

Abstract
Extremophilic archaea, both hyperthermophiles and halophiles, dominate in habitats where rather harsh conditions are encountered. Like all other organisms, archaeal cells are susceptible to viral infections, and to date, about 100 archaeal viruses have been described. Among them, there are extraordinary virion morphologies as well as the common head-tailed viruses. Although approximately half of the isolated archaeal viruses belong to the latter group, no three-dimensional virion structures of these head-tailed viruses are available. Thus, rigorous comparisons with bacteriophages are not yet warranted. In the present study, we determined the genome sequences of two of such viruses of halophiles and solved their capsid structures by cryo-electron microscopy and three-dimensional image reconstruction. We show that these viruses are inactivated, yet remain intact, at low salinity and that their infectivity is regained when high salinity is restored. This enabled us to determine their three-dimensional capsid structures at low salinity to a ∼10-Å resolution. The genetic and structural data showed that both viruses belong to the same T-number class, but one of them has enlarged its capsid to accommodate a larger genome than typically associated with a T=7 capsid by inserting an additional protein into the capsid lattice.
INTRODUCTION
Archaea dominate in extreme environments like hypersaline lakes and hot springs (1, 2) and serve, along with their viruses, as biological models in such environments. So far, about 100 archaeal viruses have been described, in contrast to over 6,000 known bacterial viruses (3–5). All the isolated archaeal viruses infect extremophiles which belong to either of the two major archaeal phyla, Crenarchaeota, containing thermophiles and hyperthermophiles, or Euryarchaeota, containing methanogens, thermophiles, hyperthermophiles, and extreme halophiles (3, 6).
Viruses infecting extremely halophilic hosts have been found to tolerate a wider range of salinities than their host cells, which are usually sensitive to lowered ionic strengths (7–9). Furthermore, the adsorption of some viruses of halophilic hosts is dependent on salinity (7, 10, 11). Thus, it would be important to determine how salinity alterations affect viruses. For a viral infection, it is crucial that host cells are energetically capable of supporting infections, and viruses might be expected to be able to sense environmental conditions which affect host cell viability. For example, Semliki Forest virus is reversibly inactivated by mildly acidic conditions but retains infectivity upon reneutralization, and it was shown that low pH inactivates the membrane fusion capability of the virus (12). Inactivation of bacteriophage T7 by ascorbic acid treatment, which causes single-stranded nicks to accumulate in the virus genome, is reversible if the host cell mechanisms are allowed to repair the nicks before the virus is plated with the host (13).
Archaeal viruses are currently classified into nine viral families (14). Although unusual morphotypes, like bottle- and droplet-shaped morphotypes, have been discovered, about half of the studied archaeal viruses resemble classical bacteriophages, with icosahedral heads and helical tails (3, 5). These viruses are classified by tail morphology into three families, Myoviridae (long, contractile tails), Siphoviridae (long, noncontractile tails), and Podoviridae (short tails), and together, they form the order Caudovirales (3, 14, 15). The host range of the known archaeal head-tailed viruses (∼50) is limited to euryarchaeal hosts, and most of them infect extremely halophilic hosts, while only five viruses have been described to infect methanogens (3, 5). Although no head-tailed viruses infecting hyperthermophilic archaea have yet been isolated, such virus-like particles have been observed in hydrothermal environments (16). So far, detailed characterization of archaeal head-tailed viruses has been limited mostly to the genome level (17–21), and, in contrast to the extensive structural research on head-tailed bacteriophages, no virion structures are available for archaeal head-tailed viruses.
Icosahedrally symmetric viruses are built up of 60 repeats of an asymmetric unit (22). In those viruses exhibiting quasiequivalence, more than one copy of a protein makes up the asymmetric unit, existing in different conformations (23). The quasiequivalence theory places the capsomers in positions that can be described by subtriangulating the icosahedral facets. The triangulation number (T-number) is a parameter used to describe the capsomer (hexamer and pentamer) arrangement and can be calculated by using the formula T = h2 + hk + k2, where h and k are positive integers defining the pentameric and hexameric positions in the lattice (23, 24).
All head-tailed bacteriophages most likely belong to the same structure-based viral lineage, sharing a common ancestor (25–27). Despite the evolutionary relationship, the capsid size and thus the genome length of these viruses vary considerably, and the size difference between the smallest and largest genome known so far is almost 50-fold (28). Two possibilities have been proposed to explain this size variation: (i) larger genomes originated from smaller ones by acquisition of new genes, or (ii) smaller genomes were derived from larger ones by gene deletion (28–30). However, the former scenario seems to be more reasonable, because icosahedrally symmetric capsids can adopt only certain sizes according to the triangulation numbers, and the reduction of the capsid size would be a more complex process than its enlargement (23, 28, 31). So far, three different mechanisms have been shown to be used by viruses to enlarge the capsid size: (i) a larger T-number, (ii) a larger major capsid protein (MCP), or (iii) a prolate instead of an isometric capsid (32–36).
Here, we report a detailed description of the siphovirus HVTV-1 (Haloarcula vallismortis tailed virus 1) and the myovirus HSTV-2 (Halorubrum sodomense tailed virus 2), infecting extremely halophilic archaea (5). We demonstrate that halophilic viruses tolerate periods of lowered salinity, which is important for their survival in nature but also for their suitability for structural analyses. Thus, in addition to genome sequences, we report three-dimensional (3D) cryo-electron microscopy (cryo-EM) reconstructions of the capsids for both viruses. Although HVTV-1 (T=13) and HSTV-2 (T=7) have different T-numbers, they belong to the same morphological class, class 2, of icosahedrally symmetric viruses (24). Furthermore, the genome size of HSTV-2 is considered large for a T=7 capsid, and we show that HSTV-2 has solved this problem by inserting a minor protein into the capsid lattice.
MATERIALS AND METHODS
Virus and archaeal strains and growth conditions.
Haloarcula vallismortis strain ATCC 29715 (37) and Halorubrum sodomense strain DSM 3755 (38) were used as hosts for HVTV-1 and HSTV-2, respectively (5). All cultures were grown aerobically at 37°C in modified growth medium (MGM) (39, 40). A 30% stock solution of artificial salt water (SW) was prepared as described by the Halohandbook website (http://www.haloarchaea.com/resources/halohandbook/Halohandbook_2008_v7.pdf) and contains 240 g NaCl, 30 g MgCl2 · 6H2O, 35 g MgSO4 · 7H2O, 7 g KCl, 5 ml of 1 M CaCl2 · 2H2O, and 80 ml of 1 M Tris-HCl (pH 7.2) per liter of water. Broth, solid, and soft-agar media contained 23, 20, and 18% SW, respectively. Virus stocks were prepared as previously described (40), using confluent plates incubated for 3 days (HSTV-2) or 5 days (HVTV-1). Titers of about 1 × 1011 and 4 × 1010 PFU/ml were obtained in HVTV-1 and HSTV-2 stocks, respectively.
Life cycle studies.
Virus adsorption to host cells was studied by infecting mid-log-phase cells, and the virus stocks in MGM were used. Approximately 3 × 108 cells and 1,500 virus particles were mixed and incubated at 37°C without aeration. Aliquots were removed at 15, 30, and 60 min after infection, and the number of free viruses was determined by using a plaque assay. The adsorption rate (k) constant was calculated by using the formula k = (2.3/Bt) × log(P0/P), where B is the cell concentration, P0 is the concentration of unadsorbed viruses at the beginning, P is the concentration of unadsorbed viruses at the end, and t is the time interval (41).
Viral life cycles were determined by infecting mid-log-phase Haloarcula vallismortis and Halorubrum sodomense cells at a multiplicity of infection (MOI) of 10. After 30 min of incubation at 37°C without aeration, the cells were collected by centrifugation (14,000 × g for 10 min at 20°C) (Sorvall SA600) and washed twice to remove unadsorbed viruses. Cultures were grown aerobically at 37°C, and turbidity and the number of free viruses were monitored. Thin sections of HVTV-1-infected Haloarcula vallismortis cells were prepared at 0.5, 10, and 12 h postinfection (p.i.). The cells were washed with MGM broth containing 1 M 4-morpholineethanesulfonic acid (MES) (pH 6.7) (MGM-MES broth) and fixed with 3% (vol/vol) glutaraldehyde (Sigma-Aldrich) at room temperature (RT) for 20 min. After fixation, the cells were washed three times with MGM-MES broth, and thin sections were prepared as previously described (42). The micrographs were taken with a Jeol 1200EX electron microscope operating at 80 kV at the Electron Microscopy Unit of the Institute of Biotechnology, University of Helsinki.
Virus purification.
Virus particles were produced by using liquid culture propagation. Culture conditions were the same as those used for the life cycle study (see above). The infected cultures were grown overnight and then treated with DNase I (70 μg ml−1; Sigma-Aldrich) for 1 h at 37°C. The cell debris was removed (11,000 × g for 30 min at 15°C) (Sorvall SLA3000), and virus particles were precipitated from the supernatant by using 10% (HVTV-1) or 11% (HSTV-2) polyethylene glycol (PEG) 6000 (wt/vol). The precipitated particles were collected (11,000 × g for 40 min at 15°C) (Sorvall SLA3000) and resuspended in 18% SW. Aggregates were removed from the dissolved PEG precipitate by centrifugation (14,000 × g for 10 min at 15°C) (Sorvall SA600).
