

Abstract
In both mammalian and viral genomes, a large proportion of sequences are transcribed and annotated as noncoding RNAs. A polyadenylated RNA of 3.0 kb (T3.0) is transcribed from the opposite strand of the open reading frame 50 (ORF50) DNA template in the genome of Kaposi's sarcoma-associated herpesvirus (KSHV) and has been annotated previously as a noncoding RNA. ORF50 encodes the replication and transcription activator (RTA), which controls the switch of the virus between the latent and lytic phases of the life cycle. Here we show that T3.0 encodes a small peptide of 48 amino acids (designated viral small peptide 1 [vSP-1]). vSP-1 interacts with RTA at the protein abundance regulatory signal (PARS) motifs, and the association prevents RTA from being subjected to degradation through the ubiquitin-proteasome pathway. As a consequence, vSP-1 facilitates KSHV gene expression and lytic replication. This finding reveals a novel mechanism of gene regulation in the viral life cycle.
INTRODUCTION
As a herpesvirus, Kaposi's sarcoma-associated herpesvirus (KSHV) has two modes of infection: latency and productive lytic replication. The switch of KSHV between latency and lytic replication is controlled by a virally encoded transcriptional activator, namely, RTA (replication and transcription activator). RTA expression is necessary and sufficient to disrupt latency and initiate viral lytic replication. It activates a number of viral and cellular promoters by different mechanisms, including (i) directly binding to specific motifs in some promoters, (ii) piggybacking on other cellular proteins bound on certain promoters, and (iii) promoting the degradation of transcriptional repressors (reviewed in reference 1). In addition, RTA has also been found to promote the degradation of several proteins, cellular and viral (including itself), through the ubiquitin-proteasome pathway by serving as an intrinsic E3 ubiquitin ligase (2). Therefore, defining the mechanism that regulates RTA expression and activity is crucial for understanding the molecular switch of the KSHV life cycle.
Genomewide analyses of the KSHV transcriptome have revealed that nearly the entire viral genome, including both DNA strands, is transcribed. Ganem and colleagues observed extensive transcription from noncoding regions, including both intergenic regions and noncoding regions antisense to known open reading frames (ORFs) (3). Currently no biological function has been demonstrated to be associated with these noncoding RNAs. The same phenomenon is also seen in mammalian genomes. Although only 2% of the total human genomic sequence consists of protein-coding genes, >90% of the human genome is transcribed, as revealed by large-scale cDNA cloning projects (4, 5) and genomewide tiling arrays (6–8). The majority of the transcripts are long RNAs with little or no protein-coding capacity (with the criterion that only open reading frames with at least 100 codons are annotated) (9). Although the functions of the majority of these noncoding RNAs have not been revealed, several potential functions are beginning to emerge, including inducing chromatin remodeling to affect gene expression either in cis on neighboring genes (10) or in trans (11), serving as antisense RNAs to generate endogenous small interfering RNA (endo-siRNA) (12, 13), binding to specific protein partners to modulate protein activity (14, 15), serving as a structural component to form an RNA-protein complex that regulates cell functions (16), and serving as precursors to small RNAs, including microRNA (miRNA) and Piwi-interacting RNA (piRNA) (17–20).
A 3.0-kb polyadenylated RNA (designated T3.0) that is transcribed from the opposite strand of ORF50 in the KSHV genome has been identified in KSHV-infected cells and has been annotated as a noncoding RNA because no large open reading frame was found in the transcript (21). Since T3.0 RNA is potentially able to base-pair with ORF50 mRNA, which specifies RTA, we wondered if T3.0 modulates RTA expression, either positively or negatively. Here we report that T3.0 indeed upregulates RTA expression. However, T3.0 exerts this function by encoding a small peptide that complexes with RTA and prevents it from being degraded by the ubiquitin-proteasome pathway, representing a novel mechanism underlying RTA regulation and KSHV reactivation. This finding also demonstrates a novel paradigm for the function of so-called noncoding RNAs in cells.
MATERIALS AND METHODS
Cells.
The primary effusion lymphoma cell line BCBL-1 was obtained from the NIH AIDS Research and Reference Reagent Program. The cells were grown in RPMI 1640 medium (Gibco-BRL, Gaithersburg, MD) supplemented with 10% fetal bovine serum (Gibco-BRL) and penicillin-streptomycin (50 U/ml). Human embryonic kidney (HEK) 293T cells were obtained from the ATCC and were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and antibiotics (penicillin-streptomycin and amphotericin B [Fungizone]).
Constructs.
The pCR3.1-ORF50 plasmid, RTA internal deletion mutants, and the promoter-luciferase reporter plasmids [pK8-DE250 and pOrilyt (R)-12F] have been described previously (22). T3.0 cDNA was amplified from a bacterial artificial chromosome (BAC)-cloned KSHV genome (BAC36) by PCR using a pair of primers adding EcoRI and XhoI restriction sites (all the primers used in this study are listed in Table 1), followed by subcloning into the pcDNA3.1+ vector (Invitrogen). vSP-1 and vSP-2 were amplified from the T3.0 construct by PCR, followed by subcloning into the pCMV-3FLAG-1 expression vector (Agilent Technologies). For the Myc-tagged RTA vector, RTA sequence was first amplified from the pCR3.1-ORF50 construct and then subcloned into the pCMV-3MYC-1 expression vector (Agilent Technologies). The sequence fidelity of all new constructs was verified by DNA sequencing. Expression was verified by Western blot analysis with antibodies against tag epitopes.
Table 1.
