

Abstract
Human scavenger receptor class B, member 2 (SCARB2), and P-selectin glycoprotein ligand-1 (PSGL1) have been identified to be the cellular receptors for enterovirus 71 (EV71). We compared the EV71 infection efficiencies of mouse L cells that expressed SCARB2 (L-SCARB2) and PSGL1 (L-PSGL1) and the abilities of SCARB2 and PSGL1 to bind to the virus. L-SCARB2 cells bound a reduced amount of EV71 compared to L-PSGL1 cells. However, EV71 could infect L-SCARB2 cells more efficiently than L-PSGL1 cells. The results suggested that the difference in the binding capacities of the two receptors was not the sole determinant of the infection efficiency and that SCARB2 plays an essential role after attaching to virions. Therefore, we examined the viral entry into L-SCARB2 cells and L-PSGL1 cells by immunofluorescence microscopy. In both cells, we detected internalized EV71 virions that colocalized with an early endosome marker. We then performed a sucrose density gradient centrifugation analysis to evaluate viral uncoating. After incubating the EV71 virion with L-SCARB2 cells or soluble SCARB2 under acidic conditions below pH 6.0, we observed that part of the native virion was converted into an empty capsid that lacked both genomic RNA and VP4 capsid proteins. The results suggested that the uncoating of EV71 requires both SCARB2 and an acidic environment and occurs after the internalization of the virus-receptor complex into endosomes. However, the empty capsid formation was not observed after incubation with L-PSGL1 cells or soluble PSGL1 under any of the tested pH conditions. These results indicated that SCARB2 is capable of viral binding, viral internalization, and viral uncoating and that the low infection efficiency of L-PSGL1 cells is due to the inability of PSGL1 to induce viral uncoating. The characterization of SCARB2 as an uncoating receptor greatly contributes to the understanding of the early steps of EV71 infection.
INTRODUCTION
Enterovirus 71 (EV71) belongs to the genus Enterovirus within the family Picornaviridae (1). The virus contains positive-sense RNA surrounded by an icosahedral capsid assembled from 60 copies of the four structural proteins VP1, VP2, VP3, and VP4 (2–4). VP1, VP2, and VP3 create a canyon on the viral surface (3, 4) that is the site of attachment to the cellular receptor on many enteroviruses (5). The first report of EV71 isolation was in patients with neurological diseases, including fatal encephalitis and aseptic meningitis, in California from 1969 to 1972 (6). Later studies reported that EV71 was a causative agent of hand, foot, and mouth disease (HFMD) in young children and infants (7, 8). The clinical symptoms of HFMD due to EV71 are generally mild and self-limiting; however, EV71 occasionally causes severe neurological diseases, such as brainstem encephalitis and acute flaccid paralysis (9). Recently, epidemic outbreaks of neurovirulent EV71 have been reported mainly in Southeast and East Asia, including Taiwan, Malaysia, Singapore, Japan, and China (10–15). From 2008 to 2011, the epidemic outbreaks of EV71 in China resulted in approximately 1,900 fatal cases (16). In 2011, the epidemic in Vietnam resulted in 98 fatal cases (http://www.wpro.who.int/vietnam/media_center/press_releases/hfmd_pr.htm).
Two molecules—human scavenger receptor class B, member 2 (SCARB2; also known as lysosomal integral membrane protein II or CD36b like-2) (17), and human P-selectin glycoprotein ligand-1 (PSGL1; also known as selectin P ligand) (18)—were reported to be the cellular receptors for EV71. SCARB2 belongs to the CD36 family and has two transmembrane domains (19). Physiologically, SCARB2 works as the receptor for β-glucocerebrosidase (β-GC) transport from the endoplasmic reticulum to the lysosome (20, 21) and plays an important role in the maintenance of lysosomes (19). Mouse cells become susceptible to all tested EV71 strains when they express human SCARB2 (17, 22). The binding of SCARB2 to EV71 occurs within the luminal domain of SCARB2 at amino acids 142 to 204 (23), and amino acids 144 to 151 were demonstrated to be particularly important (24). The EF loop region of VP1, which lines the wall of the canyon on the viral surface, was found to be important for binding to SCARB2 (24). EV71 infection via the SCARB2-dependent pathway was inhibited by a small interfering RNA (siRNA) treatment against the molecules that are involved in the clathrin-dependent endocytic pathway and by inhibitors of endosomal acidification (25, 26). In addition to EV71, coxsackievirus A7 (CVA7), CVA14, and CVA16 have utilized SCARB2 as a receptor for infection (17, 22). PSGL1 is a sialomucin leukocyte membrane protein that is expressed as a homodimer of disulfide-linked subunits and can bind to three different selectins (P, E, and L) (27–29). Physiologically, PSGL1 is expressed on myeloid cells and stimulated T lymphocytes (30) and plays a critical role in the tethering and rolling of leukocytes for the recruitment of these cells from blood vessels into inflamed tissues (30). Several EV71 strains (PSGL1-binding strain EV71-PB) bind to PSGL1, but other strains (PSGL1-nonbinding strain EV71-non-PB) do not (18). The binding of EV71-PB to PSGL1 requires tyrosine sulfations at the N-terminal region of PSGL1 (31). Mouse cells that express PSGL1 are susceptible only to EV71-PB and CVA16 (18). However, the transgenic expression of PSGL1 in mice failed to confer susceptibility to EV71 in vivo (32).