The virus concentrate was purified in a linear 5 to 20% (wt/vol) sucrose gradient in 18% SW by rate-zonal centrifugation (104,000 × g for 1 h 5 min for HVTV-1 and 1 h 40 min for HSTV-2 at 15°C) (Sorvall AH629). The light-scattering virus zone was collected and further purified in a CsCl gradient in 18% SW (mean density, 1.5 g/ml) by equilibrium centrifugation (72,000 × g for 20 h at 15°C) (Sorvall AH629). The virus zone was collected and diluted 2-fold with 18% SW without NaCl. The purified virus particles were concentrated by differential centrifugation (210,000 × g for 1 h 15 min at 15°C) (Sorvall TH641), and the pellets were resuspended in 9% SW or 20 mM Tris-HCl (pH 7.2). For density determination, the CsCl gradients were fractionated after equilibrium centrifugation (83,000 × g for 24 h at 15°C) (Sorvall TH641), and the density and A260 of the fractions were determined.
Stability assays.
HVTV-1 and HSTV-2 sensitivity to lowered ionic strength was studied by diluting the virus stocks 1,000-fold in 9, 18, and 23% SW and incubating the virus stocks for 1 h at 4°C. After the incubation, dilutions series were prepared in the corresponding SW and incubated for 15 min at RT, and the infectivity was determined by a plaque assay. The virus stocks were also diluted 100- and 1,000-fold in 20 mM Tris-HCl (pH 7.2) and incubated for 1 h at 4°C. After this, dilution series were prepared in 9% SW and 20 mM Tris-HCl (pH 7.2) and incubated for 15 min at RT, and the infectivity was determined by a plaque assay. In all plaque assays, the components were added in the following order, to avoid contact of the cells and the low-salinity buffers: host cells, soft agar, and virus dilutions.
In order to determine how fast the viruses are inactivated at lowered ionic strength, the virus stocks were diluted 100-fold (HSTV-2) or 1,000-fold (HVTV-1) in 20 mM Tris-HCl (pH 7.2) and incubated at 4°C. After 5, 15, 30, and 60 min, dilutions series were prepared in 9% SW and 20 mM Tris-HCl (pH 7.2). The dilution series were incubated for 15 min at RT, and the infectivity was determined by a plaque assay. In order to determine the reactivation time, the virus stocks were diluted 100-fold (HSTV-2) or 1,000-fold (HVTV-1) in 20 mM Tris-HCl (pH 7.2) and incubated for 60 min at 4°C. The dilution series of the samples were prepared in 9% SW and incubated for 5, 10, and 15 min at RT, and the infectivity was determined by a plaque assay.
The CsCl-purified virus particles in 20 mM Tris-HCl (pH 7.2) or 9% SW (see above) were used for the virion stability assays. The virus pellets (see differential centrifugation described above) were resuspended and incubated for 15 min at 4°C, after which the A260 was measured, the infectivity was determined by a plaque assay, and the particles were vitrified. Alternatively, the particles, which were incubated in 20 mM Tris-HCl (pH 7.2) for 15 min, were diluted with 18% SW to obtain a final concentration of 9% SW. This dilution was incubated for 15 min at RT, after which the A260 and the infectivity were measured, and the virions were vitrified.
Cryo-electron microscopy and image processing.
Freshly made virus particles were applied in 3-μl aliquots onto Quantifoil R 2/2 grids and vitrified in liquid ethane, as described previously (43). The micrographs were recorded by using a Gatan 626 cryo-holder and an FEI Tecnai F20 microscope operating at 200 keV at liquid nitrogen temperature (at the Cryo-Electron Microscopy Unit of the Institute of Biotechnology, University of Helsinki). Images were collected under low-dose conditions on a Gatan Ultrascan 4000 charge-coupled-device (CCD) camera at a magnification of ×68,000 with a 4.0-μm underfocus (virus particles from the virus stability assay described above) or in 20 mM Tris-HCl (pH 7.2) on Kodak SO163 film at a magnification of ×62 000 and with a range of underfoci of 0.4 to 2.9 μm (HVTV-1) or 0.7 to 2.9 μm (HSTV-2), as described previously (44).
The micrographs recorded on film were scanned at 7-μm samplings with a 12-bit grayscale readout by using a PhotoScan TD scanner (Z/I Imaging). The digitized micrographs were binned to a pixel size of 2.26 Å/pixel, and the contrast transfer function (CTF) parameters of the images were evaluated by using CTFFIND3 (45). Viral particles were picked by using ETHAN (46) and selected in the program BOXER of the EMAN suite (47). The particles were boxed, normalized, and apodized with the program RobEM (48) using the EMAN coordinates.
The random model computation (RMC) method (49) was used to produce 10 initial models of each virus from 150 randomly selected far-from-focus particles to about 30 Å. The best model of each virus was used as a starting model for full orientation search and origin determination of each full data set using AUTO3DEM (48). After several iterations, particles were recentered by using RobEM, and iterations were continued with these images (44). Full CTF correction was carried out with AUTO3DEM as a part of the reconstruction calculation, and the resolution was estimated by using a Fourier shell correlation cutoff of 0.5 (50). A total of 3,530 and 4,930 particles were used for the full reconstruction of HVTV-1 and HSTV-2, respectively.
The final maps were sharpened by using the program EM-Bfactor (51, 52). The reconstructions were first calculated out to the Nyquist frequency and then corrected with the estimated B-factors (407.20 for HVTV-1 and 435.42 for HSTV-2). Finally, the corrected maps were filtered back to the original resolution by using Bsoft (53). All visualization was carried out with UCSF Chimera (54).
Genome sequencing and protein analysis.
Purified virus genomic DNA was sheared hydrodynamically and repaired, and 1- to 3-kbp fragments were inserted into the plasmid pBluescript. Individual clones were sequenced to depths of 10-fold (HSTV-2) and 11-fold (HVTV-1) coverages using an ABI 3700 sequencer and assembled by using phredPhrapp and Consed (version 16) at the Pittsburgh Bacteriophage Institute. A total of 958 reads (HSTV-2) and 1,495 reads (HVTV-1) were assembled. Finished sequences were analyzed and annotated with genome editors, including DNAMaster (http://cobamide2.bio.pitt.edu/), Glimmer (55), GeneMark (56), tRNA ScanSE (57), COILS (58), and Aragorn (59), to identify genome features. Functional identifications of the genes are based on BLASTP (60) matches with expect values better than 10−7, except as noted in Results and for some functional domains which were assigned based on HHpred (61–63) data with a probability of greater than 97.2.
Protein concentrations were determined by the Coomassie blue method, using bovine serum albumin as a standard (64). Modified Tricine-SDS-polyacrylamide gel electrophoresis (Tricine-SDS-PAGE) with 4 and 14% acrylamide concentrations in the stacking and separation gels, respectively, was used to analyze the samples (65). N-terminal sequencing was performed as previously described (66), at the Protein Chemistry Core Facility of the Institute of Biotechnology, University of Helsinki.
Accession numbers.
The reconstructions have been deposited in the EMDB under accession numbers EMD-2234 (HVTV-1) and EMD-2235 (HSTV-2). The nucleotide sequences determined have been deposited in GenBank under accession numbers KC117376 (HSTV-2) and KC117377 (HVTV-1).
RESULTS
HVTV-1 and HSTV-2 have a long intracellular phase.
In order to study the life cycle of the viruses, the host cells were infected in the mid-log growth phase. The adsorption experiments showed that HVTV-1 had high and HSTV-2 low adsorption efficiencies, as the adsorption rate constants (41) calculated during the first 15 min were 3.1 × 10−10 and 1.4 × 10−11 ml/min, respectively. In the one-step growth experiment, HVTV-1 lysed the infected cells at ∼12 h p.i., and the increase in the number of extracellular viruses coincided with lysis (Fig. 1A). The viral levels reached approximately 3 × 1010 PFU/ml in the growth medium. The HSTV-2-infected cells lysed at ∼10 h p.i., and a virus yield similar to that of HVTV-1 infection was obtained (data not shown). The HVTV-1 infection cycle was also studied by using electron microscopy. Thin sections of the infected cells showed virions adsorbed onto the cell surface and, later on, production of intracellular progeny virions (Fig. 1B to D). Empty capsids, most likely procapsids preceding genome packaging, were still observed at 12 h p.i. in the lysing cells (Fig. 1D), indicating that not all the virions were filled.
Fig 1.

