

Abstract
Efavirenz (EFV), a nonnucleoside reverse transcriptase (RT) inhibitor, also inhibits HIV-1 particle release through enhanced Gag/Gag-Pol processing by protease (PR). To better understand the mechanisms of the EFV-mediated enhancement of Gag processing, we examined the intracellular localization of Gag/Gag-Pol processing products and their precursors. Confocal microscopy revealed that in the presence of EFV, the N-terminal p17 matrix (p17MA) fragment was uniformly distributed at the plasma membrane (PM) but the central p24 capsid (p24CA) and the Pol-encoded RT antigens were diffusely distributed in the cytoplasm, and all of the above were observed in puncta at the PM in the absence of EFV. EFV did not impair PM targeting of Gag/Gag-Pol precursors. Membrane flotation analysis confirmed these findings. Such uniform distribution of p17MA at the PM was not seen by overexpression of Gag-Pol and was suppressed when EFV-resistant HIV-1 was used. Forster's fluorescence resonance energy transfer assay revealed that Gag-Pol precursor dimerization occurred mainly at the PM and that EFV induced a significant increase of the Gag-Pol dimerization at the PM. Gag-Pol dimerization was not enhanced when HIV-1 contained the EFV resistance mutation in RT. Bacterial two-hybrid assay showed that EFV enhanced the dimerization of PR-RT fragments and restored the dimerization impaired by the dimerization-defective mutation in the RT tryptophan repeat motif but not that impaired by the mutation at the PR dimer interface. Collectively, our data indicate that EFV enhances Gag-Pol precursor dimerization, likely after PM targeting but before complete particle assembly, resulting in uniform distribution of p17MA to and dissociation of p24CA and RT from the PM.
INTRODUCTION
Retroviruses contain the virus-specific enzymes protease (PR), reverse transcriptase (RT), and integrase (IN). These enzymes are encoded within the pro and pol genes and are essential for viral infectivity. In the majority of retroviruses, these enzymes are produced by ribosomal frameshifting at the N and/or C terminus of PR in the context of Gag-Pro-Pol. In the case of human immunodeficiency virus type 1 (HIV-1), pro and pol are contiguous in the same reading frame and thus are synthesized as a 160-kDa Gag-Pol precursor protein, at a Gag-to-Gag-Pol ratio of 10:1 to 20:1, during Gag protein synthesis (1). HIV-1 Gag/Gag-Pol processing, that is, cleavage of Gag/Gag-Pol precursors into individual mature domains (2), occurs during or soon after viral particle budding (3) and is critical for virion maturation and infectivity (4). PR is the enzyme responsible for Gag/Gag-Pol processing and is known to be catalytically active as a dimer (5–7). HIV-1 PR is initially embedded in the Pol region, which consists of the N-terminal transframe region termed p6*, the central PR and RT, and the C-terminal IN, and is subsequently released autocatalytically from the Pol region to become a fully active PR dimer (8–10). Since dimerization of PR is required for activation of PR, dimerization of the Gag-Pol precursor is thought to be a prerequisite for PR activation.
The Gag protein is synthesized as a 55-kDa precursor that is composed of p17 matrix (p17MA), p24 capsid (p24CA), p7 nucleocapsid (p7NC), p6 domains, and the spacer peptides SP1 and SP2. Gag is the main virion structural protein and contains the signals for membrane targeting (in p17MA) (11, 12), particle assembly (in p24CA and p7NC) (13), and particle budding (in p6) (14). Thus, HIV particle assembly and release are essentially driven by the Gag protein (13, 15), and indeed, expression of Gag alone produces virus-like particles composed of uncleaved Gag (16). In contrast, the Gag-Pol protein is incorporated into HIV particles only through coassembly with Gag (17, 18), and PR embedded in Gag-Pol subsequently cleaves the Gag region into p17MA, p24CA, p7NC, and p6 domains. The expression ratio of Gag to Gag-Pol is critical for viral particle assembly, since overexpression of Gag-Pol and active PR leads to the cessation of particle production, accompanied by an enhancement of Gag processing in the cytoplasm (19–21). Recent studies have also indicated that the Gag-to-Gag-Pol ratio is important for virion RNA dimerization (22). In vitro processing studies with purified PR have indicated that the Gag domain junctions are cleaved at different rates (SP1-NC > SP2-p6 > MA-CA > CA-SP1), which is suggestive of ordered Gag processing (23–25).
Efavirenz (EFV), a nonnucleoside reverse transcriptase inhibitor (NNRTI), binds to the pocket of HIV-1 RT, which is close to but distinct from the polymerase active site, and allosterically inhibits RT-mediated DNA synthesis, likely due to displacement of the amino acid constellation involved in formation of the active site (26). Previous studies have shown that EFV acts as a chemical enhancer of RT dimerization (27–29). Recent studies have indicated that EFV also inhibits the late stage of the HIV-1 life cycle, i.e., virus particle release, through the enhancement of Gag/Gag-Pol processing (30). They have also shown enhanced dimerization of the Pol fragments containing PR-RT in the presence of EFV in a yeast two-hybrid assay (30). Mutations of the tryptophan repeat motif in RT that impaired RT dimerization and polymerase activity (31) rescued the defect of HIV-1 particle release imposed by EFV (32). However, more studies are needed to understand what is indeed happening in EFV-treated cells. Specific questions would address whether EFV enhances dimerization of the Gag-Pol precursors and, if so, where in the cells this occurs and what impact it has on the Gag/Gag-Pol processing products. To address these issues, we employed Forster's fluorescence resonance energy transfer (FRET) assays to study Gag-Pol dimerization. We also examined the intracellular localizations of Gag/Gag-Pol precursors and their processing products in EFV-treated cells. Our data indicate that EFV enhances Gag-Pol precursor dimerization after plasma membrane (PM) targeting but before complete particle assembly, resulting in an aberrant distribution of Gag/Gag-Pol processing products.
MATERIALS AND METHODS
DNA construction.
The HIV-1 proviral molecular clone pNL43 (33) was used as the wild type (WT) in this study. Its derivative clone containing an inactive form of PR (D25N amino acid substitution in PR) is referred to as PR(−) (34, 35). A pNL43 derivative expressing the nonmyristoylated form of Gag/Gag-Pol (G2A amino acid substitution in Gag) has been described previously (36) and is referred to as myr(−). A pNL43 derivative containing a stop codon at the p17MA-p24CA junction (referred to as p17MAstop) has been described previously (37). A pNL43 derivative expressing the Gag protein tagged with a FLAG sequence and the Gag-Pol protein tagged with a hemagglutinin (HA) sequence, each at the C terminus (referred to as Gag-FLAG/Pol-HA), and a derivative expressing Gag-Pol in which the gag and pol genes were placed into the same reading frame by deleting the 5 T nucleotides at the ribosomal frameshifting site were described previously (35). The HIV-1 recombinant molecular clone pNX and its derivative pNX(K103N), which contains the EFV resistance mutation K103N in RT, were generated in the pNL43 backbone in previous studies (38).
For FRET imaging, unique XbaI and NotI sites were initially introduced at the PR-RT junction in the pNL43 derivative PR(−), and the sequences encoding enhanced green fluorescent protein (EGFP) and the mStrawberry fluorescent protein (referred to as Gag/Pol-EGFP and Gag/Pol-mStrawberry, respectively) were inserted in frame between the XbaI and NotI sites. pNL43 derivatives expressing Gag-EGFP and Gag-mStrawberry (as positive controls) have been described in our previous studies (35). For disruption of PR dimerization, the amino acid substitutions D25N, T26A, D29A, and R87A (39) were introduced into PR by overlapping PCR (giving a product referred to as mtPR). For disruption of RT dimerization, the amino acid substitution W401A (27, 31) was introduced into RT (giving a product referred to as mtRT). The EFV resistance mutation K103N was similarly introduced into RT.
For two-hybrid assay in Escherichia coli, the HIV-1 cDNA fragments encoding PR, RT, PR-RT, p6*-PR-RT, PR-RT-IN, and the entire Pol region (p6* to IN) with inactive PR were cloned as “baits” in frame with the λ repressor protein λcI in the E. coli expression plasmid pBT, containing a chloramphenicol resistance gene (Stratagene). The fragments were also cloned as “targets” in frame with the α subunit of RNA polymerase in the E. coli expression plasmid pTRG, containing a tetracycline resistance gene (Stratagene). The cDNA fragments encoding mtPR-RT, mtRT, PR-mtRT, and RT(K103N) were similarly cloned into pBT and pTRG plasmids. The RT in this assay included the RNase H domain.
Cell culture and DNA transfection.