The mode of infection of picornaviruses varies for each type, and each viral receptor plays a distinct role (33, 34). Poliovirus (PV) receptor (PVR; also known as CD155) and intercellular adhesion molecule 1 (ICAM-1) are cellular receptors for PV and the major-group human rhinoviruses (HRVs), respectively (35–39). These receptors play roles in viral attachment, internalization, and uncoating (33, 34). The αvβ1, αvβ3, αvβ6, αvβ8, and α5β1 integrins are the receptors for foot-and-mouth-disease virus (FMDV) (40), and members of the low-density-lipoprotein-receptor (LDL-R) family are the receptors for the minor-group HRVs (41). These receptors have roles in viral attachment and internalization but not in uncoating (33, 34). Several strains of coxsackievirus B1 (CVB1), CVB3, and CVB5 have two receptors, coxsackievirus and adenovirus receptor (CAR) (42, 43) and decay-accelerating factor (DAF). CAR plays roles in viral attachment, internalization, and uncoating, just like PVR and ICAM-1 in PV and major-group HRV infections, respectively. DAF can bind to the virus but cannot initiate uncoating (33, 44–46). The expression of CAR is sufficient for the establishment of infection in most cultured cells. However, in polarized epithelial cells, CAR is not located on the apical surface. DAF provides an initial attachment site on the apical surface, and DAF-mediated signals permit the virus to interact with CAR in the tight junctions (47).
Conformational alterations for the uncoating of picornaviruses require their respective viral receptor and/or acidic conditions. The uncoating of PV, CVB, and the major-group HRVs is triggered by their receptors (34, 46, 48, 49). After the binding of the receptor to the virion, the native virion releases VP4 and becomes an A particle at a neutral pH. After A-particle formation, the A particle releases viral genomic RNA and becomes an empty capsid. Another typical conformational alteration of the minor-group HRVs and aphthoviruses, such as FMDV and equine rhinitis A virus (ERAV), is triggered by the acidic environment of endosomes (34, 50, 51). Under an acidic pH, the native virion is converted either into an A particle and then an empty capsid (32) or into an empty capsid and then pentameric subunits for viral RNA release (50, 52). The uncoating of HRV type 16 (HRV16) and HRV14 requires both ICAM-1 and low pH (53).
Studies have reported that both EV71 receptors, SCARB2 and PSGL1, can promote EV71 infection by acting as viral attachment factors (17, 18). Recently, Chen et al. evaluated the role of SCABR2 and PSGL1 during uncoating, and they observed a shift in the viral RNA peak to a more slowly sedimenting fraction in sucrose density gradient centrifugation after the virion was incubated with SCARB2 at an acidic pH; this was not observed after incubation with PSGL1 (24). However, the precise roles of the two receptors and the mode of uncoating have not yet been reported. In this study, we compared the infection efficiency of EV71 via the SCARB2-dependent pathway with the infection efficiency via the PSGL1-dependent pathway. Additionally, we evaluated the binding of EV71 to SCARB2 and PSGL1 and the internalization of EV71 via these receptors. We also evaluated the process of the conformational alteration of EV71 by these receptors.
MATERIALS AND METHODS
Cells.
Rhabdomyosarcoma (RD) cells, Vero cells, and L929 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma) supplemented with 5% fetal bovine serum (FBS) and a penicillin-streptomycin solution (Life Technologies) (5% FBS–DMEM). L cells expressing SCARB2 (L-SCARB2 cells) and L cells transfected with empty pCAGGS-PUR vector (L-Empty cells) (17) were cultured in 5% FBS–DMEM supplemented with puromycin (4 μg ml−1; Calbiochem).
Viruses.
The EV71 strains SK-EV006/Malaysia/97 (SK-EV006; genogroup B), BrCr/USA/70 (BrCr; genogroup A), 1095/Japan/97 (1095; genogroup C), and EV71-GFP, which expresses green fluorescent protein (GFP) upon viral replication, were used in this study (17, 23, 54). Unless otherwise noted, we used EV71 strain SK-EV006 as a representative strain of EV71. The poliovirus strain Mahoney was recovered from the infectious cDNA clone pOM (55).
Plasmids.
The cDNA fragment of PSGL1 was amplified from RD cells by reverse transcription (RT)-PCR with the primers PSGL1-Eco(+) (CAGAATTCACCATGCCTCTGCAACTCCTCC) and PSGL1-Xho(−) (CACACTCGAGCTAAGGGAGGAAGCTGTG), and the PCR product was inserted into pCAGGS-PUR (56). The resulting construct was designated pCA-PSGL1.
Establishment of L-PSGL1 cells.