Infection cycle of HVTV-1 in its host Haloarcula vallismortis. (A) Turbidity of infected (open circles) and uninfected (closed circles) cultures and number of free viruses in the infected culture (bars). The cultures were washed at 30 min p.i. to remove unadsorbed viruses. Arrows indicate time points of the thin-section samples. (B to D) Micrographs of thin sections of the infected cells observed by transmission electron microscopy at 30 min (B), 10 h (C), and 12 h (D) p.i. Bars, 200 nm (B) and 500 nm (C and D). In panel B, arrows show virions adsorbed to the cell surface. In panels C and D, arrows indicate intracellular genome-containing virions. In panel D, the arrowhead shows an empty capsid.
Liquid culture propagation was used for virus production and purification (see Materials and Methods). The specific infectivities of the purified HVTV-1 and HSTV-2 virions in 9% SW were approximately 3 × 1012 and 4 × 1012 PFU/mg of protein, respectively. The specific infectivity calculated based on the nucleic acid content was about 6 × 1010 PFU/A260 for both viruses in 9% SW. The densities of HVTV-1 and HSTV-2 in CsCl were 1.47 and 1.46 g/ml, respectively, corresponding to that of bacterial sipho- and myoviruses (15).
Low salinity causes reversible inactivation of HVTV-1 and HSTV-2.
The virus stocks contained 23% SW, corresponding to 3.15 M NaCl (3.44 M total salinity). In order to study the stability of the viruses under conditions of lowered ionic strength, the virus stocks were diluted 1,000-fold in 23%, 18% (2.47 M NaCl), and 9% (1.23 M NaCl) SW and in 20 mM Tris-HCl (pH 7.2). Both viruses remained infective in the SW buffers but were inactivated in the Tris-HCl buffer (Fig. 2A and B). However, when HVTV-1 incubated in Tris-HCl buffer was restored to 9% SW, the virus regained its infectivity (Fig. 2A). Reactivation of HSTV-2 was observed only when the virus was first diluted only 100-fold in the Tris-HCl buffer and then brought back to 9% SW (Fig. 2B). Kinetics studies indicated that inactivation and reactivation were achieved in less than 5 min (data not shown).
Fig 2.

Viruses are reversibly inactivated under conditions of low salinity without observable morphological changes. (A and B) Effect of lowered ionic strength on the infectivity of HVTV-1 (A) and HSTV-2 (B). The virus stocks (white bars) were diluted either 100-fold (black bars) or 1,000-fold (gray bars) in 23, 18, or 9% salt water (SW) or in 20 mM Tris-HCl (pH 7.2) and incubated for 1 h at 4°C. After incubation, the samples were further diluted in the same buffer, except that the Tris-HCl samples were diluted in 9% SW as well. The diluted samples were incubated for 15 min at RT, and infectivity was then determined. The bars represent the averages of data from two experiments. The infectivity scale in panels A and B is the same, and the infectivities are corrected for the dilution. (C) Cryo-electron micrographs of HVTV-1 at a 4-μm underfocus. (I) Virions incubated in 9% SW; (II) virions incubated in 20 mM Tris-HCl (pH 7.2) buffer; (III) virions which were first incubated in 20 mM Tris-HCl (pH 7.2) and then restored in 9% SW. Bar, 50 nm. (D) Specific infectivity on a logarithmic scale of HVTV-1 virions at different salinities (I to III). (E) Cryo-electron micrographs of HSTV-2 at a 4-μm underfocus showing particles as in panel A. In panel I, the arrow indicates a noncontracted tail, the arrowhead indicates a contracted tail, and the insets show longitudinal views of baseplates dissociated from the capsids. Bar, 50 nm. (F) Specific infectivity on a logarithmic scale of HSTV-2 virions at different salinities (I to III).
As inactivation was reversible, the effect of altering salinity on overall virus morphology was determined. The highly purified virions were incubated under three different conditions: (i) in 9% SW, (ii) in Tris-HCl (pH 7.2), and (iii) first in Tris-HCl (pH 7.2) and then in 9% SW. Cryo-electron micrographs show that the overall morphology remained unaltered during the salinity changes (Fig. 2C and E). The specific infectivity was the same before and after the low-salt conditions (Fig. 2D and F), which correlates well with the observation that the empty/DNA-filled capsid ratio was approximately the same (data not shown) in all three preparations, indicating that the salinity changes did not cause genome release.
Genome sequences of HVTV-1 and HSTV-2.
The genome sequences of HVTV-1 (Fig. 3) and HSTV-2 (Fig. 4) were determined, and we report our analysis of the two sequences separately below.
Fig 3.

Physical map of the HVTV-1 genome. Genes are represented as rectangles, with those above the central scale transcribed from left to right and those below transcribed from right to left, and the markers are spaced at 1-kbp and 100-bp intervals. All predicted genes are protein encoding, with the exception of gene 55, which encodes a tRNAGln. DNA in the virion has a 585-bp direct terminal repeat, indicated by arrows. HNH, histidine-asparagine-histidine homing endonuclease.
Fig 4.

Physical map of the HSTV-2 genome. Genes are represented as rectangles above or below the central scale, depending on whether they are transcribed from left to right or right to left, respectively, and the markers are spaced at 1-kbp and 100-bp intervals. All predicted genes are protein encoding, with the exception of gene 70, which encodes a tRNAGln. DNA in the virion has a 340-bp direct terminal repeat, indicated by arrows.
(i) HVTV-1 genome.
The unique genome sequence of HVTV-1 is 101,734 bp (Fig. 3). The virion DNA has discrete ends with a direct terminal repeat of 585 bp, bringing the length of the packaged DNA to 102,319 bp. The G+C content of the genome is 58%. The sequence bears no close overall similarity to any previously reported viral sequence.
Considering first the structural genes, we identified the MCP gene as gene 120, based on the N-terminal sequence of the most abundant protein in the SDS gel pattern (Fig. 5A). The mature capsid protein sequence begins at residue 103 of the predicted sequence of gp120 (gp for gene product), indicating that the capsid protein is N-terminally processed. Such processing of capsid proteins during capsid maturation is common but not universal among tailed bacteriophages. Gene product 119 has a “peptidase U35” match in the Conserved Domain Database (CDD). This maturation protease family is found in many tailed-bacteriophage genomes, including coliphage HK97. The position of this gene in the genome, particularly its relationship to the MCP gene and other identifiable head genes, together with its sequence affinity for characterized head maturation proteases make a good case that the function of this gene product is likely to be as the maturation protease of HVTV-1. The protein sequence of gene product 114 contains a C-terminal domain that shows similarity to the accessory head assembly factor of phage Mu protein F and gp7 of Bacillus phage SPP1. Gene product 110 is a homologue of the large terminase subunit genes of tailed bacteriophages.
Fig 5.

HVTV-1 and HSTV-2 structural proteins. (A) Protein pattern of virions in a Tricine-SDS-polyacrylamide gel stained with Coomassie blue. 1× indicates virions purified in a sucrose gradient by rate-zonal centrifugation, and 2× indicates virions further purified in a CsCl gradient by equilibrium centrifugation. Numbers on the left indicate molecular mass markers. (B) Amino acid sequences of the major structural proteins. The N-terminal sequence determined from the protein band is shown by boldface type and underlining, except that the sixth amino acid in HSTV-2 gp20 could not be identified. The theoretical molecular masses of the mature proteins are given in parentheses.
There are two tail genes whose encoded protein sequences give weak but convincing matches in a BLASTP+PSI search. Gene product 131 matches a large number of tail-length tape measure protein (TMP) sequences, and gp136 matches a tail baseplate protein found in some bacteriophages. The identification of the TMP is strengthened by the fact that the sequence is predicted to be largely alpha-helical by COILS, with significant coiled-coil character, as is characteristic of these proteins. TMPs are thought to span the length of the tail as an extended alpha-helix, and the length of the protein sequence, 480 amino acids (aa), predicts a tail length of 72 nm when fully alpha-helical. The measured length of the helical tail sheath is in approximate agreement at 73 nm ± 5 nm (Fig. 2C). As with HSTV-2 (see below) and most long-tailed bacteriophages, there are two overlapping open reading frames (ORFs) immediately upstream of the tape measure protein gene (genes 129 and 130) that can be related by a putative −1 translational frameshift.
Of the 174 putative protein-encoding genes that we annotate in HVTV-1, only 43 (25%) make a significant match to sequences in the GenBank database in a BLASTP search (see Table S1 in the supplemental material). Beyond the putative head and tail genes described above, many of the genes for which a possible function can be inferred appear to be involved in nucleotide metabolism or DNA replication. These are predominantly but not exclusively found in the leftward-transcribed cluster of genes bounded by genes 35 and 104, while the head and tail genes that we have identified are in the rightward-transcribed cluster bounded by genes 108 and 140. Of the genes predicted to encode protein functions other than virion structure and assembly functions, we note that there are three, genes 37, 87, and 96, predicted to encode subunits of replication factor C (RFC). RFC is a two-subunit protein that serves as the DNA replication clamp loader in archaea (67). The products of genes 87 and 96 make strong matches to the large and small subunits, respectively, of the cellular RFC, and it seems likely that they constitute a virus-encoded clamp loader. The product of gene 37 makes a strong match (BLASTP E value of 3 × 10−20) to the cellular RFC small subunit, but this match extends over only ∼20% of its length, suggesting that it may have a different function. Another intriguing match is that of gp115 to Zeta-toxin, which is a component of a toxin/antitoxin system found in bacteria and archaea (68). There is no evidence for its cognate antitoxin from sequence searches of the HVTV-1 genome. The role of this Zeta-toxin homologue in the viral life cycle is unclear. There is one tRNA predicted in the HVTV-1 genome, an apparent tRNAGln (UUG anticodon), encoded by gene 55.