HeLa cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Transfection with plasmid DNA was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. EFV and nevirapine (NVP) (Moravek Biochemicals) were dissolved in dimethyl sulfoxide (DMSO) and added to cell culture after DNA transfection. The culture supernatants of HeLa cells were subjected to quantification of HIV-1 particle yields by p24CA antigen capture enzyme-linked immunosorbent assay (ELISA) (Zeptometrix).
Western blotting.
Protein samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene difluoride membrane. The membrane was incubated with mouse anti-HIV-1 p17MA (Advanced Biotechnologies), mouse anti-p24CA (40), rabbit anti-p7NC (41), sheep anti-p6 (Aalto, Ireland), rabbit anti-PR (NIH AIDS Research and Reference Reagent Program), mouse anti-RT (ICN Pharmaceuticals), mouse anti-FLAG (Sigma), and rabbit anti-HA (Sigma) antibodies and subsequently with horseradish peroxidase-conjugated secondary antibodies (ICN Pharmaceuticals). Anti-caveolin antibody was purchased from Abcam.
Membrane flotation centrifugation.
At 24 h posttransfection, HeLa cells were resuspended in buffer containing 50 mM Tris (pH 7.5), 1 mM EDTA, 150 mM NaCl, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml pepstatin A. Following a brief sonication, the cell lysates were clarified at 500 × g for 7 min at 4°C. The supernatants were adjusted to 70% (wt/vol) sucrose and overlaid with 65% and 10% (wt/vol) sucrose step gradients in phosphate-buffered saline (PBS). Equilibrium flotation centrifugation was performed in an SW55 rotor at 100,000 × g for 16 h at 4°C (37, 42).
Confocal microscopy.
HeLa cells were fixed with 3.7% paraformaldehyde in PBS for 30 min and then permeabilized with 0.1% Triton X-100 for 10 min at room temperature. After blocking with 1% bovine serum albumin, cells were incubated with mouse anti-HIV-1 p17MA (Advanced Biotechnologies), mouse anti-p24CA (40), mouse anti-RT (ICN Pharmaceuticals), mouse anti-FLAG (Sigma), rabbit anti-FLAG (Sigma), and rabbit anti-HA (Sigma) antibodies for 2 h at room temperature. Cells were subsequently incubated with Alexa Fluor 488- or 568-conjugated anti-mouse and anti-rabbit antibodies (Molecular Probes) for 1 h. After nuclear staining with TO-PRO-3 (Molecular Probes), cells were observed with a laser scanning confocal microscope (TCS-SP5; Leica).
Electron microscopy.
HeLa cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 1 h at 4°C and then treated with 2% osmium tetroxide for 1 h at 4°C. Ultrathin sections were stained with uranyl acetate and lead citrate.
FRET imaging.
At 24 h posttransfection, HeLa cells were fixed with 3.7% paraformaldehyde in PBS for 30 min at room temperature and then subjected to laser scanning confocal microscopy (CSU-X1; Yokogawa). Images were acquired using 3 filter combinations: EGFP excitation-EGFP emission (donor channel), mStrawberry excitation-mStrawberry emission (acceptor channel), and EGFP excitation-mStrawberry emission (FRET channel). FRET was confirmed by the following changes in fluorescence signals upon photobleaching of the acceptor: a decrease in acceptor fluorescence and a proportional increase in donor fluorescence.
FRET calculations were performed using the FRET calculator program FRET SE Wizard installed in a confocal microscope (TCS-SP5; Leica). From entire microscopy fields in the FRET channel, regions of interest (fluorescent puncta) were randomly selected, and their FRET values were calculated. The apparent FRET efficiency equation, FA = [B − Aβ − C(γ − αβ)]/[C(1 − βδ)], has been described previously (43). In this formula, A, B, and C are the measured intensities and correspond to donor excitation with donor emission, donor excitation with acceptor emission (FRET), and acceptor excitation with acceptor emission, respectively. α, β, γ, and δ are the calibration factors generated by the donor-only and acceptor-only references. α, β, γ, and δ correct for acceptor cross-excitation cross talk, donor cross talk, acceptor cross-excitation, and FRET cross talk, respectively. Thirty of the double fluorescence-positive cells (3 regions of interest per cell) were subjected to measurement in each experiment. In each panel, FRET efficiency was shown using a pseudocolor map over the range of 0 to 1 with a color scale bar.
Bacterial two-hybrid assay.
An E. coli two-hybrid assay based on the BacterioMatch II two-hybrid system (Stratagene) was employed in this study. E. coli strain XL-1 Blue MR containing the HIS3 gene (the reporter gene) on an F′ episome was cotransformed with pBT and pTRG plasmids. Transformants were initially grown at 37°C in M9-based synthetic medium (complete amino acid mixtures, 0.2 mM adenine, 0.5 mM isopropyl-β-d-thiogalactopyranoside, 0.4% glucose, 1 mM thiamine, and M9 salts containing 1 mM MgSO4, 0.1 mM CaCl2, and 10 μM ZnSO4) (nonselective complete conditions). The E. coli culture (optical density at 600 nm [OD600] = 0.7) was serially 10-fold diluted, and 0.6-ml aliquots were plated on M9-based synthetic agar plates without histidine but with 5 mM 3-amino-1,2,4-triazole (3-AT), a competitive inhibitor of the imidazole glycerol phosphate dehydratase encoded by the HIS3 gene (selective conditions). The 0.6-ml aliquots were similarly plated on selective agar plates including 1 μM EFV. The plates were incubated at 37°C for 3 days until colony counting.
RESULTS
Enhanced Gag processing and reduction of particle release in EFV-treated cells.
A previous study has shown that EFV enhances Gag processing, with an increase of the Gag processing intermediate p41 and final product p24CA (30). To confirm these findings, we transfected HeLa cells with WT pNL43 and added increasing doses of EFV and NVP (Fig. 1). Both are NNRTIs, but the latter has little or no effect on Gag processing (30). Intracellular Gag processing was analyzed by Western blotting (Fig. 1B). In an assay using anti-p24CA antibody, we found that the levels of p41 and p24CA were slightly increased in EFV-treated cells, consistent with previous findings (30). These band intensities were semiquantified by NIH ImageJ software, and the ratios of p41 and p24CA to p55Gag are shown in Fig. 1B. In EFV-treated cells, we also detected the processing intermediate p34, which was rarely seen in untreated cells. These processing profiles were not observed for NVP-treated cells. To identify the p41 and p34 intermediates, we carried out Western blotting with antibodies specific for p7NC and p6. The p41 intermediate was reactive with both anti-p7NC and anti-p6 antibodies, and it likely corresponded to CA-NC-p6. The p34 intermediate was reactive only with the anti-p7NC antibody, and it likely corresponded to CA-NC (Fig. 1A). The appearance of the p41 and p34 intermediates suggests, although it does not prove, preferential cleavage at the MA-CA junction in EFV-treated cells. Similar processing profiles were observed when HIV-1-transfected 293T cells were treated with EFV (data not shown). Next, we measured the levels of particle production by p24CA antigen capture ELISA. Viral particle production from HeLa cells was reduced in an EFV dose-dependent manner, and such a significant reduction was not seen by NVP treatment (Fig. 1C). A block in particle production has previously been reported for a relatively high dose of EFV (5 μM) (30).
Fig 1.