L929 cells were transfected with pCA-PSGL1. After 24 h, the cells were passaged with 5% FBS–DMEM containing 5 μg ml−1 puromycin for selection. A total of 5 independent clones of the PSGL1-expressing transformant were tested for susceptibility to EV71-GFP. All of the PSGL1-expressing transformants were susceptible to EV71-GFP at similar levels. We selected a representative clone from these 5 clones to be the L cells expressing PSGL1 (L-PSGL1 cells).
Flow cytometry.
We performed flow cytometry as previously reported (17, 57), with some modifications. Briefly, we resuspended L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells in phosphate-buffered saline [PBS(−)] (per liter, 8.00 g NaCl, 1.15 g Na2HPO4, 0.20 g KCl, 0.20 g KH2PO4 [pH 7.4]) containing 0.02% EDTA, stained them with anti-human LIMPII/SR-B2 goat polyclonal antibody (R&D Systems) or anti-human CD162 mouse monoclonal antibody (clone KPL-1; BD Pharmingen) followed by Alexa Fluor 488 donkey anti-goat IgG (H+L) or Alexa Fluor 488 donkey anti-mouse IgG (H+L) (Life Technologies), respectively, and then analyzed the cells on a FACSCalibur flow cytometer (Becton, Dickinson and Company) with CellQuest Pro software (Becton, Dickinson).
Single-round infection assay.
L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells were infected with EV71-GFP and incubated for 24 h at 37°C. Images of the cells were acquired using an IX70 microscope (Olympus) and a DP70 charge-coupled-device camera (Olympus) and were analyzed using DP controller software (Olympus). These infected cells were detached and fixed with 4% paraformaldehyde (PFA). The cells were then analyzed with a FACSCalibur flow cytometer using CellQuest Pro software. The amount of EV71-GFP was chosen so that infected virus produces approximately 1,000 GFP-positive cells in RD cells under the same conditions.
Viral growth kinetics.
RD cells, L929 cells, L-SCARB2 cells, and L-PSGL1 cells were infected with EV71 at a multiplicity of infection (MOI) of 0.01 or 10. After incubation for 1 h at 37°C, the inoculum was removed and the cells were washed twice with 5% FBS–DMEM. The infected cells were maintained in 5% FBS–DMEM for 0, 6, 12, 24, 36, or 48 h at 37°C and then collected. The samples were freeze-thawed 3 times before use. The viral titers were determined with Vero cells and were expressed as the 50% tissue culture infectious dose (TCID50) according to the Reed-Muench method (58). After 24 h, the cells that were infected at an MOI of 0.01 or 10 were imaged.
Virus purification.
RD cells were infected with EV71 at an MOI of 5. The cells and media were frozen at 18 h postinfection. After thawing, the cell debris was removed by centrifugation in an NA-4HS rotor (Tomy) at 10,000 rpm for 20 min at 4°C. The supernatant was layered onto a 1.25-g/ml and 1.48-g/ml CsCl discontinuous step gradient in PBS(−). The empty (E) particles and native virions (filled [F] particles) were collected from a fraction on the 1.25-g/ml CsCl gradient and a fraction between the 1.25-g/ml and 1.48-g/ml CsCl gradients, respectively. The E particles and F particles were diluted with PBS(−) containing 0.1% bovine serum albumin (BSA) and pelleted by ultracentrifugation in an SW32Ti rotor (Beckman) at 32,000 rpm for 2 h at 15°C. Each pellet was suspended in PBS(−) containing 0.1% BSA and 1% SDS. Each supernatant was layered onto a 15 to 30% sucrose density gradient in PBS(−) containing 0.1% BSA and centrifuged in an SW41Ti rotor (Beckman) at 39,000 rpm for 2 h at 4°C. After fractionation (1 ml/fraction), the fractions that included the E particles and F particles were collected and pelleted by ultracentrifugation in the SW41Ti rotor at 39,000 rpm for 2 h at 4°C. The pellets were suspended in PBS(−) containing 0.1% BSA and used as purified E and F particles.
To prepare [35S]methionine- and [35S]cysteine-labeled EV71 strains SK-EV006, BrCr, and 1095 and poliovirus, RD cells were infected with EV71 or poliovirus at an MOI of 5. At 3 h postinfection, the medium was changed to methionine- and cysteine-free DMEM (Sigma) containing [35S]methionine and [35S]cysteine (EasyTag Express35S protein labeling mix, [35S]-; PerkinElmer). The labeled virus was purified as described above.
Coimmunoprecipitation assay.
A coimmunoprecipitation assay was performed as previously described (23). Briefly, the purified F particles were incubated with control Fc (Fc portion of human IgG; 3 μg; R&D Systems), SCARB2-Fc (0.1, 0.3, 1, or 3 μg; R&D Systems) or PSGL1-Fc (0.1, 0.3, 1, or 3 μg; R&D Systems), and anti-human IgG (Fc specific)-agarose (Sigma) in 1 ml of 5% FBS–DMEM for 2 h at 4°C. Then, the beads were washed twice with 5% FBS–DMEM, suspended in SDS sample buffer, and incubated for 10 min at 95°C. After the beads were removed, the samples were loaded onto 12% Mini-Protean TGX precast gels (Bio-Rad), followed by Western blotting with anti-EV71 serum (kindly provided by H. Shimizu, NIID, Japan) (54) or protein staining using an Oriole fluorescent gel stain (Bio-Rad).