(ii) HSTV-2 genome.
The HSTV-2 unique genome sequence is 68,187 bp long (Fig. 4). The DNA in the virion has discrete ends and direct terminal repeats of 340 bp, bringing the size of the virion DNA to 68,527 bp. The G+C content of the genome is 60%. The HSTV-2 genome is similar in sequence and gene arrangement to the previously characterized archaeal myoviruses HF1 and HF2 (20, 21). Dot plot comparison shows that the overall similarity of the HSTV-2 and HF1 and HF2 genomes is moderate, 70.4 and 59.8% nucleotide identities, respectively, over the length of HSTV-2, with the degree of similarity falling off at the right end of the HSTV-2 genome (data not shown). Overall, the HF1 genome is 7,372 bp longer and the HF2 genome 9,484 bp longer than the HSTV-2 genome. A striking difference between the genomes is an apparent net deletion of 12,250 bp in HSTV-2 relative to HF2, located between HSTV-2 genes 40 and 41.
Analysis of the HSTV-2 genome identified 104 putative ORFs (see Table S2 in the supplemental material). Several genes can be identified as encoding structural proteins of the virion or virion assembly factors. The most abundant protein on the SDS-PAGE gel of HSTV-2 virions (Fig. 5A) is, by that criterion, the MCP. Its N-terminal sequence identifies it as the product of gene 13. Unlike the MCP of HVTV-1 (see above) and like the MCPs of some tailed bacteriophages, HSTV-2 gp13 is not N-terminally processed, except for the first methionine. HSTV-2 gp13 shows significant sequence identity to the hypothetical protein HalHV1gp089 of HF1 (43%) and to the hypothetical proteins HF2p101 and HF2p102 of HF2 (41%). Two more abundant proteins on the gel are identified by the N-terminal sequence as the products of genes 20 and 21. Gene product 20 makes a convincing sequence match to the tail sheath subunits of contractile-tailed bacteriophages, and its high molecular weight and the position of its gene relative to the MCP gene support this identification. The tail tube subunit gene in contractile-tailed bacteriophages typically immediately follows the tail sheath gene, and we make this assignment for gene 21. This identification is supported by the high abundance of gp21 in the virion and its characteristically relatively low molecular weight (Fig. 5).
Somewhat distant but convincing sequence similarities identify the products of genes 7, 8, and 30 as the large subunit of terminase, the portal protein, and a tail baseplate component, respectively. These identifications are compatible with the gene positions expected from tailed phages. We make four additional assignments in the structural gene region based on gene position and sequence characteristics. Gene 12 encodes a protein with a high propensity for alpha-helical structure. This, together with its position immediately upstream of the MCP gene, allows a tentative identification as the head assembly scaffolding protein, based on precedents from tailed bacteriophages. Gene 24 encodes another protein with high predicted alpha-helicity as well as significant coiled-coil propensity, and this, together with gene position, suggests that this is the tail-length TMP. Gene product 24 would be 98 nm long as an alpha-helix, and the measured length of the HSTV-2 tail is 101 ± 5 nm (Fig. 2E), supporting this identification. Between the tail tube gene (gene 21) and the TMP gene (gene 24), there are two overlapping open reading frames. This pattern is seen in most long-tailed bacteriophages, where expression of the downstream ORF is accomplished by a low-frequency translational frameshift. We propose that expression in the case of HSTV-2 includes such a frameshift. A frameshift into the +1 frame would be required in this case, and we find a potential +1 “shifty” sequence in the overlap region.
Genes 41, 44, and 48 encode proteins that show strong sequence similarities to thymidylate synthase, DNA polymerase, and adenylate kinase, respectively. This small cluster of nucleotide metabolism and DNA replication genes lies adjacent to the location where HF2 has 12,250 bp more than HSTV-2, and this “extra” DNA in HF2 encodes at least two additional enzymes of nucleotide metabolism, a dCTP deaminase and a ribonucleotide reductase (alpha subunit). HSTV-2 gene product 75 has the closest sequence BLAST (E value of 0) and HHpred (100% probability) matches to bacterial XerCD members of the integrase family.
The products of genes 26, 34, 51, 52, 72, and 93 were assigned to a function based on HHpred data. Considering the position of gene 26 in the genome, its product may be a tail component. Gene 34 is predicted to encode a metal-binding protein. Genes 51 and 52 most probably encode a dual-specificity phosphatase and an ATP-dependent helicase, respectively. Gene 72 encodes a putative homing endonuclease, and gp93 is a putative heat shock protein. Finally, we find a single tRNA in the HSTV-2 genome, a tRNAGln (UUG anticodon) located just upstream from gene 69. This tRNA has the same apparent amino acid specificity and anticodon as the tRNA in HVTV-1, but the two sequences are only 62% identical.
HSTV-2 has a larger genome than expected based on the capsid T-number.
Cryo-electron micrographs showed that HVTV-1 and HSTV-2 had angular, icosahedrally symmetric capsids (Fig. 2C and E). HVTV-1 had a long, thin siphovirus-like tail structure (Fig. 2C). For HSTV-2, we observed a long, thick myovirus-like tail with tail fibers and both contracted and noncontracted tails with baseplates (Fig. 2E) as well as end-on views of isolated baseplates (Fig. 2E, inset). 3D icosahedral reconstruction was used to solve the structure of HVTV-1 and HSTV-2 DNA-filled capsids under low-salinity conditions to 10.5- and 9.8-Å resolutions, respectively (Fig. 6). The dimensions of the HVTV-1 capsid were 760 Å (facet to facet) and 955 Å (vertex to vertex). The corresponding dimensions for HSTV-2 were 679 and 738 Å (Fig. 6A and B). The central sections of the capsids showed that HSTV-2 is more spherical than HVTV-1 and revealed several highly ordered layers of concentrically packaged DNA (Fig. 6A and B). The spacing between the DNA layers was 24 Å, and the genome packaging densities calculated based on the genome length and the inner icosahedral volume were 0.51 bp/nm3 (HVTV-1) and 0.58 bp/nm3 (HSTV-2). The genome packaging efficiencies (69), calculated based on the formula ρpack = 0.34πNbp/Ωcapsid (where Nbp is the number of base pairs and Ωcapsid is the capsid volume), were 0.55 for HVTV-1 and 0.62 for HSTV-2. The genomes of both viruses were least well organized under 5-fold vertices (Fig. 6A and B), indicating the presence of a portal structure in one of the 12 vertices.
Fig 6.

Cryo-EM reconstruction of the HVTV-1 and HSTV-2 capsids. (A and B) Central section of HVTV-1 (A) and HSTV-2 (B) capsids. The icosahedral reconstructions are viewed along a 2-fold axis. The ellipse, triangle, and pentagon indicate 2-fold-, 3-fold-, and 5-fold-symmetry axes, respectively. Bar, 10 nm. (C and D) Isosurface representation of HVTV-1 (C) and HSTV-2 (D) capsid reconstructions along an icosahedral 2-fold axis. The capsids are displayed at a threshold of a sigma value of 3.0. The symmetry axes are shown as in panel A. Coloring was done using radial-depth cueing (bar, 325- to 425-Å radius) in UCSF Chimera (54).
The MCP capsomers of HVTV-1 were arranged in a T=13 lattice, and those of HSTV-2 were arranged in a T=7 lattice (Fig. 6C and D). Homology searches using I-Tasser (70) did not reveal any structural homologues for the MCPs of HVTV-1 and HSTV-2 (data not shown), and the resolution of the capsid reconstructions was too low to allow reliable fitting of known MCP structures. The HVTV-1 capsid had 5-fold-symmetric spikes projecting from the vertices and trimeric decorative structures, most likely formed by a minor capsid protein, occupying the center of each MCP hexamer (Fig. 6C). In the HSTV-2 capsid, a minor capsid protein formed additional trimeric structures between the MCP pentamers and hexamers and between the MCP hexamers, at the local 3-fold positions (Fig. 6D).
HSTV-2 was observed to deviate from the general trend when the genome sizes of head-tailed viruses and herpes simplex virus 1 (HSV-1) were compared to their capsid T-number (Fig. 7A). Among the T=7 head-tailed viruses, for which the genome sequences are available, HSTV-2 had the largest genome (Fig. 7B). The MCPs of epsilon15, T7, and lambda (71) are about the same size as the HSTV-2 MCP. However, the HSTV-2 genome is 1.4 times longer than the genome of lambda and 1.7 times longer than the genomes of epsilon15 and T7.
