deficient in TSC1 or -2, exhibit severe neurologic phenotypes, reinforcing the observation that mTORC1 hyperactivity is toxic to neurons. UCH-L1 mutations have been linked to Parkinson's disease, where the I93M mutation predisposes to early disease onset (39). Inhibition of mTORC1 activity leads to improved neurologic function in patients with tuberous sclerosis and in models of Parkinson's disease, though only in the former is mTOR inhibition routinely used (40, 41). The involvement of UCH-L1 in the mTOR signaling pathway raises the possibility that UCH-L1 expression affects the biological response to mTOR inhibition in tumors. One may imagine that the suppressive action of UCH-L1 on mTORC1 may lead to sensitivity of these tumors to inhibitors that target mTORC1. Further work is required to determine if UCH-L1 expression has an impact on the response to these agents.
ACKNOWLEDGMENTS
P.J.G. is a Harriet H. Samuelsson Foundation Pediatric Cancer Research Scientist and a former Basic Science Scholar of the American Society of Hematology and is supported by grants from the NIH (CA151351-01), Gabrielle's Angels Cancer Research Foundation, and the Mayo Foundation and by a Howard Hughes Medical Institute Physician Scientist Early Career Award.
We thank Darren Baker, Richard Bram, Jan van Deursen, Zhenkun Lou, Yuichi Machida, Liviu Malureanu, Hidde Ploegh, Robin Ricke, Christian Schlieker, and Pengbo Zhou for critical reading of the manuscript. We thank Shahram Misaghi for assistance with UCH-L1 purification. We also thank the members of the van Deursen laboratory for thoughtful discussions.
Footnotes
Published ahead of print 7 January 2013
REFERENCES
- 1.Zoncu R, Efeyan A, Sabatini DM. 2011. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12:21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hussain S, Foreman O, Perkins SL, Witzig TE, Miles RR, van Deursen J, Galardy PJ. 2010. The de-ubiquitinase UCH-L1 is an oncogene that drives the development of lymphoma in vivo by deregulating PHLPP1 and Akt signaling. Leukemia 24:1641–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson PO, Barber PC, Hamid QA, Power BF, Dhillon AP, Rode J, Day IN, Thompson RJ, Polak JM. 1988. The immunolocalization of protein gene product 9.5 using rabbit polyclonal and mouse monoclonal antibodies. Br. J. Exp. Pathol. 69:91–104 [PMC free article] [PubMed] [Google Scholar]
- 4.Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. 1999. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 23:47–51 [DOI] [PubMed] [Google Scholar]
- 5.Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Kruger R, Hattori N, Mellick GD, Quattrone A, Satoh J, Toda T, Wang J, Ioannidis JP, de Andrade M, Rocca WA. 2004. UCHL1 is a Parkinson's disease susceptibility gene. Ann. Neurol. 55:512–521 [DOI] [PubMed] [Google Scholar]
- 6.Ragland M, Hutter C, Zabetian C, Edwards K. 2009. Association between the ubiquitin carboxyl-terminal esterase L1 gene (UCHL1) S18Y variant and Parkinson's disease: a HuGE review and meta-analysis. Am. J. Epidemiol. 170:1344–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ovaa H, Kessler BM, Rolen U, Galardy PJ, Ploegh HL, Masucci MG. 2004. Activity-based ubiquitin-specific protease (USP) profiling of virus-infected and malignant human cells. Proc. Natl. Acad. Sci. U. S. A. 101:2253–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otsuki T, Yata K, Takata-Tomokuni A, Hyodoh F, Miura Y, Sakaguchi H, Hatayama T, Hatada S, Tsujioka T, Sato Y, Murakami H, Sadahira Y, Sugihara T. 2004. Expression of protein gene product 9.5 (PGP9.5)/ubiquitin-C-terminal hydrolase 1 (UCHL-1) in human myeloma cells. Br. J. Haematol. 127:292–298 [DOI] [PubMed] [Google Scholar]
- 9.Hibi K, Westra WH, Borges M, Goodman S, Sidransky D, Jen J. 1999. PGP9.5 as a candidate tumor marker for non-small-cell lung cancer. Am. J. Pathol. 155:711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miest T, Saenz D, Meehan A, Llano M, Poeschla EM. 2009. Intensive RNAi with lentiviral vectors in mammalian cells. Methods 47:298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu E, Knutzen CA, Krauss S, Schweiger S, Chiang GG. 2011. Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc. Natl. Acad. Sci. U. S. A. 108:8680–8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22:159–168 [DOI] [PubMed] [Google Scholar]