Oligonucleotide primers used in this study
| Primer target or function | Sequence |
|---|---|
| T3.0 | 5′-GGAATTCCTCAGTCACGGAAGTAATTACGC-3′ |
| 5′-CTGCTCGAGTACATGGCGCAAGATGACAAGGTAAAG-3′ | |
| T3.0 1st mut | 5′-GAG GCG ACC CGA CAT CCC AAG TTT CAG GGC CCG CTT CGT CTA ACA G-3′ |
| 5′-CTG TTA GAC GAA GCG GGC CCT GAA ACT TGG GAT GTC GGG TCG CCT C-3′ | |
| T3.0 2nd mut | 5′-GCA CAC CTT GGT CTC CGT CAA GAC CGC CCG GAA GCT CTT CGC CC-3′ |
| 5′-GGG CGA AGA GCT TCC GGG CGG TCT TGA CGG AGA CCA AGG TGT GC-3′ | |
| vSP-1 | 5′-CGGGATCCATGTTTCAGGGCCCGCTTCGTCTAACA-3′ |
| 5′-GGAATTCCCAAGGTGTGCCGTGTAGAGA-3′ | |
| vSP-2 | 5′-GGAATTCATGACCGCCCGGAAGCTCTTCGC-3′ |
| 5′-GGGCGAAGAGCTTCCGGGCGGTCTTGACGGAGACCAAGGTGTGC-3′ | |
| ORF50 (in the pCMV-cMYC-1 vector) | 5′-GGAATTCATGGCGCAAGATGACAAGGGTAAGAAG-3′ |
| 5′-CCGCTCGAGTCAGTCTCGGAAGTAATTACGCCATTG-3′ | |
| Flag tag insertion upstream of the vSP-1 AUG within the T3.0 transcript | 5′-CGACCCGACATCCCATGGACTACAAGGATGACGACGATAAGATGTTTCAGGGCCC-3′ |
| 5′-GGGCCCTGAAACATCTTATCGTCGTCATCCTTGTAGTCCATGGGATGTCGGGTCG-3′ | |
| Flag tag insertion upstream of the vSP-2 AUG within the T3.0 transcript | 5′-CCTTGGTCTCCGTCATGGACTACAAGGATGACGACGATAAGATGACCGCCCGGAA-3′ |
| 5′-TTCCGGGCGGTCATCTTATCGTCGTCATCCTTGTAGTCCATGACGGAGACCAAGG-3′ | |
| GAPDH | 5′-TGATGACATCAAGAAGGTGGTGAAG-3′ |
| 5′-TCCTTGGAGGCCATGTGGGCCAT-3′ |
Antibodies.
Mouse anti-β-actin, rabbit anti-Flag epitope, rabbit anti-Myc epitope, rabbit anti-SP1, and mouse anti-histone antibodies were purchased from Cell Signaling (Beverly, MA). A rabbit anti-ubiquitin antibody was purchased from Calbiochem (Gibbstown, NJ). A mouse monoclonal anti-RTA antibody was provided by Erle Robertson (University of Pennsylvania). A rabbit polyclonal anti-RTA antibody was provided by Charles Wood (University of Nebraska—Lincoln).
To generate an antibody against vSP-1, a keyhole limpet hemocyanin (KLH)-conjugated peptide of vSP-1 (MFQGPLRLTGRIDSDLRPVAGVTTVAGVVPPMVPQTWRHPWRAHPGGWC) was used to immunize BALB/c mice (ProMab Biotechnologies, Richmond, CA). The titers of the antibody were determined by enzyme-linked immunosorbent assays (ELISA) and by Western blotting.
Site-directed mutagenesis.
All mutations were generated using a QuikChange multisite-directed mutagenesis kit (Stratagene). Mutagenesis was performed according to the manufacturer's protocol. T3.0-1st mut and T3.0-2nd mut were generated using the pcDNA3.1-T3.0 construct as a template. The primers are shown in Table 1. The positions of the point mutations in the genome are shown in Fig. 1A.
Fig 1.

T3.0 RNA enhances the expression of RTA in cells. (A) Schematic representation of the intron-exon structure of ORF50 mRNA and the T3.0 transcript with its nucleotide positions in the KSHV genome. T3.0-1st mut and T3.0-2nd mut represent two separate point mutations. The positions of the point mutations that changed the initiation codon AUG to AAG are indicated. (B) The T3.0 expression vector and pCR3.1-ORF50 were introduced into 293T cells. RNAs were isolated at 48 h posttransfection and were subjected to Northern blot analysis for T3.0 or ORF50. 28S and 18S rRNAs served as controls to ensure the equal loading of each sample. E, empty vector. (C) 293T cells were cotransfected with 5 μg of either a T3.0 expression vector or an empty construct and 0.5 μg of pCR3.1-ORF50. Cells were analyzed by Western blotting using an anti-RTA antibody. (D) 293T cells were cotransfected with 0.1 μg of the ORF50 expression plasmid and increasing amounts of the T3.0 expression vector (0, 0.1, 2.5, or 5.0 μg). Cells were analyzed as described in the legend to panel C. The total amount of plasmids was maintained at 5.1 μg by the addition of the empty vector pCR3.1. (E) BCBL-1 cells were transfected with 3 μg of either an empty vector or the T3.0 expression vector by nucleoporation (Amaxa). After 24 h, cells were treated with 20 ng/ml of TPA for 4 h and were analyzed for the RTA protein level by Western blotting. (F) The promoter-luciferase reporter plasmids under the control of either the K8 or the ori-Lyt promoter were introduced into 293 cells with pCR3.1-ORF50 along with either an empty vector (RTA + E) or the T3.0 expression vector (RTA + T3.0). The reporter plasmid pRL-TK, encoding Renilla luciferase, was included as an internal control. Cells were lysed, and luciferase activities were measured at 48 h. The activity of firefly luciferase relative to that of the internal control, Renilla luciferase, is shown. Data are means and standard deviations for three experimental replicates.
Transient-transfection assays.
293T cells were plated into 60-mm dishes 24 h prior to transfection and were transfected with plasmid DNA using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. BCBL-1 cells were transfected by nucleoporation using the Nucleofector II device (program S002; Nucleofector kit V) (Lonza) according to the manufacturer's protocol. After 48 h, BCBL-1 cells were treated with 20 ng/ml of 12-O-tetradecanoylphorbol-13-acetate (TPA) to activate RTA expression.
Coimmunoprecipitation.
Cell lysates were clarified by high-speed centrifugation at 4°C and were subjected to immunoprecipitation with either an anti-FLAG M2 affinity gel (Sigma) or recombinant protein G (rProtein G) agarose (Invitrogen) cross-linked with antibodies against Myc epitopes (Cell Signaling) or with a monoclonal antibody against RTA overnight at 4°C. Protein complexes were washed four times in lysis buffer, eluted with a protein loading buffer, incubated at 95°C for 5 min, and resolved by immunoblotting using specific antibodies.
Western blotting.
Cell lysates or immunoprecipitates were resolved on either 4–12% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (Invitrogen) and were transferred to Hybond enhanced-chemiluminescence (ECL) nitrocellulose membranes (GE Healthcare). Membranes were blocked for 2 h in 5% nonfat dry milk containing 0.1% Tris-buffered saline (TBS)–Tween 20 and were then incubated with diluted primary antibodies overnight at 4°C. Anti-rabbit or anti-mouse immunoglobulin G antibodies conjugated to horseradish peroxidase (Amersham) were used as the secondary antibodies. An enhanced-chemiluminescence system (Amersham) was used for the detection of antibody-antigen complexes. For the RTA stability assay, cells were treated with 100 μg/ml of cycloheximide (CHX) (Sigma) 48 h after transfection and were lysed at different time points.
RNA isolation and RT-coupled quantitative PCR (RT-qPCR).