Fig 7.

Genome size and T-number comparison. (A) Genome size versus T-number. Triangles indicate HSTV-2 (GenBank accession number KC117376) and HVTV-1 (accession number KC117377), accordingly. Circles indicate epsilon15 (accession number AY150271), HK97 (accession number AF069529), T7 (accession number V01146), P22 (accession number BK000583), lambda (accession number J02459), T5 (accession number AY543070), SPO1 (accession number FJ230960), HSV-1 (accession number X14112), and phiKZ (accession number AF399011). (B) Genome sizes of T=7 head-tailed viruses.
The facet-to-facet diameter of mature lambda virions is 600 Å, and as the shell thickness is about 40 Å, the corresponding inner diameter of lambda is 520 Å (72). The inner diameter of the HSTV-2 virion (facet to facet) was considerably larger, 620 Å (Fig. 6B). A closer comparison of the HSTV-2 and lambda (73) capsids revealed that the trimeric structures of HSTV-2 occupy the same positions as the gpD trimers of lambda (Fig. 8). At this resolution, it appears that three MCP hexamers of HSTV-2 each contribute two rods of density that interact with the base of the trimeric structures (Fig. 8). Thus, without the trimers, the interaction between hexamers would most likely be too loose to hold the capsid together, indicating that the minor capsid protein has an important role in stabilizing the HSTV-2 capsid. The resolution of the HSTV-2 reconstruction was not high enough to accurately model the trimers. However, to facilitate a rough comparison to gpD (Fig. 8, magnified region), we used the “fit in map” tool in UCSF Chimera (54) to fit the lambda gpD protein structure (PDB accession number 1C5E) (74) into the position occupied by the trimeric structures in the HSTV-2 capsid lattice. The gpD protein of lambda is 11.4 kDa (74). Protein products of several genes, such as gene 14 or 15, downstream of the MCP gene are potential homologues of gpD (Fig. 4), and there were unidentified protein bands from purified virions, one of which may represent this minor capsid protein stabilizing the lattice (Fig. 5A).
Fig 8.
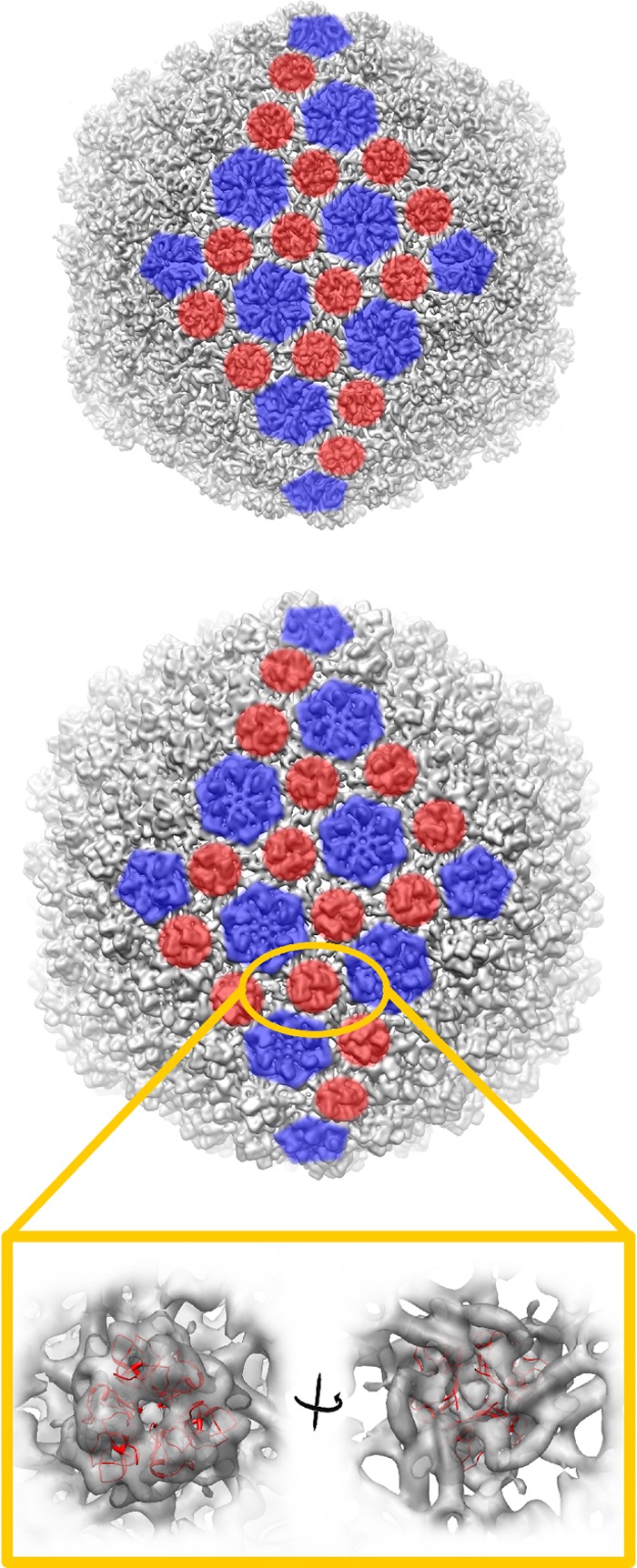
Organization of the two capsid proteins of HSTV-2, illustrated for two icosahedral facets. Shown is the capsid protein organization of bacteriophage lambda (structure under EMDB accession number EMD-5012 filtered to a 9.8-Å resolution and isosurface drawn at a threshold of a sigma value of 3.0) (73) (top) and archaeal virus HSTV-2 (isosurface drawn at a threshold of a sigma value of 3.0) (bottom). For two icosahedral facets, positions of MCP pentamers and hexamers are shown in blue, and positions of minor capsid protein trimers are shown in red. The magnified region shows cryo-EM density of the HSTV-2 minor capsid protein trimer with the bacteriophage lambda gpD protein structure (PDB accession number 1C5E) (74) fitted in and shown as a red ribbon. Visualization was done by using UCSF Chimera (54). Fitting of the gpD structure was done with the “fit in map” tool in UCSF Chimera.
DISCUSSION
In this study, we report a detailed characterization of two head-tailed viruses, HVTV-1 and HSTV-2, infecting extremely halophilic archaea—from the life cycles to the genome sequences and 3D reconstruction of the capsids. Two important aspects were observed. First, this study revealed that halophilic head-tailed viruses survive under changing salinity conditions. Second, the capsid structure of HSTV-2 introduced a new strategy for how to enlarge a capsid to accommodate a larger genome.
The infection cycles of HVTV-1 and HSTV-2 (Fig. 1) were typical of virulent head-tailed viruses, and cycles of 6 to 17 h were previously reported for several haloarchaeal head-tailed viruses (9, 11, 75–78). The adsorption efficiency of head-tailed viruses infecting halophilic prokaryotes seems to vary considerably (7, 78, 79), and it has been observed that viruses with high adsorption rates usually infect a host originating from the same geographical site as the virus (7). In contrast to this, HVTV-1 had a high adsorption efficiency, even though it was isolated from Thailand and its host was from the United States (5, 80, 81). HSTV-2, with a low adsorption rate, also infects a host isolated from a different location than the virus origin (5, 38).
Several viruses infecting halophilic hosts require high salinity to be infective, although many also tolerate low salinity (7, 9, 82, 83). For example, φN, infecting Halobacterium salinarum, survives even in distilled water (9). Salinity fluctuations occur in hypersaline environments due to evaporation as well as rainfall and freshwater streams (84, 85), and thus, viruses infecting hosts residing in such environments should be able to withstand these fluctuations. The salt-dependent infectivity inactivation/reactivation described here (Fig. 2) could be important for the survival of the viruses in their natural habitats. During low-salinity periods, viruses are inactive, as their host cell viability is poor, and when salinity and cell viability increase, viruses become infectious.
The adsorption of some halophilic head-tailed viruses is salt dependent and is most efficient at 3 to 4 M NaCl (7). Our observation that HVTV-1 and HSTV-2 are reversibly inactivated at low salinity (Fig. 2) supports a hypothesis that salt is necessary for a halophilic virus to adsorb to its host cell. Myovirus T4 requires tryptophan for adsorption: in the absence of tryptophan, the tail fibers are retracted, and in its presence, they are extended (86). Although no major structural changes were observed for HVTV-1 and HSTV-2 during the salinity changes (Fig. 2C and E), there might have been minor variations in the tail fibers. However, we did not observe T4-like, long tail fibers in either HVTV-1 or HSTV-2, nor did we identify potential homologous genes in their genome sequences.