Virus attachment assay.
L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells were mixed with various amounts of purified [35S]methionine- and [35S]cysteine-labeled F particles and incubated for 2 h at 4°C to allow viral attachment to the cell surface. After two washes with PBS(−) containing 0.1% BSA, the virus bound to the cells was recovered with 0.5% SDS solution, and the radioactivity was measured using Insta-Gel Plus liquid scintillation cocktail (PerkinElmer) and an LSC-6100 liquid scintillation counter (Aloka). The radioactivity of the virus that specifically bound to the cells was calculated by subtracting the counts obtained for L-Empty cells from the counts obtained for L-SCARB2 cells or L-PSGL1 cells.
Observation of internalized EV71 in L-SCARB2 cells and L-PSGL1 cells by indirect immunofluorescence.
A total of 2.8 × 104 cells of L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells were seeded onto 8-well CultureSlides (BD). Two days after seeding, purified EV71 F particles were added at an MOI of 300 at 4°C to allow viral attachment to the cell surface without entry. The cells were then shifted to 37°C (designated time zero). The cells were washed with PBS(−), fixed in PBS(−) containing 4% PFA for 5 or 15 min, and washed four times with cold PBS(−) before immunofluorescence processing. The fixed cells were incubated with PBS(−) containing 0.05% saponin and 5% BSA fraction V (Sigma) to permeate cells and block nonspecific reactions. The fixed cells were incubated overnight at 4°C with the following primary antibodies: a 1:1,000 dilution of the anti-EV71 serum that had been adsorbed with fixed L929 cells and a 1:300 dilution of anti-early endosome antigen 1 (EEA1) goat antibodies (N-19; Santa Cruz Biotechnology). After being washed with PBS(−), the cells were then incubated with the secondary antibodies (Alexa Fluor 488 or 568 donkey anti-rabbit or -goat IgG [H+L]; Life Technologies) for 90 min at room temperature. After another PBS(−) wash, the cells were mounted in Vectashield with DAPI (4′,6-diamidino-2-phenylindole) mounting medium (Vector Laboratories). The cells were imaged under a laser-scanning microscope (TCS SP2; Leica Microsystems). The maximum projection was created using Imaris software (Carl Zeiss, Inc.).
Quantification of viral RNA by real-time RT-PCR.
Viral RNA was extracted using a QIAamp viral RNA minikit (Qiagen), and real-time RT-PCR was performed as previously described (59, 60), with several modifications. The viral RNA was assayed in a 20-μl reaction mixture containing 2 μl of viral RNA using a one-step SYBR PrimeScript Plus RT-PCR kit (TaKaRa) with the primers EQ-1 (ACATGGTGTGAAGAGTCTATTGAGCT) and EQ-2 (CCAAAGTAGTCGGTTCCGC). The mixtures were subjected to real-time RT-PCR using a LightCycler 480 system II (Roche) with a reverse transcription step at 42°C for 5 min, followed by 45 cycles at 95°C for 5 s and at 60°C for 20 s. The plasmid pSVA-EV71 (17) was used as a control for the quantification of the copy number.
Conformation alteration assay in vitro.
[35S]methionine- and [35S]cysteine-labeled EV71 F particles from strain SK-EV006, BrCr, or 1095 (10,000 cpm) were incubated with control Fc (1 μg), SCARB2-Fc (1 μg), or PSGL1-Fc (1 μg) in PBS(−) (pH 7.4) containing 0.1% BSA for 1 h at 4°C. After a pH shift from pH 7.4 to the indicated pH by the addition of 9 volumes of PBS(−) (at the indicated pH) containing 0.1% BSA, the mixtures were incubated for 0, 0.5, 1, 2, or 3 h at 37°C, sonicated for 5 min using a Bioruptor UCD-200TM apparatus (Cosmo Bio), layered onto a 15 to 30% sucrose density gradient in PBS(−) (pH 7.4) containing 0.1% BSA, and centrifuged in the SW41Ti rotor at 39,000 rpm for 2 h at 4°C. After fractionation, the radioactivity of each fraction (0.6 ml) was measured using Insta-Gel Plus and an LSC-6100 liquid scintillation counter. In several cases, the fractions were pooled and ultracentrifuged in the SW32Ti rotor at 32,000 rpm for 2 or 3 h at 4°C. The pellets were suspended in PBS(−) including 0.1% BSA and used to quantify viral RNA or for autoradiography on a 10 to 20% Tricine gel (Life Technologies), a BAS-IP MS 2040 phosphorimaging plates (Fujifilm), and a BAS-2500 image scanner (Fujifilm). The EV71 empty capsid was obtained by heating the F particles for 15 min at 56°C. The condition is previously described as a way to generate PV empty capsids (61). The polioviruses that were incubated with 1 μg of PVR-His (R&D systems) or 1 μg of control Fc for 1 h at 37°C were used as markers of the native virion, A particle, and empty capsid (48).
Conformation alteration assay in vivo.