Cultured cells were lysed, and total RNA was prepared using TRIzol reagent (Ambion). RNA samples were treated with amplification-grade DNase I (Invitrogen). Reverse transcription (RT) was performed using SuperScript II reverse transcriptase (Invitrogen). Real-time PCRs were carried out in triplicate both with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) internal controls and with no-template controls by using cycle conditions of 95°C for 10 min, 45 cycles of 10 s at 95°C, 10 s at 55°C, and 10 s at 72°C, and 1 cycle of 15 s at 65°C and 30 s at 40°C in LightCycler capillaries on a LightCycler system (Roche). Each 20-μl PCR mixture contained 2 μl of relevant cDNA, 50 pmol of gene-specific primers, and 4 μl of LightCycler FastStart DNA Master SYBR green I PCR master mix (Roche). PCR product intensity data were normalized to those for GAPDH and were then analyzed with LightCycler software, version 4.0 (Roche), to calculate fold values for the appropriate parallel sample combinations based on exponential-phase measurements.
Northern blotting.
Total RNA isolated from transfected 293T cells with TRIzol reagent was separated by electrophoresis in a 1% agarose–6% formaldehyde gel in 20 mM morpholinepropanesulfonic acid (MOPS) buffer, pH 7.0. The RNA was transferred to a Nytran membrane (Schleicher & Schuell, Keene, NH) and was hybridized with a single-stranded 32P-labeled probe. The probe was prepared by using T4 polynucleotide kinase (New England Biolabs). Hybridization was carried out in ULTRAhyb solution (Ambion) for 4 h at 65°C.
Luciferase assays.
293T cells were cotransfected with a luciferase reporter construct and with the pCR3.1-ORF50 and T3.0 expression vectors. The pRL-TK plasmid was also included as an internal control that constitutively expresses Renilla luciferase. A dual-luciferase reporter assay system (Promega) was used to examine the responsiveness of the promoters to RTA. Transfected cells were washed once with 1× phosphate-buffered saline (PBS) and were suspended in 400 μl of 1× passive lysis buffer. Cells were broken by one freeze-thaw cycle and were centrifuged in a microcentrifuge for 1 min. Supernatants were assayed for firefly luciferase and Renilla luciferase activities by use of a TD-20/20 luminometer with a dual auto injector (Turner Designs). The luciferase assays were carried out according to the manufacturer's instructions (Promega).
Fractionation assay.
293T cells were cotransfected with the pCR3.1-ORF50 and Flag-vSP-1 expression vectors. Forty-eight hours posttransfection, cells were fractionated by using the Subcellular Protein Fractionation kit (Thermo Scientific) according to the manufacturer's instructions. Fractions were subjected to SDS-PAGE and Western blot analyses with specific antibodies.
Ubiquitination assay.
293T cells were transfected with relevant expression plasmids and were then treated with dimethyl sulfoxide (DMSO) or MG132 (0.5 μM) (Calbiochem) for 16 h. The cells were harvested, lysed, and immunoprecipitated using an anti-Myc antibody. The precipitates were subjected to 4–12% SDS-PAGE, followed by immunoblotting using a rabbit anti-ubiquitin antibody (Calbiochem).
Bioinformatics and statistical analysis.
NCBI BLASTn for T3.0, ORF49, ORF50, and vSP-1 was performed using genome sequences of KSHV (NCBI accession no. U75698) and the KSHV T3.0 mRNA sequence (NCBI accession no. AF402655). The nucleotide sequence of the Flag-tagged vSP-1 transcript and its predicted ORF were predicted using NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html).
Data were analyzed for statistical significance by a one-tailed Student t test. Differences with P values of ≤0.05 were considered statistically significant.
RESULTS
T3.0 RNA enhances RTA expression.
During the lytic phase of the KSHV life cycle, there is extensive transcription from noncoding regions, including both intergenic regions and noncoding regions antisense to known open reading frames (ORFs) (3, 21). A 3.0-kb polyadenylated RNA (designated T3.0) that is transcribed from the opposite strand of ORF50 was identified and has been annotated as a noncoding RNA because no large open reading frame was found in the transcript (Fig. 1A) (21). Since T3.0 RNA is potentially antisense to ORF50 mRNA, which encodes RTA, we asked whether RTA could be regulated by T3.0, either positively or negatively. To address this question, T3.0 was coexpressed with RTA in 293T cells (Fig. 1B), and the effect of T3.0 on RTA expression was examined by Western blot analysis. The results showed that RTA expression was significantly increased in the presence of T3.0 RNA, since the RTA protein level was found to be higher than that in the absence of T3.0 (Fig. 1C). RTA expression was enhanced by T3.0 in a dose-dependent manner (Fig. 1D). The effect of T3.0 on endogenous RTA was also seen in BCBL-1 cells latently infected by KSHV. Ectopic expression of T3.0 in BCBL-1 cells resulted in a significant increase in the RTA level after viral lytic replication was chemically induced with 12-O-tetradecanoylphorbol-13-acetate (TPA) (Fig. 1E). In addition, the activities of the K8 and ori-Lyt promoters, downstream target promoters of RTA, upon overexpression of T3.0 were examined by a luciferase reporter assay. The activities of both promoters were significantly enhanced with overexpression of T3.0 in 293T cells (Fig. 1F).
T3.0 RNA encodes small peptides, one of which is responsible for RTA upregulation.
Although T3.0 was annotated as a noncoding RNA, the possibility that the T3.0 transcript encodes small peptides was not excluded. The NCBI and Expasy open reading frame prediction programs suggest that there are at least three predicted small open reading frames (sORFs) with AUG start codons within the T3.0 transcript. Xu and Ganem recently reported that small peptides are synthesized from sORFs in T3.0 RNA and showed that the T3.0 transcript associates with the polyribosomes (23). We also independently investigated whether these potential small ORFs are translatable in cells by inserting a Flag tag at the N terminus of each of the potential sORFs, followed by Western blot analysis of transfected cells with an anti-Flag antibody. The results indicated that two potential ORFs, encompassing nucleotides 74356 to 74035 and 74029 to 73912, respectively, could be translated after T3.0 was introduced into 293T cells (Fig. 2A). However, the third ORF appeared not to be translated. It is worth noting that the third ORF is identical to ORF49, which has been reported not to be translated from T3.0 but is expressed by translation from a short transcript of 1.2 kb (annotated as ORF49 mRNA) (24).
Fig 2.