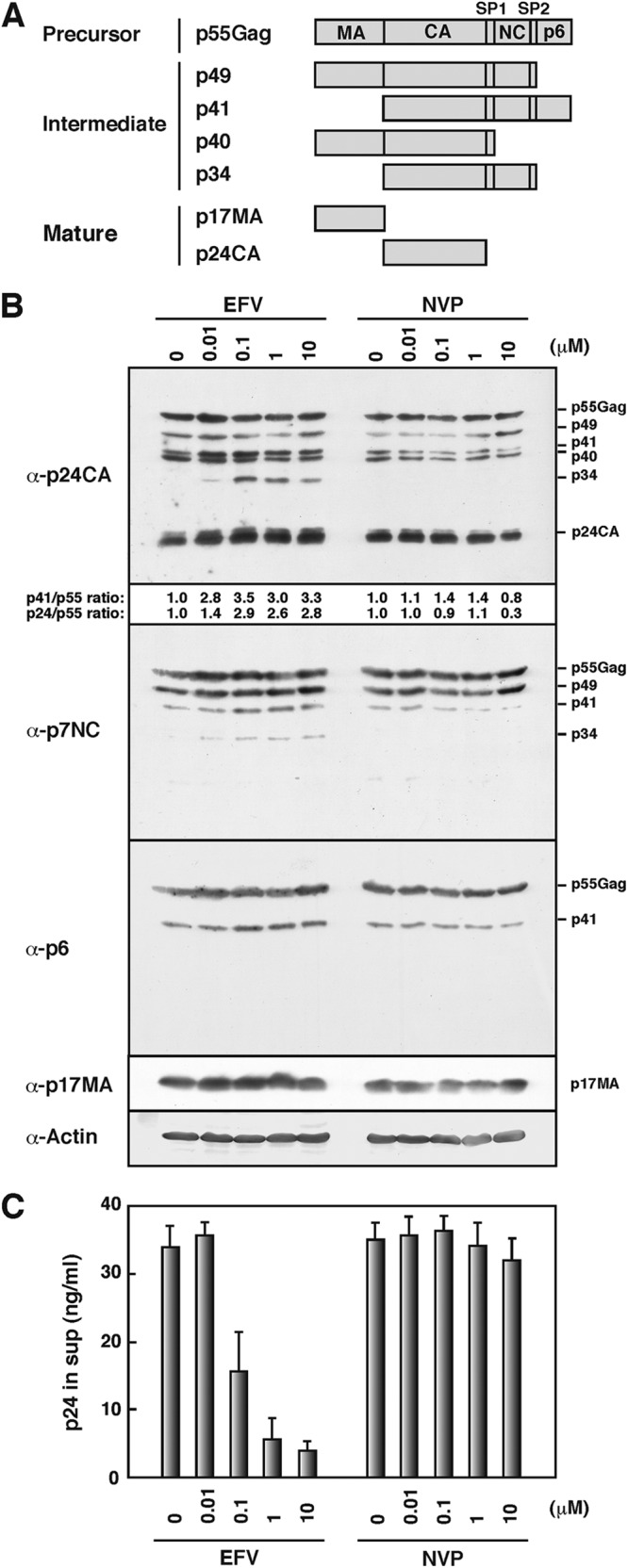
Gag processing and viral particle production. (A) Schematic representation of Gag precursor protein, processing intermediates, and mature Gag domains. (B) Intracellular Gag processing. HeLa cells were transfected with WT pNL43 and then cultured with increasing doses of EFV and NVP. DMSO was used as a control. At 48 h posttransfection, cells were collected and subjected to Western blotting using anti-p24CA, anti-p17MA, anti-p7NC, anti-p6, and anti-actin antibodies. (C) Virus particle production. Culture media at 48 h posttransfection were subjected to p24CA antigen capture ELISA. The levels in the media were normalized to the Gag antigen levels in the cells. Data are shown as means with standard deviations for 3 independent experiments, in which all samples included 0.1% DMSO.
PM targeting of Gag/Gag-Pol precursors in EFV-treated cells.
It has repeatedly been shown that enhanced Gag processing by overexpression of PR or Gag-Pol fails to produce viral particles (19–21), suggesting that earlier cleavage of Gag in the cytoplasm is unfavorable for efficient particle release. To examine whether EFV imposes similar cytoplasmic Gag processing, we carried out confocal microscopy. We first observed the localizations of Gag/Gag-Pol precursors. To this end, we used the pNL43 derivative Gag-FLAG/Pol-HA, which allows discrimination between Gag and Gag-Pol by addition of two distinct epitope tags (FLAG and HA) to the C termini of Gag and Gag-Pol, respectively. This construct shows normal intracellular transport of Gag/Gag-Pol and produces equivalent levels of viral particles compared with WT pNL43 (35). HeLa cells were transfected with the PR(−)/Gag-FLAG/Pol-HA construct, containing inactive PR, and were stained with anti-FLAG and anti-HA antibodies (Fig. 2). Three patterns of antigen distribution (diffuse cytoplasmic alone, punctate at the PM, and punctate in the cytoplasm) were observed (Fig. 2A), as reported previously, and it has been suggested that the puncta at the PM represent particle assembly and that the puncta in the cytoplasm may correspond to internalized antigens (36, 44, 45) or endosomal targeting (46, 47). Representative confocal images of EFV-treated and untreated cells suggest Gag/Gag-Pol accumulation at the PM (Fig. 2B). We observed antigen-positive cells for each dose of EFV and sorted them into three categories based on the antigen distribution patterns. Our data indicated that EFV did not impair PM targeting of Gag/Gag-Pol precursors (Fig. 2B, right panel). We also observed a slightly suppressed appearance of cytoplasmic puncta with increasing doses of EFV (Fig. 2B, right panel), but this was not seen with the Gag-FLAG construct lacking the Pol region (Fig. 2D). Next, we used the Gag-FLAG/Pol-HA construct, containing active PR, and both Gag/Gag-Pol precursors and their processing products were detected with the anti-FLAG and anti-HA antibodies (Fig. 2C). We expected that the construct containing active PR would display a distinct antigen distribution in EFV-treated cells. However, confocal microscopy and subsequent quantification of the antigen distributions revealed that the antigens accumulated at the PM in both EFV-treated and untreated cells, in patterns which were very similar to those of the PR(−)/Gag-FLAG/Pol-HA construct (compare Fig. 2B and C), although in this case the signals were likely a mixture of precursors and processed products.
Fig 2.

Intracellular localization of Gag/Gag-Pol precursor proteins. HeLa cells transfected with the Gag-FLAG/Pol-HA construct, containing either inactive or active PR, and the Gag-FLAG construct were cultured with increasing doses of EFV. DMSO was used as a control. At 24 h posttransfection, cells were immunostained with anti-FLAG and anti-HA antibodies. Nuclei were stained with TO-PRO-3. All micrographs are shown at the same magnification. (A) Representative patterns of intracellular localization of antigens. (B and C) Confocal images of Gag/Gag-Pol localizations in cells transfected with the pNL43 derivative with inactive PR (B) or active PR (C). For semiquantification of Gag/Gag-Pol localization, antigen-positive cells (approximately 80 cells) were observed for each dose of EFV treatment, and the number of cells with each pattern of Gag/Gag-Pol distribution (accumulation at the PM with diffuse cytoplasmic distribution, only diffuse distribution in the cytoplasm, or accumulation at cytoplasmic puncta) was counted. (D) Confocal images of Gag localizations in cells transfected with the pNL43 derivative expressing Gag-FLAG. Semiquantification of Gag localization was carried out similarly to that described above, using approximately 80 antigen-positive cells for each dose of EFV.
Aberrant localization of Gag/Gag-Pol processing products in EFV-treated cells.
Intracellular localization of Gag/Gag-Pol processing products was similarly observed by confocal microscopy (Fig. 3 and 4). Previous authors as well as our own group have previously shown that some anti-p17MA antibodies (against the C-terminal region of p17MA) recognize the final processing product p17MA but not Gag precursors or processing intermediates (47–49). When cells were stained with this type of anti-p17MA antibody, EFV-untreated cells showed punctate staining at the PM, confirming that Gag processing occurs mostly at the PM. In contrast, cells treated with >0.1 μM EFV displayed a uniform distribution of p17MA at the PM (Fig. 3). In the majority of Gag-positive cells (stained with anti-FLAG antibody), p17MA was evenly distributed at nearly all areas of the PM, like a fringed cell periphery (Fig. 3A). This PM staining was often accompanied by diffuse staining in the cytoplasm. We observed approximately 80 Gag-positive cells for each dose of EFV and counted the numbers of cells showing the different p17MA distribution patterns (punctate accumulation at the PM, uniform distribution at the PM, and no signal) (Fig. 3B). The data indicated that upon EFV treatment, the mature p17MA domain became evenly scattered at the PM. These confocal images were essentially identical when WT pNL43-transfected cells were treated with EFV (see Fig. 6A).
Fig 3.

Intracellular localization of mature p17MA domain. HeLa cells were transfected with the Gag-FLAG/Pol-HA construct containing active PR and were cultured with increasing doses of EFV. DMSO was used as a control. At 24 h posttransfection, cells were immunostained with rabbit anti-FLAG and anti-p17MA antibodies. Nuclei were stained with TO-PRO-3. (A) Confocal images of mature p17MA localization. All micrographs are shown at the same magnification. (B) Semiquantification of p17MA localization. Gag-positive cells probed with anti-FLAG antibody (approximately 80 cells) for each dose of EFV were subjected to observation for p17MA localization, and the number of cells with each pattern of p17MA distribution (punctate accumulation at the PM, uniform distribution at the PM, or no signal) was counted.
Fig 4.

Intracellular localizations of p24CA and RT products and electron microscopy of cells. HeLa cells were transfected with WT pNL43 and were cultured with increasing doses of EFV. DMSO was used as a control. (A) Confocal images of distributions of p24CA and RT products. At 24 h posttransfection, cells were immunostained with anti-p24CA and anti-RT antibodies. Nuclei were stained with TO-PRO-3. All micrographs are shown at the same magnification. (B) Semiquantification of p24CA and RT antigen localization patterns. Antigen-positive cells (approximately 30 cells) for each dose of EFV were subjected to counting for antigen localization patterns (accumulation at the PM, only diffuse distribution in the cytoplasm, or accumulation at cytoplasmic puncta). (C) Electron microscopy. At 24 h posttransfection, cells were subjected to electron microscopy. Micrographs are shown at the same magnification.