- 13.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. 2007. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25:903–915 [DOI] [PubMed] [Google Scholar]
- 14.Walters BJ, Campbell SL, Chen PC, Taylor AP, Schroeder DG, Dobrunz LE, Artavanis-Tsakonas K, Ploegh HL, Wilson JA, Cox GA, Wilson SM. 2008. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol. Cell Neurosci. 39:539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ. 2001. Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J. Cell Sci. 114:4385–4395 [DOI] [PubMed] [Google Scholar]
- 16.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. 1996. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15:6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 18.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14:1296–1302 [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Stevens PD, Gao T. 2011. mTOR-dependent regulation of PHLPP expression controls the rapamycin sensitivity in cancer cells. J. Biol. Chem. 286:6510–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF. 2004. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166:213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. 2006. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66:1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. 2002. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 16:1472–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh P, Wu M, Zhang H, Sun H. 2008. mTORC1 signaling requires proteasomal function and the involvement of CUL4-DDB1 ubiquitin E3 ligase. Cell Cycle 7:373–381 [DOI] [PubMed] [Google Scholar]
- 25.Li J, Wang QE, Zhu Q, El-Mahdy MA, Wani G, Praetorius-Ibba M, Wani AA. 2006. DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res. 66:8590–8597 [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Zhou P. 2007. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 26:775–780 [DOI] [PubMed] [Google Scholar]
- 27.Hodgins RR, Ellison KS, Ellison MJ. 1992. Expression of a ubiquitin derivative that conjugates to protein irreversibly produces phenotypes consistent with a ubiquitin deficiency. J. Biol. Chem. 267:8807–8812 [PubMed] [Google Scholar]
- 28.Colland F. 2010. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem. Soc. Trans. 38:137–143 [DOI] [PubMed] [Google Scholar]
- 29.Love KR, Catic A, Schlieker C, Ploegh HL. 2007. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat. Chem. Biol. 3:697–705 [DOI] [PubMed] [Google Scholar]
- 30.Nag DK, Finley D. 2012. A small-molecule inhibitor of deubiquitinating enzyme USP14 inhibits Dengue virus replication. Virus Res. 165:103–106 [DOI] [PubMed] [Google Scholar]
- 31.Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, Harpon J, Battaglia V, Vivat V, Sippl W, Colland F. 2012. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 19:467–477 [DOI] [PubMed] [Google Scholar]
- 32.Zhao Y, Xiong X, Jia L, Sun Y. 2012. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 3:e386 doi:10.1038/cddis.2012.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. 2011. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44:325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101 [DOI] [PubMed] [Google Scholar]
- 35.Copp J, Manning G, Hunter T. 2009. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 69:1821–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. 2010. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J. Biol. Chem. 285:7866–7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plank TL, Logginidou H, Klein-Szanto A, Henske EP. 1999. The expression of hamartin, the product of the TSC1 gene, in normal human tissues and in TSC1- and TSC2-linked angiomyolipomas. Mod. Pathol. 12:539–545 [PubMed] [Google Scholar]
- 38.Sampson JR. 2009. Therapeutic targeting of mTOR in tuberous sclerosis. Biochem. Soc. Trans. 37:259–264 [DOI] [PubMed] [Google Scholar]
- 39.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. 1998. The ubiquitin pathway in Parkinson's disease. Nature 395:451–452 [DOI] [PubMed] [Google Scholar]
- 40.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. 2008. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 14:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. 2009. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 12:1129–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]