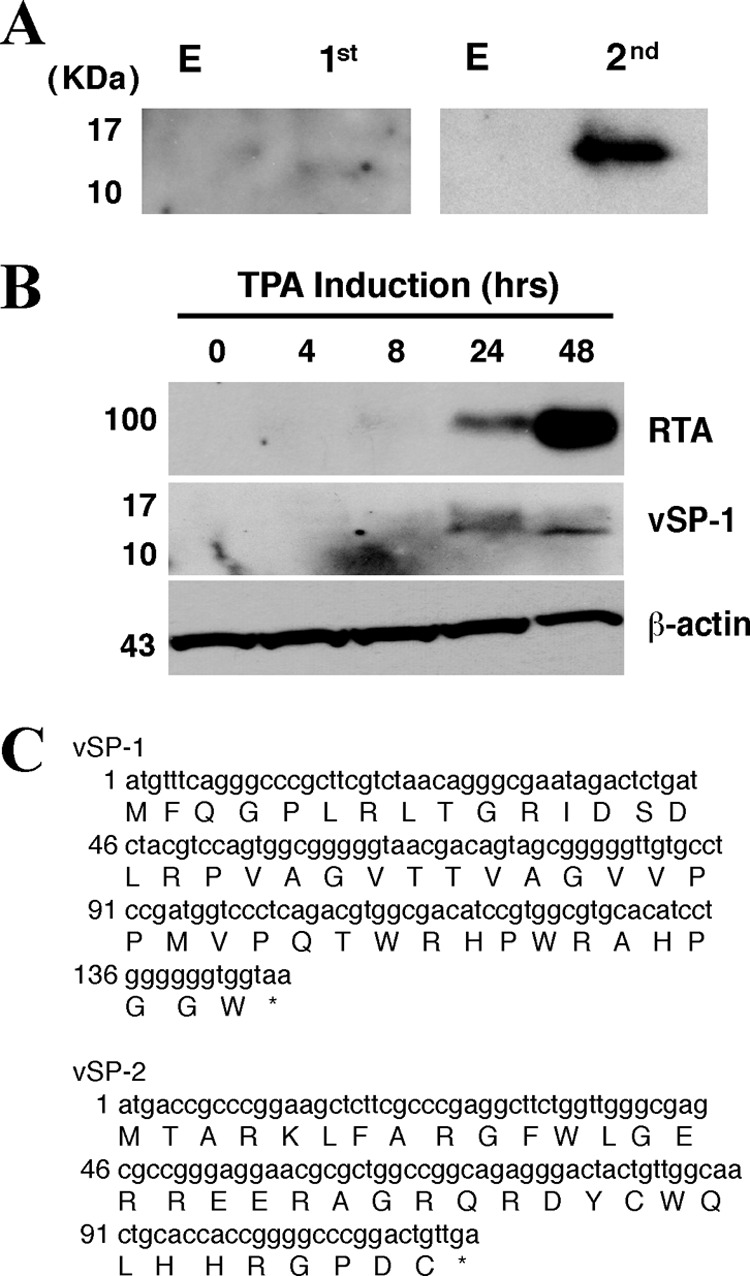
T3.0 RNA encodes two small peptides. (A) A Flag tag sequence was inserted into the T3.0 transcript just upstream of the AUG codon at position 74356 (left) or 74028 (right). The resultant constructs were introduced into 293T cells. Forty-eight hours posttransfection, cells were subjected to Western blotting using an anti-Flag antibody. (B) BCBL-1 cells were treated with 20 ng/ml of TPA and were analyzed at different time points, as indicated, by Western blotting with an anti-RTA or anti-vSP-1 antibody. (C) Nucleotide sequences of the predicted sORFs, obtained by using the NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). The amino acid sequences of vSP-1 and vSP-2 are given below the nucleotide sequences.
Furthermore, we also attempted to raise antibodies in mice against these two potential small peptides by using synthetic peptides of 48 and 38 amino acids (aa) as immunogens. A mouse antiserum reacted with the 48-aa synthetic peptide from the first AUG and with an ectopically expressed peptide in cells transfected with vectors expressing this small peptide. BCBL-1 cells were treated with TPA to induce lytic replication of KSHV and were subjected to kinetic analysis by Western blotting with the same antibody. The results showed that at 24 and 48 h postinduction, the antibody could detect a peptide at the same position as that of the Flag-tagged peptide shown in Fig. 2A, suggesting that the small peptide is indeed translated from T3.0 in cells during viral reactivation (Fig. 2B). The 48-aa small peptide, translated by using the first AUG codon of T3.0 RNA, is designated viral small peptide 1 (vSP-1), and the 38-aa small peptide, translated using the second AUG codon of T3.0 RNA, is designated vSP-2. The predicted amino acid sequences are shown in Fig. 2C.
To determine if either of these two potential small peptides regulates RTA, as seen with T3.0, two lines of experiments were carried out. First, we generated two T3.0 mutants in which the initiation codons for vSP-1 and vSP-2 were altered from AUG to AAG, respectively (Fig. 1A). When the mutant and wild-type T3.0 constructs were introduced into 293T cells along with the ORF50 expression vector, the mutation at the initial codon of vSP-1 (T3.0-1stmut, with a mutation at position 74356) abolished RTA enhancement activity, while the mutant bearing the mutation at the initial codon of vSP-2 (T3.0-2ndmut, with a mutation at position 74028) and wild-type T3.0 retained their RTA upregulation activities (Fig. 3A).
Fig 3.

vSP-1 enhances the expression of RTA. (A) 293T cells were cotransfected with pCR3.1-ORF50 and T3.0 mutant constructs as indicated, followed by Western blotting for the RTA expression level. T3.0-1st mut is T3.0 with a point mutation at position 74356, and T3.0-2nd mut is T3.0 with a point mutation at position 74028 (as diagramed in Fig. 1A). Lane E, empty vector. (B) 293T cells were cotransfected with pCR3.1-ORF50 and either vSP-1, vSP-2, or an empty vector. Then the expression of RTA and vSP-1/2 was analyzed by Western blotting. (C) Effect of vSP-1 on the expression of the KSHV lytic genes downstream of RTA in the RTA-initiated lytic gene expression cascade. BCBL-1 cells were transfected with the vSP-1 expression vector. Twenty-four hours posttransfection, cells were induced by TPA for 16 h. (Top) Cell lysates were subjected to Western blotting for the expression levels of the different KSHV viral proteins by using specific antibodies. (Bottom) The band intensities are plotted graphically. Data are means and standard deviations for three experimental replicates. *, P ≤ 0.05. (D) A Flag-vSP-1 or empty vector was introduced into 293T cells with either a K8α or an ORF45 expression vector. Transfected cells were lysed and were analyzed by Western blotting using antibodies against K8α, ORF45, and β-actin.