Fig 6.
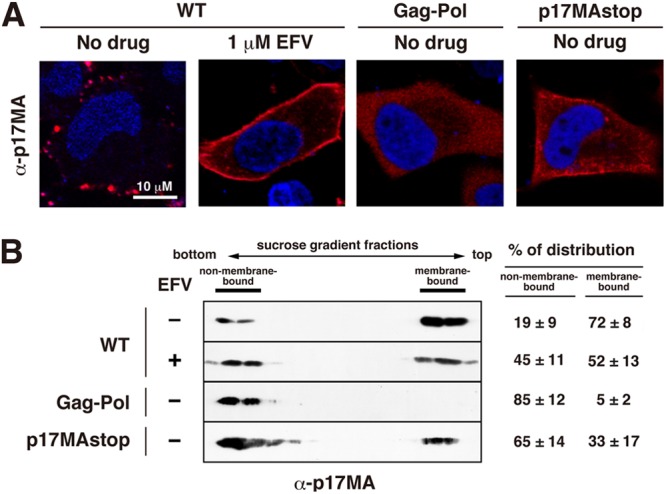
Membrane association of mature p17MA domain. HeLa cells were transfected with WT pNL43 and were cultured in the absence or presence of 1 μM EFV. For comparison, HeLa cells were transfected with a pNL43 derivative expressing Gag-Pol (containing active PR) alone via a frameshift mutation or a pNL43 derivative expressing p17MA but no other gag gene products. At 24 h posttransfection, cells were subjected to confocal microscopy and membrane flotation centrifugation. (A) Confocal images of p17MA localization. Cells were immunostained with anti-p17MA antibody. Nuclei were stained with TO-PRO-3. All micrographs are shown at the same magnification. (B) Membrane flotation analysis. Cell lysates were clarified, and supernatants were subjected to equilibrium flotation centrifugation with 70%-65%-10% (wt/vol) sucrose step gradients. The gradient fractions were subjected to Western blotting using anti-p17MA antibody. Data from 3 independent experiments were subjected to semiquantification using NIH ImageJ software, and the mean % antigen distributions to membrane-bound and non-membrane-bound fractions are shown with standard deviations.
Although antibodies that recognize only the final processing product p24CA are currently not available, it has been reported that some antibodies are skewed toward preferential detection of cytosolic monomeric Gag and the cleaved p24CA domain over highly assembled Gag precursors, due to inaccessibility of the antibodies under nondenatured conditions (50, 51). When cells transfected with WT pNL43 but untreated with EFV were stained with anti-p24CA antibody, antigens were observed in puncta at the PM, accompanied by diffuse staining in the cytoplasm. In contrast, immunostaining with the same antibody revealed no or little punctate staining at the PM but a very diffuse cytosolic distribution in WT pNL43-transfected and EFV-treated cells (Fig. 4A). We observed 30 p24CA antigen-positive cells for each dose of EFV and categorized them into three groups based on the antigen distribution patterns (diffuse cytoplasmic alone, punctate at the PM, and punctate in the cytoplasm) (Fig. 4B). Our results indicated that upon EFV treatment, p24CA-related antigens became more diffusely distributed in the cytoplasm than at the PM. Such relocation was much highlighted when cells were immunostained with anti-RT antibody. The anti-RT antibody preferentially detected punctate antigens at the PM in EFV-untreated cells. In contrast, antigens detected with this antibody were diffusely distributed in the cytoplasm in EFV-treated cells (Fig. 4A). Quantitative analysis of the antigen distribution similarly suggested that EFV treatment led to alteration of the RT-related antigen distribution to the cytoplasm from the PM (Fig. 4B).
Electron microscopy was carried out to examine whether budding structures were present at the cell surface. As shown in Fig. 4C, numerous extracellular particles were observed in EFV-untreated cells. In contrast, EFV-treated cells showed steep reductions in extracellular particles and budding structures. Small patches with an electron-dense layer were observed on the cell surface at a low frequency, but the majority of cells rarely displayed complete spherical budding structures.
Dissociation of Gag/Gag-Pol processing products from the membrane in EFV-treated cells.
To examine the membrane affinity of the processing products, membrane flotation experiments were carried out using sucrose step gradients. The gradient fractions were subjected to Western blotting using anti-p24CA, anti-p17MA, and anti-RT antibodies (Fig. 5, left panels). Antigen distribution to the membrane-bound and non-membrane-bound fractions was semiquantified using NIH ImageJ software (Fig. 5, right panels). As expected, relatively large fractions of mature p17MA, p24CA and p51/p66RT were distributed to the membrane-bound fractions in EFV-untreated cells (75%, 60%, and 69%, respectively). Significant reductions in their distributions to membrane-bound fractions were observed in 1 μM EFV-treated cells (49%, 21%, and 39%, respectively) (Fig. 5A). However, the distribution of p55Gag and p160Gag-Pol precursors to membrane-bound fractions was not largely affected by EFV treatment (Fig. 5A and B). These results suggested that the mature domains were easily dissociated from the membrane in EFV-treated cells. It is known that the membrane affinity of the p55Gag precursor is much stronger than those of C-terminally truncated Gags (52, 53). When a nonmyristoylated pNL43 derivative containing active PR was used, the p55Gag precursor and processing products were distributed exclusively to the non-membrane-bound fractions (Fig. 5C), consistent with a previous study (42). Similar membrane flotation profiles were observed for EFV-treated cells (Fig. 5C).
Fig 5.
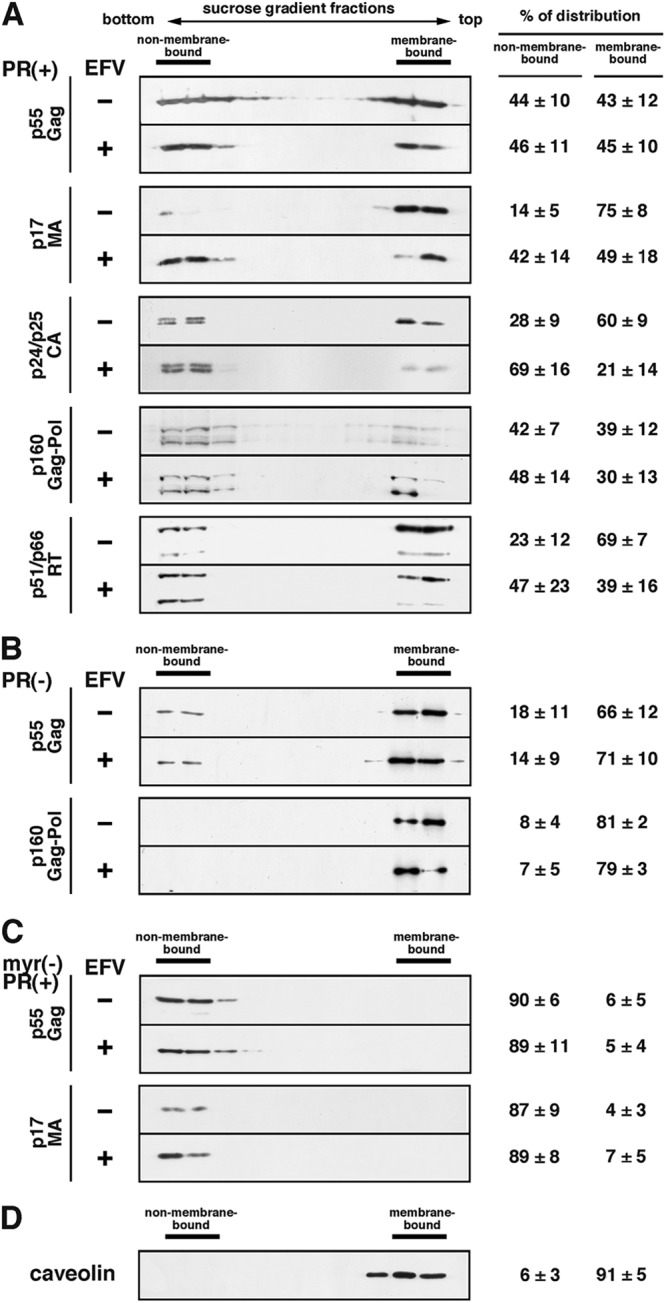
Membrane associations of Gag/GagPol precursors and their processing products. HeLa cells were transfected with WT pNL43 (A), a pNL43 derivative containing inactive PR (B), or a nonmyristoylated pNL43 derivative containing active PR (C) and were cultured in the absence or presence of 1 μM EFV. At 24 h posttransfection, cell lysates were clarified and supernatants were subjected to equilibrium flotation centrifugation with 70%-65%-10% (wt/vol) sucrose step gradients in PBS. Fractions of the gradients were subjected to Western blotting using anti-p17MA, anti-p24CA, and anti-RT antibodies. Representative blots are shown. All data from 3 independent experiments were subjected to semiquantification using NIH ImageJ software, and the mean % Gag distributions to membrane-bound and non-membrane-bound fractions are shown with standard deviations. (D) The gradient fractions were also subjected to Western blotting using anti-caveolin antibody.