[35S]methionine- and [35S]cysteine-labeled EV71 F particles (5 × 105 cpm) were incubated with 2 × 106 L-SCARB2 cells or L-PSGL1 cells in PBS(−) (pH 7.4) containing 0.1% BSA for 2 h at 4°C. After being washed twice with PBS(−) (pH 7.4) containing 0.1% BSA, the cells were incubated for 0.5, 1, or 2 h at 37°C in 5% FBS–DMEM, lysed with PBS(−) containing 1% Triton X-100, sonicated for 5 min using a Bioruptor UCD-200TM apparatus, and centrifuged at 20,000 × g for 1 min at 4°C. The supernatants (approximately 6 × 103 and 1 × 104 cpm for L-SCARB2- and L-PSGL1-associated virus, respectively) were layered onto a 15 to 30% sucrose density gradient in PBS(−) containing 0.1% BSA and centrifuged in the SW41Ti rotor at 39,000 rpm for 2 h at 4°C. After fractionation, the radioactivity of each fraction (0.6 ml) was measured using Insta-Gel Plus and an LSC-6100 liquid scintillation counter.
RESULTS
Comparison of EV71 infections in L-SCARB2 cells and L-PSGL1 cells.
To compare the efficiency of EV71 infection in L929 cells that expressed SCARB2 and PSGL1, we established L-SCARB2 cells and L-PSGL1 cells that constitutively expressed human SCARB2 and PSGL1, respectively. Then, we confirmed the cell surface expression of human SCARB2 and human PSGL1 on L-SCARB2 cells and L-PSGL1 cells by flow cytometry (Fig. 1A). Human SCARB2 and human PSGL1 were detected on the surfaces of their respective cell lines. We therefore infected L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells with an equal amount of EV71-GFP. EV71-GFP, which has capsid proteins that are identical to those of the EV71-PB strain SK-EV006/Malaysia/97 (SK-EV006), can enter cells via the SCARB2-dependent pathway (17, 22) and the PSGL1-dependent pathway (18). EV71-GFP requires a longer time for a single round of replication than the wild-type EV71 strain SK-EV006 and does not effectively spread within 24 h (23). The infected cells were observed at 24 h postinfection (Fig. 1B). GFP-positive cells were not found in the L-Empty cells. A large number of GFP-positive cells were detected in L-SCARB2 cells, whereas a small number of GFP-positive cells were detected in L-PSGL1 cells. According to the flow cytometric analysis, the number of GFP-positive L-SCARB2 and L-PSGL1 cells was significantly greater than that of the L-Empty cells (P = 0.0000044 and P = 0.0090, respectively). The number of GFP-positive cells was significantly greater in the L-SCARB2 cells than in the L-PSGL1 cells (P = 0.0000049) (Fig. 1C). These results indicate that SCARB2 and PSGL1 mediate EV71-GFP infection in L929 cells and that the infection efficiency via the two receptors is significantly different.
Fig 1.

Efficiency of EV71 infection in L-SCARB2 cells and L-PSGL1 cells. (A) Expression of human SCARB2 (top) and PSGL1 (bottom) on the cell surface. L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells were stained with an anti-SCARB2 antibody (solid lines), a normal goat IgG (dotted lines), an anti-PSGL1 antibody (solid lines), or a normal mouse IgG (dotted lines) and analyzed by flow cytometry. (B and C) EV71-GFP infection of L-SCARB2 cells and L-PSGL1 cells. L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells were infected with EV71-GFP. These cells were imaged with a fluorescence microscope (B) and analyzed by flow cytometry (C). The flow cytometry data are shown as the mean counts with the SDs (n = 3). **, P < 0.01 according to Student's t test. (D and E) RD cells, L929 cells, L-SCARB2 cells, and L-PSGL1 cells were infected with EV71 at MOIs of 10 (D) and 0.01 (E), and the viral titers were determined at 0, 12, 24, 36, and 48 h and at 0, 6, 12, 24, and 48 h after infection, respectively. The data are shown as the mean viral titers with the SDs (n = 3).
To validate the results that were obtained in the experiments using EV71-GFP, we examined cytopathic effect (CPE) induction and viral spread using wild-type EV71. Using L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells, the growth kinetics of EV71 were evaluated in infections at MOIs of 10 and 0.01 (Fig. 1D and E). EV71 grew in L-SCARB2 cells as efficiently as in RD cells (Fig. 1D and E). The viral growth in L-PSGL1 cells was better than that in L929 cells. However, the growth in L-PSGL1 cells was less efficient than that in L-SCARB2 cells and RD cells (Fig. 1D and E). The viral titer at the last time point in the L-SCARB2 cells was approximately 100-fold or 10,000-fold higher than the titer that was obtained in L-PSGL1 cells. EV71 propagated minimally in the negative-control L929 cells. At 24 h postinfection with an MOI of 10, we observed a clear CPE in all of the L-SCARB2 cells and the RD cells; however, only a small fraction of the L-PSGL1 cells showed a CPE (data not shown). In addition, a clear CPE was observed in L-SCARB2 cells at 24 h postinfection at an MOI of 0.01 but not in L-PSGL1 cells (data not shown). These results indicate that EV71 infects and spreads more efficiently via SCARB2 than PSGL1.