Second, the sequences of peptides vSP-1 and vSP-2 were cloned into pCMV-3FLAG-1 vectors, allowing expression of the small peptides with Flag tags fused at the N-terminal site. Western blot analysis of the cells transfected with these two expression vectors by use of an anti-Flag antibody showed bands between 10- and 17-kDa markers (Fig. 3B). It is not clear why the band positions of vSP-1 and vSP-2 on SDS-PAGE gels are not consistent with their calculated molecular sizes of 5.3 kDa for vSP-1 and 4.7 kDa for vSP-2. The discrepancy might be caused by the unusual secondary or tertiary structure of the peptides or by covalent modifications. The Flag-vSP-1 and Flag-vSP-2 expression vectors were used to transfect 293T cells along with the ORF50 expression vector. The results showed that vSP-1 expression increased RTA expression but vSP-2 expression did not (Fig. 3B). These two lines of evidence strongly suggest that T3.0 regulates RTA expression through a small peptide encoded by T3.0.
To further understand the role of vSP-1 in the context of viral lytic reactivation, a vSP-1 expression vector was introduced into a latently KSHV infected BCBL-1 cell line, followed by TPA induction for 24 h. The endogenous RTA protein level was higher in cells overexpressing vSP-1 than in cells transfected with an empty vector. As a consequence, the expression levels of RTA downstream target gene products, such as K8 and ORF45 proteins, were also increased in the presence of vSP-1 (Fig. 3C). The expression of K8 and ORF45 was not directly affected by the presence of vSP-1 in cells (Fig. 3D).
It appears that the degrees of upregulation of RTA and downstream viral gene expression by vSP-1 are not dramatic but subtle. This is probably the nature of regulation by small peptides, resembling that by microRNAs.
vSP-1 stabilizes RTA protein expression.
To characterize the subcellular localization of vSP-1 in cells, 293T cells cotransfected with an ORF50-expressing vector and either an empty vector or a Flag-vSP-1 vector were fractionated into cytosolic, nuclear, and chromatin-bound fractions. The presence of Flag-tagged vSP-1 in each fraction was analyzed by immunoblotting with specific antibodies against the Flag tag as well as against the proteins from various cellular compartments. The fractionation of the cells revealed the cytoplasmic localization of vSP-1 (Fig. 4A), which suggests that the small peptide is not likely to act at the level of RTA transcription. This notion was confirmed by the fact that no significant differences in RTA mRNA levels were found between cells that express vSP-1 and those that do not (Fig. 4B and 1B). In contrast, cotransfection of 293T cells with vSP-1 and RTA expression vectors resulted in elevated RTA protein levels and increased stability of the protein relative to those in cells that did not express vSP-1 (Fig. 4C). Taking these results together, we conclude that vSP-1 regulates the RTA level at a posttranscriptional level in the cytoplasm.
Fig 4.
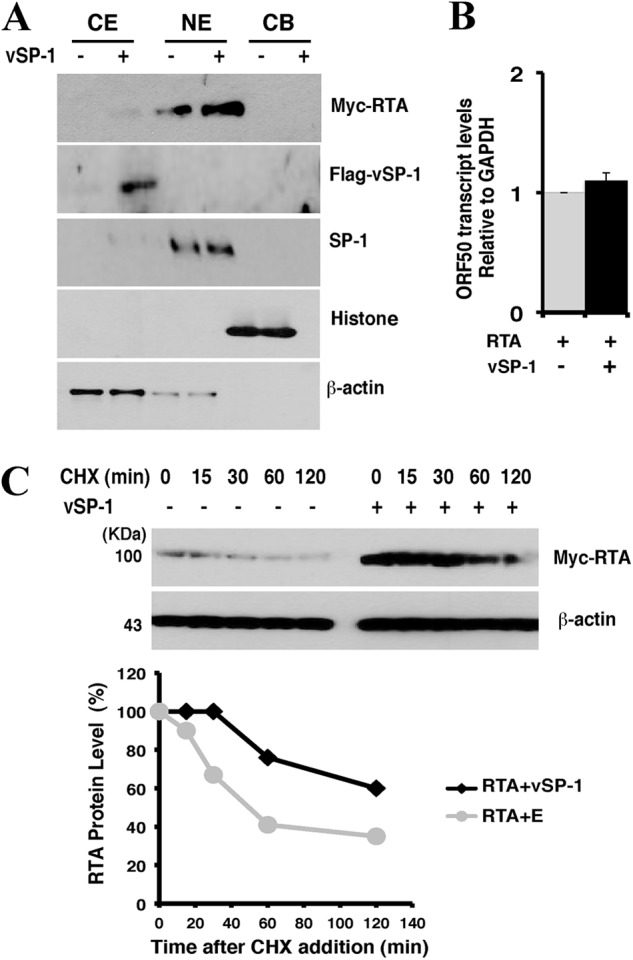
vSP-1 stabilizes RTA protein. (A) Subcellular localization of vSP-1. 293T cells were cotransfected with the Flag-vSP-1 and RTA expression vectors. Cells were fractionated using the Subcellular Protein Fractionation kit. Normalized portions of each extract were analyzed by Western blotting using specific antibodies against Flag (vSP-1) and Myc (RTA). Antibodies against proteins from various cellular compartments, including β-actin (cytoplasmic soluble proteins [CE]), SP-1 (nuclear soluble proteins [NE]), and histone (chromatin-bounded proteins [CB]), were included as references. (B) 293T cells were cotransfected with an RTA expression vector and either a Flag-vSP-1 vector (filled bar) or an empty vector (shaded bar) for 48 h. RTA mRNA was detected using RT-qPCR with specific primers. Data are means and standard deviations for three experimental replicates. (C) Measurement of RTA stability in the absence and presence of vSP-1. pCR3.1-ORF50 was introduced into 293T cells with either a Flag-vSP-1 vector or an empty vector (E). Forty hours posttransfection, cells were treated with 100 μg/ml of cycloheximide (CHX). (Top) Cells were analyzed by immunoblotting for RTA at different time points, as indicated. β-Actin was used as a control for equivalent sample loading. (Bottom) The band intensities on the exposed film are plotted graphically.
vSP-1 complexes with RTA protein.
To reveal the mechanism underlying the upregulation of RTA by vSP-1, we used a coimmunoprecipitation (co-IP) assay to determine whether vSP-1 interacts with RTA. 293T cells were cotransfected with expression vectors for RTA and Flag-tagged vSP-1. Cell extracts were immunoprecipitated with an anti-Flag antibody. Western blot analysis detected RTA in the precipitate, indicating that vSP-1 is complexed with RTA in cells where these proteins are coexpressed (Fig. 5A). Then RTA was mapped for the domain(s) that interacts with vSP-1 by use of a series of deletion mutants of RTA (D1 to D8 [Fig. 5B, top]). In a co-IP assay, mutant D7 (RTAΔ626–641) displayed a significant reduction in interaction with vSP-1, suggesting that the region missing in these deletion clones is the target of vSP-1 in the RTA (Fig. 5B, bottom).
Fig 5.