Uniform distribution of p17MA at the PM in EFV-treated cells.
It has long been known that overexpression of Gag-Pol and active PR induces enhanced Gag/Gag-Pol processing in the cytoplasm (19–21). Thus, we compared the p17MA distribution pattern in WT pNL43-transfected and EFV-treated cells with the pattern for cells overexpressing Gag-Pol containing active PR (Fig. 6A). For cells expressing Gag-Pol with active PR, confocal microscopy revealed only a diffuse cytosolic distribution of p17MA, with no uniform distribution at the PM, as reported previously (35). We also employed a pNL43 derivative expressing p17MA but no other gag gene products (referred to as p17MAstop) (37) for comparison. This construct generates cleaved p17MA in the cytoplasm, and then the p17MA is targeted to the PM. Expression of p17MA by this construct showed an uneven staining with some accumulations in the cytoplasm, possibly representing promiscuous membrane targeting of p17MA. Staining at the cell periphery was also seen but was weak compared with the PM staining in EFV-treated cells (Fig. 6A). EFV-treated cells were strongly fringed with p17MA in all z-stack sections. These data suggested that the uniform distribution of p17MA at the PM observed in EFV-treated cells was not fully ascribable to cytosolic processing and subsequent targeting of p17MA to the PM.
Membrane flotation analysis was carried out to examine the membrane association of p17MA (Fig. 6B). The p17MA distribution to membrane-bound and non-membrane-bound fractions was semiquantified as described above (Fig. 6B, right panel). Our independent experiments repeatedly showed that the % distribution of p17MA to the membrane-bound fraction was as follows: EFV-untreated cells > EFV-treated cells > p17MAstop > Gag-Pol with active PR.
Enhancement of Gag-Pol dimerization at the PM in EFV-treated cells.
Because dimerization of PR is required for activation of PR and PR is synthesized as a Gag-Pol precursor, dimerization of the Gag-Pol precursor must occur before PR activation. We previously established FRET assays with the pNL43 derivatives Gag/Pol-EGFP and Gag/Pol-mStrawberry, in which the fluorescent proteins were placed in frame at the PR-RT junction containing inactive PR (35). These constructs expressed authentic Gag and GagPol tagged with EGFP or mStrawberry. We utilized the FRET assay for direct measurement of Gag-Pol precursor dimerization in this study (Fig. 7). We quantified the mean FRET efficiencies for fluorescent puncta in 30 cells. The pNL43 derivatives Gag-EGFP and Gag-mStrawberry were used as positive controls and displayed FRET signals at the PM (FA = 0.54) (Fig. 7A), consistent with previous FRET analyses of Gag-Gag interactions (54–57). Negative controls with a combination of the pNL43 derivative Gag/Pol-EGFP and cytosolic mStrawberry fluorescent protein showed no FRET signals anywhere (FA = 0.03). When FRET analysis was carried out using a combination of the Gag/Pol-EGFP and Gag/Pol-mStrawberry constructs, FRET signals were detected predominantly at the PM in EFV-untreated cells. It was possible that if EFV enhanced Gag-Pol precursor dimerization, FRET occurred in the cytoplasm as well as at the PM. However, we found that Gag-Pol precursor dimerization occurred at the PM in EFV-treated cells, as observed in untreated cells (Fig. 7A). We observed approximately 60 double fluorescence-positive cells and confirmed FRET localization at the PM (Fig. 7B, top panel). When Gag/Pol-EGFP and Gag/Pol-mStrawberry constructs containing active PR, which were uncleavable at the PR-fluorescent protein junction but cleavable at the fluorescent protein-RT junction, were used, the FRET signals were similarly observed at the PM (data not shown).
Fig 7.

Dimerization of Gag-Pol precursor in HeLa cells. HeLa cells were cotransfected with the Gag/Pol-EGFP and Gag/Pol-mStrawberry constructs, both of which had an inactive PR. At 24 h posttransfection, cells were fixed and subjected to FRET analysis by confocal microscopy. A combination of the pNL43 derivatives expressing Gag-EGFP and Gag-mStrawberry was used as a positive control, and a combination of the pNL43 derivative expressing Gag/Pol-EGFP (containing inactive PR) and a plasmid expressing mStrawberry (not fused to any other protein) was used as a negative control. mStrawb, mStrawberry. (A) FRET efficiencies were calculated from confocal images obtained with 3 filter combinations: EGFP excitation-EGFP emission (donor), mStrawberry excitation-mStrawberry emission (acceptor), and EGFP excitation-mStrawberry emission (FRET). FRET efficiencies were color coded with color scale bars over a range of 0 to 1 in confocal images. (B) (Top) Semiquantification of FRET localization. Double fluorescence (EGFP and mStrawberry)-positive cells (approximately 60 cells) were observed for each experiment. FRET signals were categorized based on their intracellular localization (at the PM or in the cytoplasm). N.A., not applicable. (Bottom) FRET efficiencies. Thirty double fluorescence-positive cells (3 regions of interest per cell) for each combination were subjected to calculation for FRET efficiencies in 3 independent experiments, and the mean FRET efficiencies are shown with standard deviations. Statistical analysis was performed with Student's t test, and P values of <0.05 are considered significant. **, P < 0.01.
The FRET efficiency for Gag-Pol dimerization in EFV-treated cells (FA = 0.41) was significantly higher than that for untreated cells (FA = 0.2), demonstrating that dimerization of Gag-Pol precursors was enhanced by EFV (Fig. 7B, bottom panel). We introduced the mutations for disruption of PR dimerization (mtPR) (39) and the W401A mutation for disruption of RT dimerization (mtRT) (31) and then examined the FRET efficiencies of the resulting constructs. In EFV-untreated cells, the FRET efficiencies were similar to or slightly lower than that for Gag-Pol without these mutations. When cells were treated with EFV, the FRET efficiency for Gag-Pol with mtRT increased (FA = 0.31), whereas that for Gag-Pol with mtPR did not (Fig. 7B, bottom panel). All of these FRET efficiencies were significantly greater than that of the negative control (Gag/Pol-EGFP plus mStrawberry). Together, these data suggested that (i) the dimerization-defective mutations mtPR and mtRT were suppressed in the context of Gag-Pol, possibly through dimerization of other domains; (ii) EFV substantially enhanced dimerization of Gag-Pol with mtRT; and (iii) the mutations in the PR dimer interface abrogated the enhancement of Gag-Pol dimerization by EFV. In our FRET system, a combination of the nonmyristoylated pNL43 derivatives myr(−)Gag/Pol-EGFP and myr(−)Gag/Pol-mStrawberry displayed no FRET signals (FA = 0.04), similar to the case for myr(−)Gag-fluorescent proteins (56, 57). However, it is likely that at least in the case of nonmyristoylated Gag-Pol, dimerization of Gag-Pol in the cytosol can occur below the detection limit of the FRET assay, because the nonmyristoylated pNL43 derivative containing active PR produced processing products (42) (also see Fig. 5C).
Efficient dimerization of PR-RT is further enhanced by EFV, but the enhancement is abrogated by dimerization-defective mutations in PR.
In the context of Gag-Pol, there are several oligomerization domains: domains for trimerization in MA, multimerization in CA-NC (15), dimerization in PR (5, 6) and RT (58), and tetramerization in IN (59, 60). To test whether the dimerization of Gag-Pol precursors observed by the FRET assay was likely through the Pol-Pol interaction, we employed two-hybrid assays. Previous studies have shown enhancement of the homodimerization of RT and PR-RT fragments by EFV in yeast two-hybrid assays (27, 28, 30). We initially used yeast two-hybrid assay systems with pAD-GAL4 and pBD-GAL4 plasmids (Stratagene) but did not see detectable levels of RT expression (data not shown), as suggested previously (61). As an alternative method, we employed a bacterial two-hybrid assay with pBT and pTRG plasmids (Stratagene) (Fig. 8A). The genes encoding PR, RT, and PR-RT, all of which contained inactive PR, were cloned into the pBT and pTRG plasmids and were expressed as λcI fusion proteins and RNA polymerase (α subunit) fusion proteins, respectively. PR-RT with an N- or C-terminal extension (p6*-PR-RT and PR-RT-IN) and the entire Pol region (p6* to IN) were similarly expressed in this system. E. coli containing the HIS3 reporter gene was cotransformed with a combination of the pBT and pTRG plasmids and then grown in M9 complete medium (nonselective conditions). Western blotting using anti-PR and anti-RT antibodies showed that the expression levels of the constructs varied but that the observed doublet band likely corresponded to both fusion proteins (Fig. 8B). To measure the dimerization of the constructs, the E. coli culture was serially diluted and plated on histidine-negative M9 medium (selective conditions) (Fig. 8C, top panel). PR and RT showed significant dimerization activities. Interestingly, despite resulting in the lowest expression level of PR-RT, this culture produced the largest number of colonies, suggesting that the dimerization of PR-RT fragments was the most efficient among the constructs used in this assay. The N-terminal p6* extension and, similarly, the C-terminal IN extension to PR-RT additively weakened the PR-RT dimerization activity. When the dimerization-defective mutations were introduced, the dimerization of mtPR-RT was significantly reduced (ca. 100-fold). The dimerization of PR-mtRT was more severely impaired (ca. 1,000-fold) but was not abolished (compare with the negative control). Thus, the measurement on the basis of colony numbers in bacterial two-hybrid assays allowed us to evaluate the extents of a broad range of protein-protein interactions.