Purification of EV71 native virion F particles.
It has been reported that EV71 in cell culture medium is a mixture of noninfectious empty (E) particles and infectious filled (F) particles (62). E particles are composed of VP0 (VP4-VP2), VP1, and VP3 and have no infectivity, whereas F particles are composed of VP1, VP2, VP3, VP4, and viral genomic RNA and are infective (62). To evaluate the interaction between EV71 and the receptors, we purified F particles as described in Materials and Methods. Fractions 8 to 10 (Fig. 2A, left) and fractions 3 to 5 (Fig. 2A, right) of the 15 to 30% sucrose gradient ultracentrifugation contained E particles and F particles, respectively. We then confirmed the purity and the protein components of the E and F particles by autoradiography (Fig. 2B) or Western blotting using anti-VP2 and anti-EV71 antibodies (Fig. 2C). The deduced molecular masses of VP0, VP1, VP2, VP3, and VP4 are 35, 33, 28, 27, and 7 kDa, respectively. The autoradiogram suggested that the purified E particles were composed of VP0, VP1, and VP3, whereas the purified F particles were composed of VP1, VP2, VP3, and VP4 (Fig. 2B). In agreement with the results presented above, Western blotting using the anti-VP2 antibody detected both the 35-kDa VP0 and the 28-kDa VP2 in the prepurified sample (Fig. 2C, lane Pre), while only VP0 or VP2 was detected in the purified E particles or F particles, respectively (Fig. 2C, lanes E particles and F particles). These data demonstrate that we successfully purified the native virion F particles without contamination with the E particles.
Fig 2.

Purification of EV71 F particles. (A) Sucrose density gradient purification of [35S]methionine- and [35S]cysteine-labeled EV71 E particles and F particles. The prepurified E and F particles from the stepwise CsCl gradient centrifugation were layered onto a 15 to 30% sucrose density gradient for ultracentrifugation at 39,000 rpm for 2 h at 4°C. The gradients were fractionated from the top. The E particles (left) and F particles (right) were collected from the indicated fractions. (B) Autoradiography of purified E and F particles. [35S]methionine- and [35S]cysteine-labeled EV71 E particles and F particles (10,000 cpm) were resolved by electrophoresis, and the bands were visualized by autoradiography. (C) Unlabeled E and F particles were resolved by electrophoresis and detected by Western blotting with an anti-VP2 antibody (top) and an anti-EV71 serum that predominantly recognized VP1 (bottom). The positions of the viral proteins are indicated at the sides of the blots. IB, immunoblotting.
Binding of EV71 to SCARB2 and PSGL1.
We speculated that the infection efficiency of the cells might be dependent on the amount of virus bound to the cells. We therefore determined the amount of the virus bound to L-SCARB2 cells and L-PSGL1 cells (Fig. 3A). L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells were mixed with 1 × 104, 3 × 104, or 1 × 105 cpm of the 35S-labeled EV71 F particles. After the unbound virions were removed by washing, the cells were lysed with 0.5% SDS and the radioactivity was measured using a liquid scintillation counter. EV71 bound to L-SCARB2 cells and L-PSGL1 cells; however, the amount of EV71 that bound to L-PSGL1 cells was severalfold greater than that bound to L-SCARB2 cells under all tested conditions. These data indicate that more EV71 virion binds to L-PSGL1 cells than to L-SCARB2 cells.
Fig 3.
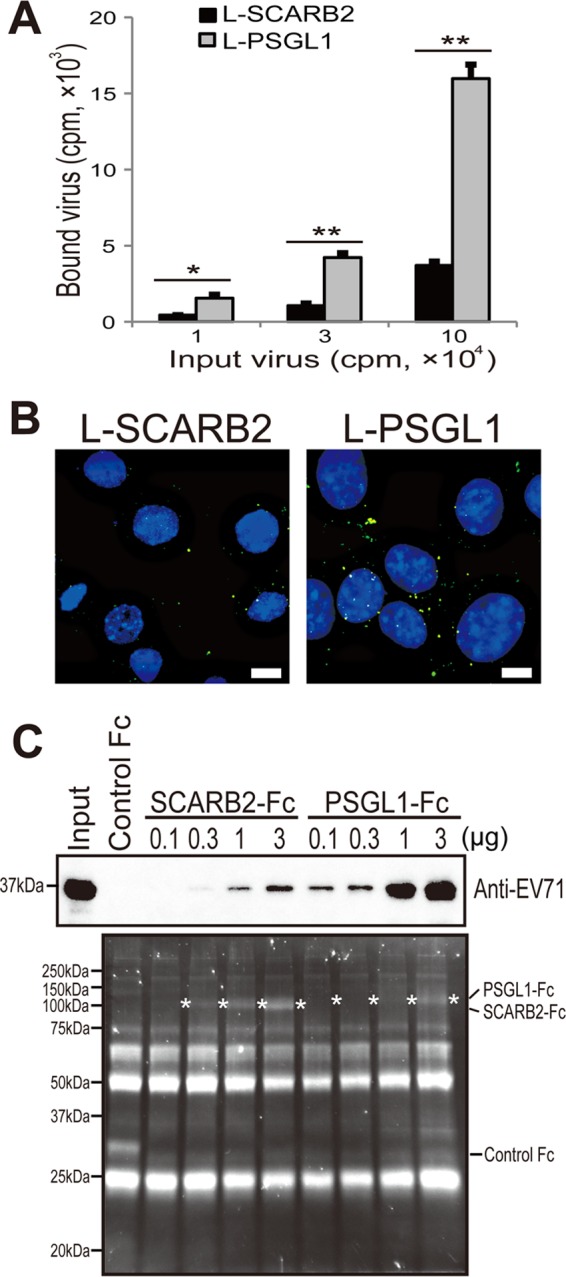