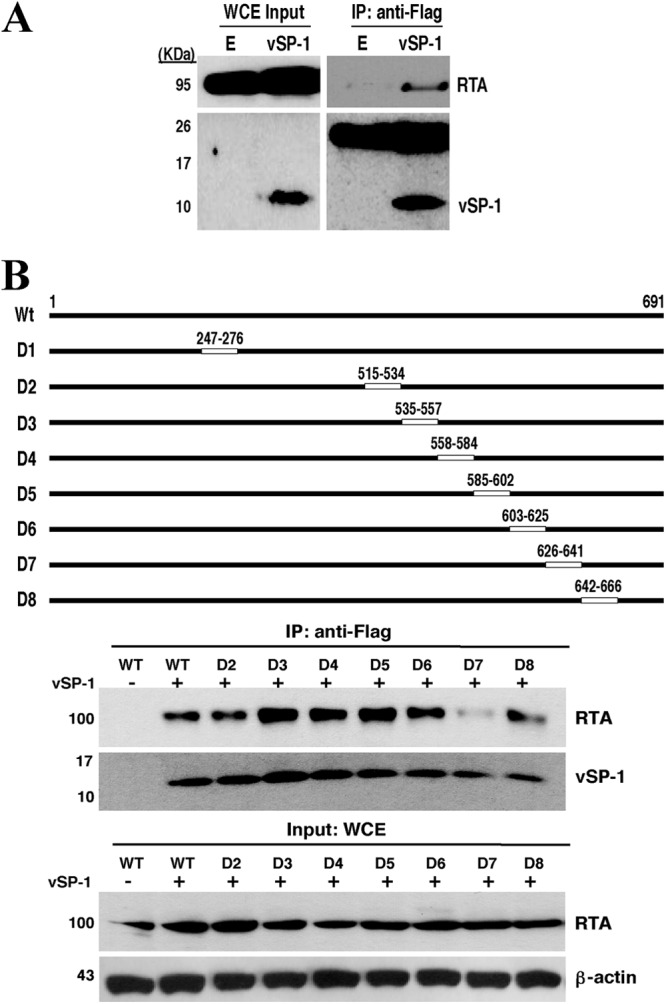
vSP-1 interacts with RTA at the PARS domain. (A) vSP-1 complexes with RTA protein. 293T cells were cotransfected with an RTA expression vector and a Flag-vSP-1 or empty (E) vector, followed by immunoprecipitation (IP) with an anti-Flag antibody and Western blotting for RTA. WCE, whole-cell extracts. (B) Mapping of RTA for the vSP-1 binding region. (Top) A set of deletion mutants of pCR3.1-ORF50 was constructed. The positions of deleted amino acids for each construct are given on the maps. (Bottom) The deletion mutants were used for coimmunoprecipitation as described in the legend to panel A. WT, wild type.
The region that is missing in mutant D7 has been reported to be part of the regulatory domains called the protein abundance regulatory signal (PARS), which consist of two components: PARS I (aa 490 to 535) and PARS II (aa 590 to 650). Mutation or deletion of either component results in abundant expression of RTA protein (25, 26). We also found that deletion of the region around the D6 and D7 deletions, or of the region around the D2 and D3 deletions, renders RTA highly and constitutively expressed. These mutants are no longer regulated by vSP-1 but are constitutively expressed at levels much higher than that of wild-type RTA (Fig. 6). Our data are consistent with the previous finding and suggest the hypothesis that vSP-1 might stabilize RTA by interacting with the PARS II domain and abrogating PARS-associated regulation.
Fig 6.
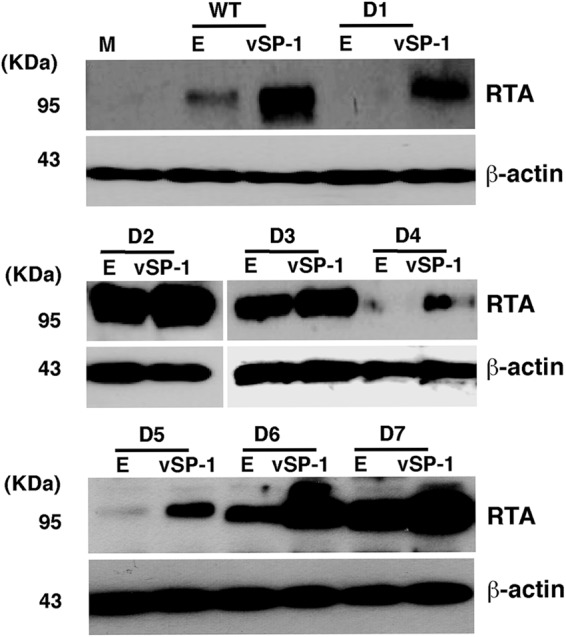
The PARS-I and PARS-II domains control the stability of RTA. 293T cells were cotransfected with the RTA expression vector pCR3.1-ORF50 and its deletion mutants as shown in Fig. 5B, with or without a vSP-1 expression construct, for 48 h. RTA expression levels were examined by Western blotting using an anti-RTA antibody. Lane M, mock transfection; lanes E, empty vector. β-Actin was used as a control for equivalent sample loading.
vSP-1 inhibits the proteasome-mediated degradation of RTA.
The PARS I and PARS II domains in RTA have been hypothesized to serve consecutively as an acceptor site for ubiquitination and a dock site for ubiquitin ligase (26). Thus, it is likely that vSP-1 stabilizes RTA by interacting with PARS domains, thereby preventing the autoubiquitination and proteasome-associated degradation of RTA. To investigate this hypothesis, 293T cells cotransfected with RTA and vSP-1 expression vectors were treated with the proteasome inhibitor MG132. In the absence of vSP-1, the steady-state level of RTA was significantly increased in cells exposed to MG132. However, in the presence of vSP-1, the abundance of RTA was dramatically enhanced regardless of the presence or absence of MG132 (Fig. 7A). To further study the effect of vSP-1 on the ubiquitination status of RTA, the cell extracts were immunoprecipitated with an antibody that brought down Myc-tagged RTA, followed by Western blotting with an anti-ubiquitin antibody. In cells that did not express vSP-1 and were treated with MG132, we observed a slowly migrating high-molecular-weight polyubiquitin-RTA smear, which was absent from cells expressing vSP-1 (Fig. 7B), suggesting a role for vSP-1 in inhibiting the ubiquitination of RTA. These data provide evidence that vSP-1 enhances the stability of RTA by preventing the degradation of RTA by the ubiquitin-proteasome pathway.
Fig 7.