Fig 8.

Dimerization of Pol fragments. (A) Schematic representation of E. coli two-hybrid system (Stratagene). E. coli containing the HIS3 gene was doubly transformed with pBT (for a bait) and pTRG (for a target) plasmids, both of which expressed the same pol gene fragments (indicated) with inactive PR. In this expression system, the bait is fused to λ phage repressor λcI (237 amino acids) containing the N-terminal DNA binding domain, whereas the target is fused to the N-terminal domain of the α subunit of RNA polymerase (248 amino acids). When the bait and the target interact, RNA polymerase is recruited to the promoter and activates the transcription of the HIS3 gene, which allows E. coli to grow in the absence of histidine. A combination of pBT expressing the dimerization domain of the yeast transcriptional activator Gal4 (LFG) and pTRG expressing the domain mutant Gal11 protein (Gal11P) was used as a positive control, and a combination of pBT and pTRG (empty vectors) was used as a negative control, according to the instructions for the E. coli two-hybrid system (Stratagene). (B) Expression of Pol fragments in E. coli. E. coli transformants were grown in M9 complete medium (nonselective medium), and cells (OD600 of 0.2) were subjected to Western blotting using anti-PR and anti-RT antibodies. (C) (Top) Colony formation in E. coli two-hybrid assays. E. coli transformants were grown in M9 complete medium. After being washed, the E. coli cells (OD600 of 2.1 × 10−4, 2.1 × 10−5, and 2.1 × 10−6) were plated onto M9 histidine-dropout medium containing 5 mM 3-AT in the absence or presence of 1 μM EFV and were incubated at 37°C for 3 days. (Bottom) Relative increases of colony numbers upon addition of EFV. Data shown are means with standard deviations for 3 to 5 independent experiments. Statistical analysis was performed by Student's t test, and P values of <0.05 were considered significant. *, P < 0.05; **, P < 0.01. N.A., not applicable.
To evaluate the effect of EFV on the dimerization of each construct, dilutions of E. coli cultures were similarly plated on histidine-negative M9 medium, with and without EFV, and the relative increases in colony numbers were calculated (Fig. 8C, bottom panel). EFV at 1 μM enhanced homodimerization of RT and PR-RT 2-fold, consistent with previous results of a yeast two-hybrid assay (27, 28, 30). PR dimerization was not enhanced by EFV, confirming that EFV targets RT but not PR. The dimerization of mtRT and PR-mtRT was restored by treatment with EFV, consistent with the observation that the W401A mutation counteracts an EFV-imposed block of virus production (32). In contrast, the dimerization of mtPR-RT was not restored by EFV, consistent with the results observed by our FRET assays. These results suggested that the mutations in the PR dimerization interface abrogated the EFV-mediated enhancement of RT dimerization in the context of PR-RT as well as Gag-Pol. The N-terminal p6* extension and the C-terminal IN extension to PR-RT similarly abolished the EFV effect. In sum, our two-hybrid data suggested that (i) EFV enhanced PR-RT dimerization through RT dimerization, (ii) EFV restored the impaired dimerization of PR-RT with the W401A mutation in RT, and (iii) the enhancement of PR-RT dimerization by EFV was suppressed by PR dimerization-defective mutations or an N- or C-terminal extension.
An EFV resistance mutation in RT suppresses aberrant phenotypes and enhances PR-RT dimerization.
The amino acid substitution K103N in RT is the most common and important NNRTI resistance mutation (http://hivdb.stanford.edu/pages/drugSummaries.html), and it has been reported to restore virus production in the presence of EFV (32, 62). We tested whether the aberrant phenotypes observed here were suppressed when HIV-1 contained the K103N mutation in RT. Western blotting using anti-p24CA antibody confirmed that in parental pNX-transfected cells, the levels of p41 and p24CA were slightly increased upon EFV treatment. In contrast, these levels were largely unchanged, despite increasing doses of EFV, in pNX(K103N)-transfected cells. More characteristic was the absence of appearance of the processing intermediate p34 in pNX(K103N)-transfected cells (Fig. 9A).
Fig 9.

Gag processing and p17MA localization of EFV-resistant HIV-1. HeLa cells were transfected with the parental strain pNX and its EFV-resistant variant, pNX(K103N), and were cultured with increasing doses of EFV. (A) Intracellular Gag processing. At 48 h posttransfection, cells were collected and subjected to Western blotting using anti-p24CA antibody. (B) Intracellular localization of the mature p17MA domain. At 24 h posttransfection, cells were immunostained with anti-p17MA antibody. Nuclei were stained with TO-PRO-3. All micrographs are shown at the same magnification.
Our study used the intracellular distribution pattern of p17MA as an indicator of the EFV effect. Thus, we observed the distribution of p17MA by confocal microscopy with an anti-p17MA antibody that specifically recognizes the mature p17MA domain, as described above (Fig. 9B). The pNX(K103N)-transfected cells showed punctate staining at the PM even in the presence of 1 μM EFV, whereas the pNX-transfected cells displayed uniform distribution at the PM in the presence of 1 μM EFV.
Finally, we assessed the dimerization of Gag-Pol with the K103N mutation by FRET analysis. The majority of FRET signals were observed at the PM in EFV-treated as well as untreated cells (Fig. 7B, top panel). When the FRET efficiencies were quantified, no enhancement of Gag-Pol dimerization upon EFV treatment was observed (Fig. 7B, bottom panel). Taken together, the data indicated that the aberrant phenotypes and enhanced dimerization observed upon EFV treatment were suppressed in the EFV-resistant HIV-1 variant.
DISCUSSION
EFV enhances Gag-Pol dimerization after PM targeting but before particle assembly.
EFV has been reported to reduce HIV-1 particle release through enhanced Gag/Gag-Pol processing (28, 30). In this study, we demonstrated by FRET analysis that EFV enhanced the dimerization of Gag-Pol precursors (Fig. 7). This enhancement was observed predominantly at the PM. When we examined the intracellular localizations of Gag/Gag-Pol precursors, we found that EFV had no effect on the PM targeting of Gag/Gag-Pol precursors, regardless of whether its PR was active or inactive (Fig. 2). In contrast, the intracellular localizations of the processing products (p17MA, p24CA, and RT antigens) were significantly altered upon EFV treatment. In EFV-treated cells, the N-terminal Gag domain, p17MA, was distributed uniformly at the PM, and the p24CA and RT antigens were distributed more diffusely in the cytoplasm (Fig. 4). Consistent with these findings, the processing products showed weaker membrane binding affinities and/or dissociated from the membrane in EFV-treated cells (Fig. 5). In consideration of all these findings, we suggest that the likely scenario in EFV-treated cells is that (i) Gag-Pol precursors are normally targeted to the PM through interaction with Gag precursors; (ii) EFV enhances the stability of Gag-Pol dimerization before complete particle assembly at the PM, leading to earlier activation of PR; and (iii) p17MA remains weakly associated with the PM but the p24CA and RT antigens are dissociated from the membrane, as the PR activation in EFV-treated cells occurred prior to wrapping of the Gag/Gag-Pol capsid by the membrane.
Uniform distribution of p17MA at the PM is imposed by EFV.