Binding of EV71 to SCARB2 and PSGL1. (A) More EV71 attached to L-PSGL1 cells than to L-SCARB2 cells. L-Empty cells, L-SCARB2 cells, or L-PSGL1 cells were incubated with 1 × 104, 3 × 104, or 1 × 105 cpm of [35S]methionine- and [35S]cysteine-labeled EV71 at 4°C for 2 h. After being washed, the cells were lysed with 0.5% SDS and the amount of bound virion was measured using a liquid scintillation counter. The data are shown as the mean counts with the SDs (n = 2). * and **, P < 0.05 and P < 0.01, respectively, according to Student's t test. (B) EV71 attached to L-PSGL1 cells more efficiently than to L-SCARB2 cells, as confirmed by immunofluorescence. EV71 F particles were attached to L-SCARB2 cells or L-PSGL1 cells at 4°C and then at 37°C for 5 min. The cells were washed and fixed before undergoing immunofluorescence microscopy to detect EV71. We analyzed the cells in three-dimensional image stacks by confocal fluorescence microscopy and created a maximum projection. Green indicates EV71, and blue DAPI staining indicates the location of the nuclei. Bars, 10 μm. (C) EV71 bound to SCARB2-Fc and PSGL1-Fc. A total of 10 μg of control Fc and 0.1, 0.3, 1, and 3 μg of SCARB2-Fc and PSGL1-Fc were bound to anti-human Fc-agarose and incubated with EV71. The precipitated proteins were analyzed by Western blotting with the anti-EV71 serum or Oriole gel staining. White asterisks indicate the bands of SCARB2-Fc or PSGL1-Fc.
Next, to confirm that EV71 binds to L-PSGL1 cells more efficiently than L-SCARB2 cells, we performed immunofluorescence microscopy to compare the amount of EV71 bound to these cells (Fig. 3B). After the purified EV71 F particles were attached to L-SCARB2 cells or L-PSGL1 cells, the cells were fixed and stained with anti-EV71 serum to detect EV71. Using series of z-stack images, we created maximum projections to show three-dimensional information in a plane image. We observed EV71 puncta in L-SCARB2 cells and L-PSGL1 cells, with most of the EV71 being on the surface of the cells. The viral puncta were more intense and more numerous by severalfold in the L-PSGL1 cells than in the L-SCARB2 cells. We did not observe any virus dots in the cells without EV71 (data not shown). These results confirm that EV71 F particles bind to L-PSGL1 cells more efficiently than to L-SCARB2 cells.
The results presented above suggested that the amount of PSGL1 expressed on the surface of L-PSGL1 cells is greater than that of SCARB2 expressed on the surface of L-SCARB2 cells and/or the affinity of PSGL1 for EV71 (EV71-PB strain) is greater than that of SCARB2. We then performed a coimmunoprecipitation assay using soluble receptors. EV71 was incubated with various amounts of SCARB2-Fc (extracellular region of SCARB2 fused to the Fc region of human IgG), PSGL1-Fc (extracellular region of PSGL1 fused to the Fc region of human IgG), or control Fc (Fc region of human IgG) bound to anti-Fc agarose beads, and the precipitated proteins were analyzed by Western blotting with an anti-EV71 serum (Fig. 3C, top) and protein staining (Fig. 3C, bottom). The EV71-VP1 band was detected in the 0.3-, 1-, and 3-μg SCARB2-Fc lanes in a concentration-dependent manner and in all of the PSGL1-Fc lanes in a concentration-dependent manner; however, the band was not detected in the control Fc lane or in the 0.1-μg SCARB2-Fc lane. PSGL1-Fc precipitated a larger amount of EV71-VP1 than SCARB2-Fc by severalfold at each concentration of the receptors that we tested. These data suggest that the binding affinity of PSGL1 to EV71 is stronger than that of SCARB2. Taken together, the results indicate that L-PSGL1 cells bind more virus than L-SCARB2 cells due to a higher affinity of PSGL1 for the virus, but the amount of the virus that attached to the cell surface does not solely determine the infection efficiency.
EV71 internalization in L-SCARB2 cells and L-PSGL1 cells.