The genomes of bacterial head-tailed viruses are typically mosaic and share a large common gene pool (87–89). This mosaic nature was also observed for the genomes of HVTV-1 and HSTV-2 (Fig. 3 and 4). The MCP-encoding genes were identified in both genomes. Furthermore, genes involved in tail structure, virion assembly, nucleotide metabolism, and DNA replication were detected. Our findings indicate that HVTV-1 uses the same maturation process as coliphage HK97 (90). A gene encoding a putative maturation protease was recognized in the HVTV-1 genome, and its MCP was proteolytically cleaved similarly to the HK97 MCP, gp5 (91). In contrast to HK97, HVTV-1 assembles with a T=13 lattice of hexamers and pentamers of the MCP, similar to phage T5 (92). The center of the MCP hexamers is decorated by a small, apparently trimeric, auxiliary protein. This position in SPP1 is occupied by gp12, which has been shown to stabilize the capsid (93). It is likely that the auxiliary protein in HVTV-1 performs a similar role.
Interestingly, the HSTV-2 genome showed sequence identity to the genomes of haloarchaeal myoviruses HF1 and HF2, which are close relatives with 94.4% sequence identity (20, 21). The identity was detected in the region where the HF1 and HF2 genomes differ from each other. Thus, there might have been recombination between HSTV-2-like and HF-like ancestors. Remarkably, HF1 and HF2 have been isolated from Australia, and HSTV-2 was isolated from Israel (5, 39), and the relationship of these viruses indicates that such viruses are able to spread over wide distances. Furthermore, it seems that the MCPs of all three viruses were inherited from a recent common ancestor. In contrast to the previously studied icosahedrally symmetric archaeal viruses Serpentine Lake hispanica virus 1 (SH1) and Sulfolobus turreted icosahedral virus 1 (STIV1) and STIV2, the MCP is not predicted to be formed of β-barrels but rather with an αβ secondary structure (94–96).
The observation that viruses infecting hosts from all three domains of life share the same MCP fold has led to the hypothesis that such viruses are related and form structure-based viral lineages. Within a lineage, all viruses share the same capsid architecture inherited from an ancestor which most probably existed before the domains of cellular life separated (25, 26, 32, 97, 98). Remarkably, our examination of the gene sequences of these two archaeal viruses shows similarity in inferred protein sequences and in relative position along the genome between a small subset of the archaeal viral genes and a corresponding number of genes of the tailed bacteriophages. In the phages, where more functions have been identified, these genes include, among others, the genes encoding the terminase large subunit, the portal protein, the head maturation protease, the tail-length tape measure protein, and the pair of overlapping genes just upstream of the tape measure gene that are expressed through a programmed translational frameshift. These phage genes all have roles in producing the virion structure, and we have suggested that some of the archaeal viral genes may have functions similar to those of their apparent bacteriophage analogues (28, 87, 99). If so, these observations suggest that there may be common ancestry, at least for these few structural genes, between the head-tailed archaeal viruses and the tailed bacteriophages. This would agree with and reinforce inferences of a shared MCP lineage across host domains based on shared MCP architecture.
Returning to the MCP-based lineage, although the architecture is the same within the lineage, the capsid size may differ considerably. It has been observed that the capsid size correlates with the genome size. One strategy to enlarge the capsid is to have a higher copy number of MCP, i.e., a larger T-number (32). The other one is to use a larger building block, i.e., to have a larger MCP. For example, human adenovirus and bacterial virus PRD1 have the same MCP fold and T-number, but adenovirus has expanded its capsid by having an extra domain and a loop between the two β-barrels of the trimeric MCP (33). This, and the lack of a lipid membrane, helps adenovirus to enclose its genome, which is 2.4 times larger than the genome of PRD1. The third mechanism is to retain the T-number but change the head morphology from isometric to prolate. For example, bacteriophages T4 and T5 have the same MCP fold, and the T-number of the midsection of the prolate T4 capsid is the same as that of the isometric T5 capsid (34–36).
Our results show that HSTV-2 uses a new, fourth approach to enlarge its capsid. The HSTV-2 capsid and genome are larger than those of lambda (Fig. 7). The genome packaging efficiency of HSTV-2, however, is rather close to that reported for lambda (ρpack = 0.56) (100). Because HSTV-2 and lambda are isometric T=7 viruses and their MCPs have about the same molecular mass, this excludes the three above-mentioned enlargement strategies. Besides, according to the hypothesis put forward previously by Mannige and Brooks (24), geometrical constraints are imposed on icosahedral capsids belonging to class 2, so that T=9 (class 1) and T=12 (class 3) capsid arrangements are not easily available for a virus with a T=7 arrangement (24). There are, however, a number of incidences in the literature where other assemblies have been reported; for instance, in the system of bacteriophages P2 (T=7) and P4 (T=4), the Sid protein of P4 forces the shell into a T=4 instead of a T=7 arrangement (101). In addition, bacteriophage P22 (T=7) produces T=4 capsids in the absence of the scaffolding protein (102, 103).
We propose that HSTV-2 uses an additional, stabilizing protein in the protein lattice to make its capsid larger (Fig. 8). Several head-tailed viruses such as lambda, epsilon15, and T4 as well as HSV-1 use decorating, auxiliary proteins to stabilize their capsids (71, 73). The stabilizing protein gpD of lambda forms trimers decorating the capsid between hexamers (72–74, 104, 105), and the HSTV-2 stabilizing protein structures resemble these structures (Fig. 6 and 8). In order to enlarge the capsid, T=7 viruses such as lambda may use gpD-type helper proteins to increase their capsid size (73, 74). Based on the analysis of the HSTV-2 capsid, MCP, and genome sizes, we propose that HSTV-2 is a more extreme case of such development, as it uses a lambda gpD-type helper protein in an attempt to reach even further over the chasm of geometric constraints that separates T=7 and T=13, both belonging to class 2, in the realm of available capsid architectures (24). Remarkably, the genomes of haloarchaeal head-tailed viruses HF1 and HF2 (20, 21) are even larger than the HSTV-2 genome, and as the MCPs of these viruses share significant sequence identity, HF1 and HF2 could be T=7 viruses having even larger capsids.
Supplementary Material
ACKNOWLEDGMENTS
We thank Terhi Kemppinen, Sari Korhonen, Päivi Hannuksela, Eevakaisa Vesanen, and Helin Veskiväli for excellent technical assistance. We acknowledge Gunilla Rönnholm for the protein sequencing (Protein Chemistry Unit, Institute of Biotechnology, University of Helsinki). We thank the University of Helsinki for the support to the EU ESFRI Instruct Centre for Virus Production and Purification used in this study. We thank the National Biocenter Finland Cryo-Electron Microscopy unit and the CSC for use of facilities.
This research was supported by Academy Professor (Academy of Finland) funding grants 255342 and 256518 (D.H.B.) and Academy of Finland grants 1129684, Centre of Excellence in Virus Research 2006-2011, to S.J.B. and D.H.B. and 1129684 to S.J.B.
Footnotes
Published ahead of print 2 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03397-12.