Because the p17MA distribution pattern in EFV-treated cells (uniform at the PM) was very unique, we compared this distribution pattern with that in cells overexpressing Gag-Pol with active PR. It is well known that overexpression of Gag-Pol with active PR leads to premature Gag processing in the cytoplasm and to a failure of particle production (19–21), suggesting that if too much Gag-Pol is synthesized, it is cleaved in the cytoplasm before particle assembly. For comparison, we also used the p17MAstop construct (37) as a p17MA domain that is synthesized alone, without its C-terminal domains. We expected that these two constructs would show very similar distribution patterns of p17MA, because in both cases the mature p17MA domain is produced in the cytoplasm. Overexpression of Gag-Pol with active PR led to only a diffuse cytosolic distribution of p17MA, whereas expression of p17MAstop led to diffuse but not cytosolic staining in the cytoplasm. A fraction of p17MAstop was localized near or at the PM. However, neither of these constructs displayed preferential accumulation of p17MA at the PM (Fig. 6A). The membrane binding affinities of these p17MA molecules, as analyzed by membrane flotation analysis, were consistent with these observations. That is, nearly half (52%) of the p17MA population was distributed to membrane-bound fractions in EFV-treated cells, whereas less p17MA was distributed to membrane-bound fractions in p17MAstop-transfected cells (33%) and PR-active Gag-Pol-transfected cells (5%) (Fig. 6B). From these data, we suggest that the p17MA observed at the PM in EFV-treated cells did not consist of p17MA molecules that were generated in the cytoplasm and then reached the PM but rather that p17MA molecules were generated by Gag processing after PM targeting.
Since Gag processing normally occurs during or soon after particle budding (3), the mature p17MA domain appears at puncta on the PM. Thus, p17MA signals at puncta on the PM have been thought to be sites where HIV-1 particles are budding (47–49) and likely correspond to highly assembled p17MA in nascent particles. In contrast, the p17MA signals in EFV-treated cells did not accumulate but were evenly distributed at the PM. The p24CA signals were considerably dissociated from the PM (Fig. 4 and 5). Taking these results together, we consider that in EFV-treated cells, p17MA may have lost the association with p24CA and p7NC, composed of Gag assembly domains after Gag processing, and may have spread evenly at the PM. The preferential cleavage at the MA-CA junction observed in EFV-treated cells (Fig. 1) may represent earlier cleavage, before Gag multimerization through Gag assembly domains.
Dimerization efficiency of Pol fragments.
We previously developed FRET systems for Gag-Pol precursor dimerization by using pNL43 derivatives in which a fluorescent protein was inserted at the Pol domain junction (35). In the present study, we used these FRET systems and showed that EFV enhanced the dimerization of Gag-Pol precursors (Fig. 7B, bottom panel), but unexpectedly, most of the FRET-positive sites were at the PM, not in the cytoplasm (Fig. 7B, top panel). Since enhanced Gag/Gag-Pol processing by EFV was still observed in a nonmyristoylated HIV-1 mutant (30), cytosolic Gag-Pol dimerization is likely below the threshold for detecting Gag-Pol dimerization in our FRET assay. However, it is possible that the dimerization of Gag-Pol precursors is generally suppressed before targeting of Gag-Pol to the PM, similar to Gag PM targeting before multimerization (55, 57). An alternative possibility is that EFV may be concentrated at the PM because NNRTIs are lipophilic compounds.
We used three Gag-Pol mutants (with PR dimerization-defective mutations, an RT dimerization-defective mutation, or an EFV resistance mutation) in FRET assays (Fig. 7B, bottom panel). We found that their Gag-Pol dimerization activities were not severely impaired by the mutations. When RT contained the EFV resistance mutation K103N, dimerization of the Gag-Pol precursors was not enhanced by EFV. However, when the dimerization-defective mutation W401A was introduced into RT, the dimerization of Gag-Pol was enhanced by EFV. The EFV binding pocket and the dimer interface formed by the tryptophan repeat motif (31, 63) were spatially apart in RT, suggesting that EFV allosterically enhances RT dimerization and rescues the dimerization defect of RT with the W401A mutation. In contrast, when dimerization-defective mutations were introduced into PR, the dimerization of Gag-Pol precursors was not enhanced by EFV (Fig. 8C, bottom panel). These data suggest that dimerization of the N-terminal PR extension may be necessary for enhancement of RT dimerization by EFV in the context of Gag-Pol.
In this study, we employed E. coli two-hybrid assays to monitor dimerization of the Pol fragments. Some studies have used yeast two-hybrid assays for this purpose and have shown that an N-terminal extension of p6* significantly inhibits PR dimerization, whereas further extension of NC restores the dimerization (64). The cis-acting inhibitory effect of p6* has also been reported in other studies (65–67). Although the dimerization efficiencies of different constructs cannot simply be compared in two-hybrid assays because their expression levels vary, we found that the dimerization efficiency of p6*-PR-RT, as judged by colony numbers in our system, was approximately 4-fold lower than that of PR-RT, consistent with the results of the above studies. The C-terminal extension of IN also significantly reduced the number of colonies that appeared upon PR-RT dimerization. Interestingly, we observed that the number of colonies that appeared upon PR-RT dimerization was 4- to 5-fold higher than that with RT dimerization, regardless of a much lower expression level of PR-RT than of RT, suggesting that unlike C-terminal extension, N-terminal extension of PR may stabilize RT dimerization. These data suggest the possibility that the dimerization efficiencies of Pol fragments continually change during ordered Gag-Pol processing, which has been reported as follows: SP1-NC > RT-IN > NC-p6*-PR > PR-RT (68). To date, there are no structural data for PR-RT or full-length Pol precursors, and only individual domain oligomers, such as PR homodimers (5, 6), RT heterodimers (58), and IN homotetramers (59, 60), have been solved by nuclear magnetic resonance and crystallography. p6* has been suggested to be unstructured by nuclear magnetic resonance spectroscopy (69). Our data may provide clues for understanding the structures of Pol processing intermediates.
ACKNOWLEDGMENTS
We thank Eric O. Freed for the supply of the p17MAstop construct and Takeshi Noda for instruction in electron microscopy.
This work was supported by a human science grant from the Ministry of Health, Labor, and Welfare of Japan and by a grant-in-aid for scientific research from the Japan Society for the Promotion of Science.
Footnotes
Published ahead of print 9 January 2013
REFERENCES
- 1. Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. 1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331:280–283 [DOI] [PubMed] [Google Scholar]
- 2. Vogt VM. 1996. Proteolytic processing and particle maturation. Curr. Top. Microbiol. Immunol. 214:95–131 [DOI] [PubMed] [Google Scholar]
- 3. Kaplan AH, Manchester M, Swanstrom R. 1994. The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J. Virol. 68:6782–6786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gottlinger HG, Sodroski JG, Haseltine WA. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 86:5781–5785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL, Springer JP. 1989. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 337:615–620 [DOI] [PubMed] [Google Scholar]
- 6. Skalka AM. 1989. Retroviral proteases: first glimpses at the anatomy of a processing machine. Cell 56:911–913 [DOI] [PubMed] [Google Scholar]
- 7. Wlodawer A, Miller M, Jaskólski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J, Kent SB. 1989. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 245:616–621 [DOI] [PubMed] [Google Scholar]
- 8. Wondrak EM, Nashed NT, Haber MT, Jerina DM, Louis JM. 1996. A transient precursor of the HIV-1 protease. Isolation, characterization, and kinetics of maturation. J. Biol. Chem. 271:4477–4481 [DOI] [PubMed] [Google Scholar]
- 9. Louis JM, Clore GM, Gronenborn AM. 1999. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 6:868–875 [DOI] [PubMed] [Google Scholar]
- 10. Tang C, Louis JM, Aniana A, Suh Clore J-YGM. 2008. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature 455:693–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chukkapalli V, Ono A. 2011. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J. Mol. Biol. 410:512–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ghanam RH, Samal AB, Fernandez TF, Saad JS. 2012. Role of the HIV-1 matrix protein in Gag intracellular trafficking and targeting to the plasma membrane for virus assembly. Front. Microbiol. 3:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ganser-Pornillos BK, Yeager M, Sundquist WI. 2008. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 18:203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morita E, Sundquist WI. 2004. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20:395–425 [DOI] [PubMed] [Google Scholar]
- 15. Morikawa Y. 2003. HIV capsid assembly. Curr. HIV Res. 1:1–14 [DOI] [PubMed] [Google Scholar]
- 16. Gheysen D, Jacobs E, de Foresta F, Thiriart C, Francotte M, Thines D, de Wilde M. 1989. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 59:103–112 [DOI] [PubMed] [Google Scholar]
- 17. Park J, Morrow CD. 1992. The nonmyristylated Pr160gag-pol polyprotein of human immunodeficiency virus type 1 interacts with Pr55gag and is incorporated into virus-like particles. J. Virol. 66:6304–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith AJ, Srinivasakumar N, Hammarskjold ML, Rekosh D. 1993. Requirements for incorporation of Pr160gag-pol from human immunodeficiency virus type 1 into virus-like particles. J. Virol. 67:2266–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park J, Morrow CD. 1991. Overexpression of the Gag-Pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J. Virol. 65:5111–5117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krausslich HG. 1991. Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. U. S. A. 88:3213–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, Moss B. 1993. Overexpression of the HIV-1 gag-pol polyprotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virology 193:661–671 [DOI] [PubMed] [Google Scholar]