vSP-1 inhibits ubiquitin (Ub)-proteasome-mediated degradation of RTA. (A) (Top) 293T cells were cotransfected with the indicated expression constructs and were treated with 0.5 μM MG-132 or 0.1% DMSO. After 16 h, cells were lysed and were used for Western blotting with antibodies against Myc, Flag, or β-actin. (Bottom) The band intensities are plotted graphically. Data are representative of a minimum of three experimental replicates with standard deviations. (B) The same lysates were subjected to immunoprecipitation using an anti-Myc antibody, followed by Western blotting (WB) for anti-ubiquitin and anti-Myc antibodies.
In addition, the treatment of cells with MG132 dramatically increased the abundance of vSP-1 (Fig. 7A). This finding suggests that vSP-1 is also under the regulation of the ubiquitin-proteasome pathway, adding an additional layer of regulation of RTA expression and the viral life cycle.
DISCUSSION
In the current study, we demonstrated that a previously annotated noncoding RNA of KSHV, namely, T3.0, modulates RTA stability, thereby facilitating viral lytic replication. Further investigation discovered that the upregulation of RTA by T3.0 is actually mediated by a small peptide translated from T3.0. This small peptide, vSP-1, binds to RTA at the PARS motif and blocks the degradation of RTA by the ubiquitin-proteasome pathway. The salient features of our finding are as follows.
First, the mammalian genome is extensively transcribed, giving rise to thousands of noncoding transcripts (4, 6). The same phenomenon is also seen in viral genomes. In the KSHV genome, Ganem and colleagues observed extensive transcription from noncoding regions, including both intergenic regions and noncoding regions antisense to known open reading frames (3, 27). These noncoding RNAs have been found to regulate a wide range of biological processes through different mechanisms. Accumulating data also suggest that some of the noncoding RNAs may encode small open reading frames (sORFs) that were previously ignored in gene annotation, since they are shorter than 100 codons (28). This notion is in accordance with (i) computational methods that determine coding potential through evolutionary information on potential sORFs conserved across species and (ii) an experimental approach, such as ribosomal profiling, that provides information on ribosome occupancy on RNA. These methods have predicted that sORFs are likely to be very abundant, and they are believed to represent an untapped source of important biology (29). However, this prediction can be proven only by clear demonstrations of the functions of the products encoded by the sORFs. Recently, a study on Drosophila showed that a set of small peptides produced from the noncoding RNA gene polished rice (pri) “control epidermal differentiation in Drosophila by modifying the transcription factor Shavenbaby (Svb)” (30). Here we present the first example of a viral sORF-encoded small peptide that regulates the viral life cycle switch from latency to lytic replication by modifying a viral transcription activator in human cells. Therefore, it is possible that mammalian cells and herpesviruses encode large numbers of small peptides that regulate various biological processes. A proteomic study on human cytomegalovirus (HCMV) virions identified 12 small peptides, ranging from 22 to 60 amino acids, associated with purified HCMV virion particles. All the sequences identified were present in all the HCMV strains (TR, PH, FIX, Merlin, Toledo, and Towne) and were 97 to 100% identical at the DNA level (31). Interestingly, the nucleotide and amino acid sequences of vSP-1 are conserved in four independent isolates of KSHV (23).
It is worth noting that there are multiple sORFs in the T3.0 transcript of KSHV. At least two of them (vSP-1 and vSP-2) have been proven to be translated, giving rise to peptides. It is possible that multicistronic expression is a common feature for sORF-carrying “noncoding” RNA in viruses and cells.
Second, the results obtained in the current study suggest a novel type of regulation of RTA expression and, as a consequence, of regulation of the lytic phase of the viral life cycle. RTA is encoded by an immediate-early gene, ORF50, and the expression of its mRNA is detected as early as 2 h during reactivation (21, 32). However, as shown in Fig. 2B, the protein level of RTA peaks at 48 h after the induction of reactivation by TPA. The inconsistency between the transcription and protein levels of RTA reflects regulation of its expression at several levels—transcription and posttranscription/protein stability. Our study led to a model for posttranscriptional regulation of RTA as follows. During the onset of KSHV reactivation, RTA expression is tightly controlled by ubiquitin-proteasomal degradation mechanisms and the cellular and viral repressors, frequently leading to the abortion of lytic replication. However, when RTA expression is robust enough or reaches a level above a threshold at a later stage, it activates certain early-gene promoters, including T3.0. The T3.0-encoded small peptide vSP-1 interacts with RTA at the PARS domain and stabilizes RTA by interfering with the interaction of RTA with its negative regulators and preventing its degradation. The elevated level of RTA overcomes its suppression and induces lytic replication.
The detailed mechanism underlying the vSP-1-mediated inhibition of RTA degradation through the ubiquitin-proteasome pathway has not yet been revealed. It has been reported that RTA ubiquitination involves both PARS I and PARS II (25, 26). There are several lysines in PARS I that may function as ubiquitination binding sites, but none in PARS II. Chang et al. (26) have proposed that one or more lysines in component I of the PARS motif may function as an acceptor site for ubiquitination and that a ubiquitin ligase (E3) may dock on component II. Since vSP-1 has been found to interact with component II of the PARS motif, it is likely that vSP-1 binds to the E3 docking site, thereby blocking RTA ubiquitination. We hypothesize further that vSP-1 may function as a ubiquitin mimic so as to occupy dock sites for ubiquitin ligase or even so as to be conjugated to the acceptor sites for ubiquitination. As a result, vSP-1, a virally encoded ubiquitin-like peptide, prevents the ubiquitination and degradation of RTA. This model is proposed based on the following observations: (i) the vSP-1 binding site in RTA has been suggested to be the dock site for ubiquitin E3 ligase (26); (ii) vSP-1 shares some common features with ubiquitin and ubiquitin-like proteins, including small size and the signature carboxyl-terminal diglycine motif, which is essential for the formation of covalent bonds between ubiquitin or ubiquitin-like proteins and their target substrates (33, 34). Further investigation is needed in order to comprehend this novel mechanism of regulation.
ACKNOWLEDGMENTS
We thank Erle Robertson (University of Pennsylvania) and Charles Wood (University of Nebraska—Lincoln) for providing anti-RTA antibodies.
This work was supported by research grant R01AI052789 from the National Institutes of Health (NIH). T.J. was supported by an NIH training grant in tumor virology (T32CA115299).