REFERENCES
- 1. Chaban B, Ng SYM, Jarrell KF. 2006. Archaeal habitats—from the extreme to the ordinary. Can. J. Microbiol. 52:73–116 [DOI] [PubMed] [Google Scholar]
- 2. DeLong EF, Pace NR. 2001. Environmental diversity of bacteria and archaea. Syst. Biol. 50:470–478 [PubMed] [Google Scholar]
- 3. Pina M, Bize A, Forterre P, Prangishvili D. 2011. The archeoviruses. FEMS Microbiol. Rev. 35:1035–1054 [DOI] [PubMed] [Google Scholar]
- 4. Ackermann HW, Prangishvili D. 2012. Prokaryote viruses studied by electron microscopy. Arch. Virol. 157:1843–1849 [DOI] [PubMed] [Google Scholar]
- 5. Atanasova NS, Roine E, Oren A, Bamford DH, Oksanen HM. 2012. Global network of specific virus-host interactions in hypersaline environments. Environ. Microbiol. 14:426–440 [DOI] [PubMed] [Google Scholar]
- 6. Winker S, Woese CR. 1991. A definition of the domains Archaea, Bacteria and Eucarya in terms of small subunit ribosomal RNA characteristics. Syst. Appl. Microbiol. 14:305–310 [DOI] [PubMed] [Google Scholar]
- 7. Kukkaro P, Bamford DH. 2009. Virus-host interactions in environments with a wide range of ionic strengths. Environ. Microbiol. Rep. 1:71–77 [DOI] [PubMed] [Google Scholar]
- 8. Pietilä MK, Atanasova NS, Manole V, Liljeroos L, Butcher SJ, Oksanen HM, Bamford DH. 2012. Virion architecture unifies globally distributed pleolipoviruses infecting halophilic archaea. J. Virol. 86:5067–5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vogelsang-Wenke H, Oesterhelt D. 1988. Isolation of a halobacterial phage with a fully cytosine-methylated genome. Mol. Gen. Genet. 211:407–414 [Google Scholar]
- 10. Mei Y, Chen J, Sun D, Chen D, Yang Y, Shen P, Chen X. 2007. Induction and preliminary characterization of a novel halophage SNJ1 from lysogenic Natrinema sp. F5. Can. J. Microbiol. 53:1106–1110 [DOI] [PubMed] [Google Scholar]
- 11. Torsvik T, Dundas ID. 1980. Persisting phage infection in Halobacterium salinarum str1. J. Gen. Virol. 47:29–36 [Google Scholar]
- 12. Waarts BL, Smit JM, Aneke OJ, McInerney GM, Liljestrom P, Bittman R, Wilschut J. 2005. Reversible acid-induced inactivation of the membrane fusion protein of Semliki Forest virus. J. Virol. 79:7942–7948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richter HE, Loewen PC. 1982. Rapid inactivation of bacteriophage T7 by ascorbic acid is repairable. Biochim. Biophys. Acta 697:25–30 [DOI] [PubMed] [Google Scholar]
- 14. King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed). 2012. Virus taxonomy. Ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, London, United Kingdom [Google Scholar]
- 15. Ackermann HW. 1998. Tailed bacteriophages: the order Caudovirales. Adv. Virus Res. 51:135–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rachel R, Bettstetter M, Hedlund BP, Häring M, Kessler A, Stetter KO, Prangishvili D. 2002. Remarkable morphological diversity of viruses and virus-like particles in hot terrestrial environments. Arch. Virol. 147:2419–2429 [DOI] [PubMed] [Google Scholar]
- 17. Klein R, Baranyi U, Rössler N, Greineder B, Scholz H, Witte A. 2002. Natrialba magadii virus φCh1: first complete nucleotide sequence and functional organization of a virus infecting a haloalkaliphilic archaeon. Mol. Microbiol. 45:851–863 [DOI] [PubMed] [Google Scholar]
- 18. Pagaling E, Haigh RD, Grant WD, Cowan DA, Jones BE, Ma Y, Ventosa A, Heaphy S. 2007. Sequence analysis of an archaeal virus isolated from a hypersaline lake in Inner Mongolia, China. BMC Genomics 8:410 doi:10.1186/1471-2164-8-410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pfister P, Wasserfallen A, Stettler R, Leisinger T. 1998. Molecular analysis of Methanobacterium phage psiM2. Mol. Microbiol. 30:233–244 [DOI] [PubMed] [Google Scholar]
- 20. Tang SL, Nuttall S, Dyall-Smith M. 2004. Haloviruses HF1 and HF2: evidence for a recent and large recombination event. J. Bacteriol. 186:2810–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang SL, Nuttall S, Ngui K, Fisher C, Lopez P, Dyall-Smith M. 2002. HF2: a double-stranded DNA tailed haloarchaeal virus with a mosaic genome. Mol. Microbiol. 44:283–296 [DOI] [PubMed] [Google Scholar]
- 22. Crick FH, Watson JD. 1956. Structure of small viruses. Nature 177:473–475 [DOI] [PubMed] [Google Scholar]
- 23. Caspar DL, Klug A. 1962. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 27:1–24 [DOI] [PubMed] [Google Scholar]
- 24. Mannige RV, Brooks CL., III 2010. Periodic table of virus capsids: implications for natural selection and design. PLoS One 5:e9423 doi:10.1371/journal.pone.0009423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abrescia NGA, Bamford DH, Grimes JM, Stuart DI. 2012. Structure unifies the viral universe. Annu. Rev. Biochem. 81:795–822 [DOI] [PubMed] [Google Scholar]
- 26. Abrescia NGA, Grimes JM, Fry EE, Ravantti JJ, Bamford DH, Stuart DI. 2010. What does it take to make a virus: the concept of the viral ‘self’, p 35–58 In Stockley PG, Twarock R. (ed), Emerging topics in physical virology. Imperial College Press, London, United Kingdom [Google Scholar]
- 27. Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. 2000. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289:2129–2133 [DOI] [PubMed] [Google Scholar]
- 28. Krupovic M, Prangishvili D, Hendrix RW, Bamford DH. 2011. Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 75:610–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iyer LM, Balaji S, Koonin EV, Aravind L. 2006. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 117:156–184 [DOI] [PubMed] [Google Scholar]
- 30. Filée J, Chandler M. 2008. Convergent mechanisms of genome evolution of large and giant DNA viruses. Res. Microbiol. 159:325–331 [DOI] [PubMed] [Google Scholar]
- 31. Hendrix RW. 2009. Jumbo bacteriophages. Curr. Top. Microbiol. Immunol. 328:229–240 [DOI] [PubMed] [Google Scholar]
- 32. Bamford DH, Grimes JM, Stuart DI. 2005. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 15:655–663 [DOI] [PubMed] [Google Scholar]
- 33. Benson SD, Bamford JK, Bamford DH, Burnett RM. 1999. Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures. Cell 98:825–833 [DOI] [PubMed] [Google Scholar]
- 34. Fokine A, Chipman PR, Leiman PG, Mesyanzhinov VV, Rao VB, Rossmann MG. 2004. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. U. S. A. 101:6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fokine A, Leiman PG, Shneider MM, Ahvazi B, Boeshans KM, Steven AC, Black LW, Mesyanzhinov VV, Rossmann MG. 2005. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. U. S. A. 102:7163–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Effantin G, Boulanger P, Neumann E, Letellier L, Conway JF. 2006. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 361:993–1002 [DOI] [PubMed] [Google Scholar]
- 37. Torreblanca M, Rodriquez-Valera F, Juez G, Ventosa A, Kamekura M, Kates M. 1986. Classification of non-alkaliphilic halobacteria based on numerical taxonomy and polar lipid composition, and description of Haloarcula gen. nov. and Haloferax gen. nov. Syst. Appl. Microbiol. 8:88–99 [Google Scholar]
- 38. Oren A. 1983. Halobacterium sodomense sp. nov., a Dead Sea halobacterium with an extremely high magnesium requirement. Int. J. Syst. Bacteriol. 33:381–386 [Google Scholar]
- 39. Nuttall SD, Dyall-Smith ML. 1993. HF1 and HF2: novel bacteriophages of halophilic archaea. Virology 197:678–684 [DOI] [PubMed] [Google Scholar]
- 40. Pietilä MK, Roine E, Paulin L, Kalkkinen N, Bamford DH. 2009. An ssDNA virus infecting archaea: a new lineage of viruses with a membrane envelope. Mol. Microbiol. 72:307–319 [DOI] [PubMed] [Google Scholar]
- 41. Adams MH. 1959. Bacteriophages. Interscience Publishers, Inc, New York, NY [Google Scholar]
- 42. Bamford DH, Mindich L. 1980. Electron microscopy of cells infected with nonsense mutants of bacteriophage phi 6. Virology 107:222–228 [DOI] [PubMed] [Google Scholar]
- 43. Adrian M, Dubochet J, Lepault J, McDowall AW. 1984. Cryo-electron microscopy of viruses. Nature 308:32–36 [DOI] [PubMed] [Google Scholar]
- 44. Seitsonen J, Susi P, Heikkilä O, Sinkovits RS, Laurinmäki P, Hyypiä T, Butcher SJ. 2010. Interaction of αvβ3 and αvβ6 integrins with human parechovirus 1. J. Virol. 84:8509–8519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142:334–347 [DOI] [PubMed] [Google Scholar]
- 46. Kivioja T, Ravantti J, Verkhovsky A, Ukkonen E, Bamford D. 2000. Local average intensity-based method for identifying spherical particles in electron micrographs. J. Struct. Biol. 131:126–134 [DOI] [PubMed] [Google Scholar]
- 47. Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97 [DOI] [PubMed] [Google Scholar]
- 48. Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM—an automated and high throughput program for image reconstruction of icosahedral particles. J. Struct. Biol. 157:73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yan X, Dryden KA, Tang J, Baker TS. 2007. Ab initio random model method facilitates 3D reconstruction of icosahedral particles. J. Struct. Biol. 157:211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van Heel M, Schatz M. 2005. Fourier shell correlation threshold criteria. J. Struct. Biol. 151:250–262 [DOI] [PubMed] [Google Scholar]
- 51. Fernandez JJ. 2008. High performance computing in structural determination by electron cryomicroscopy. J. Struct. Biol. 164:1–6 [DOI] [PubMed] [Google Scholar]
- 52. Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333:721–745 [DOI] [PubMed] [Google Scholar]
- 53. Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 157:3–18 [DOI] [PubMed] [Google Scholar]