- 22. Shehu-Xhilaga M, Crowe SM, Mak J. 2001. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 75:1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tritch RJ, Cheng YE, Yin FH, Erickson-Viitanen S. 1991. Mutagenesis of protease cleavage sites in the human immunodeficiency virus type 1 gag polyprotein. J. Virol. 65:922–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, Klein CA, Swanstrom R. 1994. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 68:8017–8027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hill M, Tachedjian G, Mak J. 2005. The packaging and maturation of the HIV-1 Pol proteins. Curr. HIV Res. 3:73–85 [DOI] [PubMed] [Google Scholar]
- 26. Sluis-Cremer N, Tachedjian G. 2008. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 134:147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tachedjian G, Orlova M, Sarafianos SG, Arnold E, Goff SP. 2001. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 98:7188–7193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tachedjian G, Moore KL, Goff SP, Sluis-Cremer N. 2005. Efavirenz enhances the proteolytic processing of an HIV-1 pol polyprotein precursor and reverse transcriptase homodimer formation. FEBS Lett. 579:379–384 [DOI] [PubMed] [Google Scholar]
- 29. Venezia CF, Howard KJ, Ignatov ME, Holladay LA, Barkley MD. 2006. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry 45:2779–2789 [DOI] [PubMed] [Google Scholar]
- 30. Figueiredo A, Moore KL, Mak J, Sluis-Cremer N, de Bethune MP, Tachedjian G. 2006. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2:e119 doi:10.1371/journal.ppat.0020119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tachedjian G, Aronson HG, de los Santos M, Seehra J, McCoy JM, Goff SP. 2003. Role of residues in the tryptophan repeat motif for HIV-1 reverse transcriptase dimerization. J. Mol. Biol. 326:381–396 [DOI] [PubMed] [Google Scholar]
- 32. Chiang CC, Wang SM, Tseng YT, Huang KJ, Wang CT. 2009. Mutations at human immunodeficiency virus type 1 reverse transcriptase tryptophan repeat motif attenuate the inhibitory effect of efavirenz on virus production. Virology 383:261–270 [DOI] [PubMed] [Google Scholar]
- 33. Adachi A, Koenig S, Gendelman HE, Daugherty D, Gattoni-Celli S, Fauci AS, Martin MA. 1987. Productive, persistent infection of human colorectal cell lines with human immunodeficiency virus. J. Virol. 61:209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang M, Orenstein JM, Martin MA, Freed EO. 1995. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 69:6810–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haraguchi H, Sudo S, Noda T, Momose F, Kawaoka Y, Morikawa Y. 2010. Intracellular localization of human immunodeficiency virus type 1 Gag and GagPol products and virus particle release: relationship with the Gag-to-GagPol ratio. Microbiol. Immunol. 54:734–746 [DOI] [PubMed] [Google Scholar]
- 36. Kawada S, Goto T, Haraguchi H, Ono A, Morikawa Y. 2008. Dominant negative inhibition of human immunodeficiency virus particle production by the nonmyristoylated form of Gag. J. Virol. 82:4384–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gatanaga H, Hachiya A, Kimura S, Oka S. 2006. Mutations other than 103N in human immunodeficiency virus type 1 reverse transcriptase (RT) emerge from K103R polymorphism under non-nucleoside RT inhibitor pressure. Virology 344:354–362 [DOI] [PubMed] [Google Scholar]
- 39. Koh Y, Matsumi S, Das D, Amano M, Davis DA, Li J, Leschenko S, Baldridge A, Shioda T, Yarchoan R, Ghosh AK, Mitsuya H. 2007. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 282:28709–28720 [DOI] [PubMed] [Google Scholar]
- 40. Tsunetsugu-Yokota Y, Ishige M, Murakami M. 2007. Oral attenuated Salmonella enterica serovar Typhimurium vaccine expressing codon-optimized HIV type 1 Gag enhanced intestinal immunity in mice. AIDS Res. Hum. Retroviruses 23:278–286 [DOI] [PubMed] [Google Scholar]
- 41. Ohishi M, Nakano T, Sakuragi S, Shioda T, Sano K, Sakuragi J. 2011. The relationship between HIV-1 genome RNA dimerization, virion maturation and infectivity. Nucleic Acids Res. 39:3404–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paillart JC, Gottlinger HG. 1999. Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of gag membrane targeting. J. Virol. 73:2604–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Rheenen J, Langeslag M, Jalink K. 2004. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys. J. 86:2517–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4:e435 doi:10.1371/journal.pbio.0040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Krausslich HG. 2007. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 3:e36 doi:10.1371/journal.ppat.0030036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nydegger S, Foti M, Derdowski A, Spearman P, Thali M. 2003. HIV-1 egress is gated through late endosomal membranes. Traffic 4:902–910 [DOI] [PubMed] [Google Scholar]
- 47. Ono A, Freed EO. 2004. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J. Virol. 78:1552–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ono A, Orenstein JM, Freed EO. 2000. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J. Virol. 74:2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suyama M, Daikoku E, Goto T, Sano K, Morikawa Y. 2009. Reactivation from latency displays HIV particle budding at plasma membrane, accompanying CD44 upregulation and recruitment. Retrovirology 6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ono A, Waheed AA, Joshi A, Freed EO. 2005. Association of human immunodeficiency virus type 1 Gag with membrane does not require highly basic sequences in the nucleocapsid: use of a novel Gag multimerization assay. J. Virol. 79:14131–14140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. LeBlanc JJ, Perez O, Hope TJ. 2008. Probing the structural states of HIV-1 Pr55gag by using monoclonal antibodies. J. Virol. 82:2570–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sandefur S, Varthakavi V, Spearman P. 1998. The I domain is required for efficient plasma membrane binding of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 72:2723–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ono A, Demirov D, Freed EO. 2000. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J. Virol. 74:5142–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Derdowski A, Ding L, Spearman P. 2004. A novel fluorescence resonance energy transfer assay demonstrates that the human immunodeficiency virus type 1 Pr55Gag I domain mediates Gag-Gag interactions. J. Virol. 78:1230–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. 2007. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 81:12596–12607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li H, Dou J, Ding L, Spearman P. 2007. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J. Virol. 81:12899–12910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hogue IB, Hoppe A, Ono A. 2009. Quantitative fluorescence resonance energy transfer microscopy analysis of the human immunodeficiency virus type 1 Gag-Gag interaction: relative contributions of the CA and NC domains and membrane binding. J. Virol. 83:7322–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. 1992. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256:1783–1790 [DOI] [PubMed] [Google Scholar]
- 59. Wang JY, Ling H, Yang W, Craigie R. 2001. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 20:7333–7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tachedjian G, Aronson HG, Goff SP. 2000. Analysis of mutations and suppressors affecting interactions between the subunits of the human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 97:6334–6339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bacheler LT, Anton ED, Kudish P, Baker D, Bunville J, Krakowski K, Bolling L, Aujay M, Wang XV, Ellis D, Becker MF, Lasut AL, George HJ, Spalding DR, Hollis G, Abremski K. 2000. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob. Agents Chemother. 44:2475–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mulky A, Sarafianos SG, Jia Y, Arnold E, Kappes CJC. 2005. Identification of amino acid residues in the human immunodeficiency virus type-1 reverse transcriptase tryptophan-repeat motif that are required for subunit interaction using infectious virions. J. Mol. Biol. 349:673–684 [DOI] [PubMed] [Google Scholar]
- 64. Zybarth G, Carter C. 1995. Domains upstream of the protease (PR) in human immunodeficiency virus type 1 Gag-Pol influence PR autoprocessing. J. Virol. 69:3878–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Partin K, Zybarth G, Ehrlich L, DeCrombrugghe M, Wimmer E, Carter C. 1991. Deletion of sequences upstream of the proteinase improves the proteolytic processing of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 88:4776–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tessmer U, Krausslich HG. 1998. Cleavage of human immunodeficiency virus type 1 proteinase from the N-terminally adjacent p6* protein is essential for efficient Gag polyprotein processing and viral infectivity. J. Virol. 72:3459–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Louis JM, Wondrak EM, Kimmel AR, Wingfield PT, Nashed NT. 1999. Proteolytic processing of HIV-1 protease precursor, kinetics and mechanism. J. Biol. Chem. 174:23437–23442 [DOI] [PubMed] [Google Scholar]
- 68. Pettit SC, Lindquist JN, Kaplan AH, Swanstrom R. 2005. Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology 2:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Beissinger M, Paulus C, Bayer P, Wolf H, Rosch P, Wagner R. 1996. Sequence-specific resonance assignments of the 1H-NMR spectra and structural characterization in solution of the HIV-1 transframe protein p6. Eur. J. Biochem. 237:383–392 [DOI] [PubMed] [Google Scholar]