Footnotes
Published ahead of print 9 January 2013
REFERENCES
- 1. Guito J, Lukac DM. 2012. KSHV Rta promoter specification and viral reactivation. Front. Microbiol. 3:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yu Y, Wang SE, Hayward GS. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59–70 [DOI] [PubMed] [Google Scholar]
- 3. Chandriani S, Xu Y, Ganem D. 2010. The lytic transcriptome of Kaposi's sarcoma-associated herpesvirus reveals extensive transcription of noncoding regions, including regions antisense to important genes. J. Virol. 84:7934–7942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, Chalk AM, Chiu KP, Choudhary V, Christoffels A, Clutterbuck DR, Crowe ML, Dalla E, Dalrymple BP, de Bono B, Della Gatta G, di Bernardo D, Down T, Engstrom P, Fagiolini M, Faulkner G, Fletcher CF, Fukushima T, Furuno M, Futaki S, Gariboldi M, Georgii-Hemming P, Gingeras TR, Gojobori T, Green RE, Gustincich S, Harbers M, Hayashi Y, Hensch TK, Hirokawa N, Hill D, Huminiecki L, Iacono M, Ikeo K, Iwama A, Ishikawa T, Jakt M, Kanapin A, Katoh M, Kawasawa Y, Kelso J, Kitamura H, Kitano H, Kollias G, Krishnan SP, Kruger A, Kummerfeld SK, Kurochkin IV, Lareau LF, Lazarevic D, Lipovich L, Liu J, Liuni S, McWilliam S, Madan Babu M, Madera M, Marchionni L, Matsuda H, Matsuzawa S, Miki H, Mignone F, Miyake S, Morris K, Mottagui-Tabar S, Mulder N, Nakano N, Nakauchi H, Ng P, Nilsson R, Nishiguchi S, Nishikawa S, Nori F, Ohara O, Okazaki Y, Orlando V, Pang KC, Pavan WJ, Pavesi G, Pesole G, Petrovsky N, Piazza S, Reed J, Reid JF, Ring BZ, Ringwald M, Rost B, Ruan Y, Salzberg SL, Sandelin A, Schneider C, Schönbach C, Sekiguchi K, Semple CA, Seno S, Sessa L, Sheng Y, Shibata Y, Shimada H, Shimada K, Silva D, Sinclair B, Sperling S, Stupka E, Sugiura K, Sultana R, Takenaka Y, Taki K, Tammoja K, Tan SL, Tang S, Taylor MS, Tegner J, Teichmann SA, Ueda HR, van Nimwegen E, Verardo R, Wei CL, Yagi K, Yamanishi H, Zabarovsky E, Zhu S, Zimmer A, Hide W, Bult C, Grimmond SM, Teasdale RD, Liu ET, Brusic V, Quackenbush J, Wahlestedt C, Mattick JS, Hume DA, Kai C, Sasaki D, Tomaru Y, Fukuda S, Kanamori-Katayama M, Suzuki M, Aoki J, Arakawa T, Iida J, Imamura K, Itoh M, Kato T, Kawaji H, Kawagashira N, Kawashima T, Kojima M, Kondo S, Konno H, Nakano K, Ninomiya N, Nishio T, Okada M, Plessy C, Shibata K, Shiraki T, Suzuki S, Tagami M, Waki K, Watahiki A, Okamura-Oho Y, Suzuki H, Kawai J, Hayashizaki Y; FANTOM Consortium; RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group) 2005. The transcriptional landscape of the mammalian genome. Science 309:1559–1563 [DOI] [PubMed] [Google Scholar]
- 5. Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engstrom PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B, Wahlestedt C. 2005. Antisense transcription in the mammalian transcriptome. Science 309:1564–1566 [DOI] [PubMed] [Google Scholar]
- 6. Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, Gerstein M, Snyder M. 2004. Global identification of human transcribed sequences with genome tiling arrays. Science 306:2242–2246 [DOI] [PubMed] [Google Scholar]
- 7. Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR. 2002. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296:916–919 [DOI] [PubMed] [Google Scholar]
- 8. The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, Bell I, Cheung E, Drenkow J, Dumais E, Patel S, Helt G, Ganesh M, Ghosh S, Piccolboni A, Sementchenko V, Tammana H, Gingeras TR. 2007. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316:1484–1488 [DOI] [PubMed] [Google Scholar]
- 10. Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF. 1991. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349:38–44 [DOI] [PubMed] [Google Scholar]
- 11. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, Hannon GJ. 2008. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 453:534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, Surani MA, Sakaki Y, Sasaki H. 2008. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453:539–543 [DOI] [PubMed] [Google Scholar]
- 14. Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL. 2010. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142:409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wutz A, Rasmussen TP, Jaenisch R. 2002. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 30:167–174 [DOI] [PubMed] [Google Scholar]
- 16. Prasanth KV, Spector DL. 2007. Eukaryotic regulatory RNAs: an answer to the ‘genome complexity’ conundrum. Genes Dev. 21:11–42 [DOI] [PubMed] [Google Scholar]
- 17. Cai X, Hagedorn CH, Cullen BR. 2004. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10:1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fejes-Toth K, Sotirova V, Sachidanandam R, Assaf G, Hannon GJ, Kapranov P, Foissac S, Willingham AT, Duttagupta R, Dumais E, Gingersa TR. 2009. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 457:1028–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. 2004. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23:4051–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilusz JE, Freier SM, Spector DL. 2008. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135:919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saveliev A, Zhu F, Yuan Y. 2002. Transcription mapping and expression patterns of genes in the major immediate-early region of Kaposi's sarcoma-associated herpesvirus. Virology 299:301–314 [DOI] [PubMed] [Google Scholar]
- 22. Wang Y, Tang Q, Maul GG, Yuan Y. 2006. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: dual role of replication and transcription activator. J. Virol. 80:12171–12186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu Y, Ganem D. 2010. Making sense of antisense: seemingly noncoding RNAs antisense to the master regulator of Kaposi's sarcoma-associated herpesvirus lytic replication do not regulate that transcript but serve as mRNAs encoding small peptides. J. Virol. 84:5465–5475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. González CM, Wong EL, Bowser BS, Hong GK, Kenney S, Damania B. 2006. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 80:3062–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang PJ, Miller G. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 78:10657–10673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang PJ, Shedd D, Miller G. 2008. A mobile functional region of Kaposi's sarcoma-associated herpesvirus ORF50 protein independently regulates DNA binding and protein abundance. J. Virol. 82:9700–9716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chandriani S, Ganem D. 2010. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 84:5565–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frith MC, Forrest AR, Nourbakhsh E, Pang KC, Kai C, Kawai J, Carninci P, Hayashizaki Y, Bailey TL, Grimmond SM. 2006. The abundance of short proteins in the mammalian proteome. PLoS Genet. 2:e52 doi:10.1371/journal.pgen.0020052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng H, Chan WS, Li Z, Wang D, Liu S, Zhou Y. 2011. Small open reading frames: current prediction techniques and future prospect. Curr. Protein Pept. Sci. 12:503–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y. 2010. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science 329:336–339 [DOI] [PubMed] [Google Scholar]
- 31. Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kerscher O, Felberbaum R, Hochstrasser M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22:159–180 [DOI] [PubMed] [Google Scholar]
- 34. Welchman RL, Gordon C, Mayer RJ. 2005. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 6:599–609 [DOI] [PubMed] [Google Scholar]