- 54. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 55. Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Borodovsky M, McIninch J. 1993. Recognition of genes in DNA sequence with ambiguities. Biosystems 30:161–171 [DOI] [PubMed] [Google Scholar]
- 57. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lupas A, Van Dyke M, Stock J. 1991. Predicting coiled coils from protein sequences. Science 252:1162–1164 [DOI] [PubMed] [Google Scholar]
- 59. Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 61. Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248 doi:10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Remmert M, Biegert A, Hauser A, Söding J. 2012. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9:173–175 [DOI] [PubMed] [Google Scholar]
- 63. Söding J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960 [DOI] [PubMed] [Google Scholar]
- 64. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 65. Schägger H, von Jagow G. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368–379 [DOI] [PubMed] [Google Scholar]
- 66. Bamford DH, Ravantti JJ, Rönnholm G, Laurinavičius S, Kukkaro P, Dyall-Smith M, Somerharju P, Kalkkinen N, Bamford JK. 2005. Constituents of SH1, a novel lipid-containing virus infecting the halophilic euryarchaeon Haloarcula hispanica. J. Virol. 79:9097–9107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Davey MJ, Jeruzalmi D, Kuriyan J, O'Donnell M. 2002. Motors and switches: AAA+ machines within the replisome. Nat. Rev. Mol. Cell Biol. 3:826–835 [DOI] [PubMed] [Google Scholar]
- 68. Mutschler H, Meinhart A. 2011. ϵ/ζ systems: their role in resistance, virulence, and their potential for antibiotic development. J. Mol. Med. 89:1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Purohit PK, Inamdar MM, Grayson PD, Squires TM, Kondev J, Phillips R. 2005. Forces during bacteriophage DNA packaging and ejection. Biophys. J. 88:851–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parent KN, Khayat R, Tu LH, Suhanovsky MM, Cortines JR, Teschke CM, Johnson JE, Baker TS. 2010. P22 coat protein structures reveal a novel mechanism for capsid maturation: stability without auxiliary proteins or chemical crosslinks. Structure 18:390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dokland T, Murialdo H. 1993. Structural transitions during maturation of bacteriophage lambda capsids. J. Mol. Biol. 233:682–694 [DOI] [PubMed] [Google Scholar]
- 73. Lander GC, Evilevitch A, Jeembaeva M, Potter CS, Carragher B, Johnson JE. 2008. Bacteriophage lambda stabilization by auxiliary protein gpD: timing, location, and mechanism of attachment determined by cryo-EM. Structure 16:1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang F, Forrer P, Dauter Z, Conway JF, Cheng N, Cerritelli ME, Steven AC, Pluckthun A, Wlodawer A. 2000. Novel fold and capsid-binding properties of the lambda-phage display platform protein gpD. Nat. Struct. Biol. 7:230–237 [DOI] [PubMed] [Google Scholar]
- 75. Wais AC, Kon M, MacDonald RE, Stollar BD. 1975. Salt-dependent bacteriophage infecting Halobacterium cutirubrum and H. halobium. Nature 256:314–315 [DOI] [PubMed] [Google Scholar]
- 76. Schnabel H, Zillig W, Pfaffle M, Schnabel R, Michel H, Delius H. 1982. Halobacterium halobium phage φH. EMBO J. 1:87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Witte A, Baranyi U, Klein R, Sulzner M, Luo C, Wanner G, Krüger DH, Lubitz W. 1997. Characterization of Natronobacterium magadii phage φCh1, a unique archaeal phage containing DNA and RNA. Mol. Microbiol. 23:603–616 [DOI] [PubMed] [Google Scholar]
- 78. Pauling C. 1982. Bacteriophages of Halobacterium halobium: isolated from fermented fish sauce and primary characterization. Can. J. Microbiol. 28:916–921 [DOI] [PubMed] [Google Scholar]
- 79. Daniels LL, Wais AC. 1998. Virulence in phage populations infecting Halobacterium cutirubrum. FEMS Microbiol. Ecol. 25:129–134 [Google Scholar]
- 80. Gonzalez C, Gutierrez C, Ramirez C. 1978. Halobacterium vallismortis sp. nov. An amylolytic and carbohydrate-metabolizing, extremely halophilic bacterium. Can. J. Microbiol. 24:710–715 [DOI] [PubMed] [Google Scholar]
- 81. Torreblanca M, Rodriguez-Valera F, Juez G, Ventosa A, Kamekura M, Kates M. 1986. Classification of non-alkaliphilic halobacteria based on numerical taxonomy and polar lipid composition, and description of Haloarcula gen. nov. and Haloferax gen. nov. Syst. Appl. Microbiol. 8:89–99 [Google Scholar]
- 82. Pietilä MK, Laurinavičius S, Sund J, Roine E, Bamford DH. 2010. The single-stranded DNA genome of novel archaeal virus Halorubrum pleomorphic virus 1 is enclosed in the envelope decorated with glycoprotein spikes. J. Virol. 84:788–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Porter K, Kukkaro P, Bamford JK, Bath C, Kivelä HM, Dyall-Smith ML, Bamford DH. 2005. SH1: a novel, spherical halovirus isolated from an Australian hypersaline lake. Virology 335:22–33 [DOI] [PubMed] [Google Scholar]
- 84. Walsh DA, Papke RT, Doolittle WF. 2005. Archaeal diversity along a soil salinity gradient prone to disturbance. Environ. Microbiol. 7:1655–1666 [DOI] [PubMed] [Google Scholar]
- 85. DasSarma S, DasSarma P. 2012. Halophiles. In eLS John Wiley & Sons, Ltd, Chichester, United Kingdom: doi:10.1002/9780470015902.a0000394.pub3 [Google Scholar]
- 86. Kellenberger E, Bolle A, Boydelatour E, Epstein RH, Franklin NC, Jerne NK, Reale Scafati A, Sechaud J. 1965. Functions and properties related to the tail fibers of bacteriophage T4. Virology 26:419–440 [DOI] [PubMed] [Google Scholar]
- 87. Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc. Natl. Acad. Sci. U. S. A. 96:2192–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Jr, Hendrix RW, Hatfull GF. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182 [DOI] [PubMed] [Google Scholar]
- 89. Juhala RJ, Ford ME, Duda RL, Youlton A, Hatfull GF, Hendrix RW. 2000. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J. Mol. Biol. 299:27–51 [DOI] [PubMed] [Google Scholar]
- 90. Gertsman I, Gan L, Guttman M, Lee K, Speir JA, Duda RL, Hendrix RW, Komives EA, Johnson JE. 2009. An unexpected twist in viral capsid maturation. Nature 458:646–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Conway JF, Duda RL, Cheng N, Hendrix RW, Steven AC. 1995. Proteolytic and conformational control of virus capsid maturation: the bacteriophage HK97 system. J. Mol. Biol. 253:86–99 [DOI] [PubMed] [Google Scholar]
- 92. Huet A, Conway JF, Letellier L, Boulanger P. 2010. In vitro assembly of the T=13 procapsid of bacteriophage T5 with its scaffolding domain. J. Virol. 84:9350–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. White HE, Sherman MB, Brasiles S, Jacquet E, Seavers P, Tavares P, Orlova EV. 2012. Capsid structure and its stability at the late stages of bacteriophage SPP1 assembly. J. Virol. 86:6768–6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jäälinoja HT, Roine E, Laurinmäki P, Kivelä HM, Bamford DH, Butcher SJ. 2008. Structure and host-cell interaction of SH1, a membrane-containing, halophilic euryarchaeal virus. Proc. Natl. Acad. Sci. U. S. A. 105:8008–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rice G, Tang L, Stedman K, Roberto F, Spuhler J, Gillitzer E, Johnson JE, Douglas T, Young M. 2004. The structure of a thermophilic archaeal virus shows a double-stranded DNA viral capsid type that spans all domains of life. Proc. Natl. Acad. Sci. U. S. A. 101:7716–7720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Happonen LJ, Redder P, Peng X, Reigstad LJ, Prangishvili D, Butcher SJ. 2010. Familial relationships in hyperthermo- and acidophilic archaeal viruses. J. Virol. 84:4747–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bamford DH. 2003. Do viruses form lineages across different domains of life? Res. Microbiol. 154:231–236 [DOI] [PubMed] [Google Scholar]
- 98. Benson SD, Bamford JK, Bamford DH, Burnett RM. 2004. Does common architecture reveal a viral lineage spanning all three domains of life? Mol. Cell 16:673–685 [DOI] [PubMed] [Google Scholar]
- 99. Krupovič M, Forterre P, Bamford DH. 2010. Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of bacteria. J. Mol. Biol. 397:144–160 [DOI] [PubMed] [Google Scholar]
- 100. Roos WH, Radtke K, Kniesmeijer E, Geertsema H, Sodeik B, Wuite GJ. 2009. Scaffold expulsion and genome packaging trigger stabilization of herpes simplex virus capsids. Proc. Natl. Acad. Sci. U. S. A. 106:9673–9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dearborn AD, Laurinmäki P, Chandramouli P, Rodenburg CM, Wang S, Butcher SJ, Dokland T. 2012. Structure and size determination of bacteriophage P2 and P4 procapsids: function of size responsiveness mutations. J. Struct. Biol. 178:215–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King JA, Chiu W. 2011. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. U. S. A. 108:1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Thuman-Commike PA, Greene B, Malinski JA, King J, Chiu W. 1998. Role of the scaffolding protein in P22 procapsid size determination suggested by T=4 and T=7 procapsid structures. Biophys. J. 74:559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Katsura I. 1978. Structure and inherent properties of the bacteriophage lambda head shell. I. Polyheads produced by two defective mutants in the major head protein. J. Mol. Biol. 121:71–93 [DOI] [PubMed] [Google Scholar]
- 105. Imber R, Tsugita A, Wurtz M, Hohn T. 1980. Outer surface protein of bacteriophage lambda. J. Mol. Biol. 139:277–295 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.