To examine the internalization of purified EV71 virions by L-SCARB2 cells and L-PSGL1 cells, we performed immunofluorescence microscopy (Fig. 4). L-Empty cells, L-SCARB2 cells, or L-PSGL1 cells were incubated with purified EV71 F particles at 4°C, shifted to 37°C, and fixed. The cells were then analyzed by immunofluorescence microscopy to observe EV71 and the early endosome marker EEA1. In the z-axis planes where we could observe the internalized virus, we detected EV71 puncta in all L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells at 15 min after the temperature shift to 37°C (Fig. 4A, left). We observed approximately 100 cells for each cell line. There were fewer EV71 signals in the L-Empty cells than in the L-SCARB2 cells and L-PSGL1 cells (average numbers of EV71 puncta in one cell, 0.28, 1.06, and 1.56 for L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells, respectively). The results suggested that the internalization of the virus was enhanced by SCARB2 and PSGL1. In addition, we found that some of the EV71 puncta were colocalized with EEA1 (Fig. 4A, arrowheads), and the number of EV71 puncta that colocalized with EEA1 in L-SCARB2 cells was greater than that in L-PSGL1 cells. For a quantitative comparison of the colocalization of virus puncta with EEA1, we analyzed one z-axis midplane per cell, and the numbers of virus puncta colocalized with EEA1 per all virus puncta were as follows: 1 of 33 for L-Empty cells, 8 of 100 for L-SCARB2 cells, and 4 of 89 for L-PSGL1 cells (Fig. 4B). The analysis showed that virus was internalized into the cells and delivered to the early endosome in L-SCARB2 cells, even though a smaller amount of virus attached to L-SCARB2 cells than L-PSGL1 cells.
Fig 4.
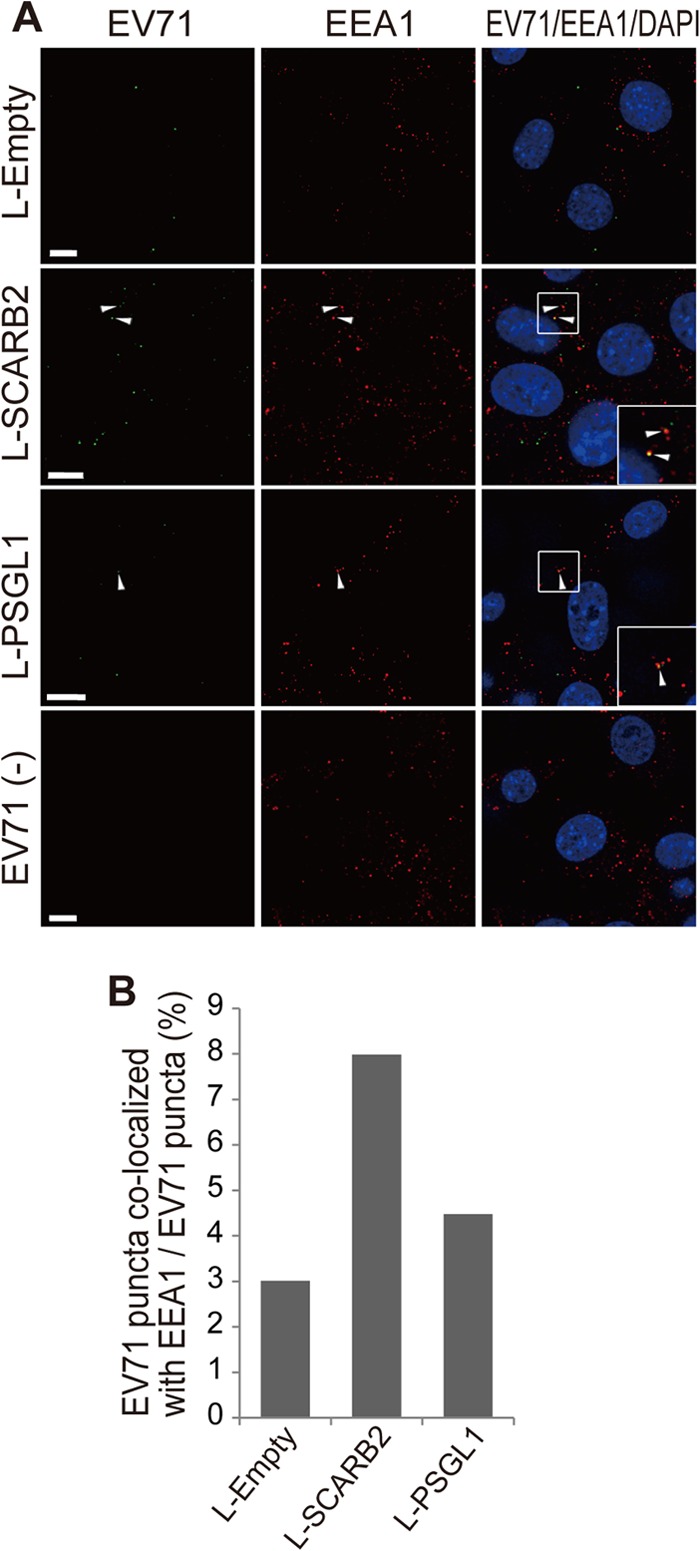
Internalization of EV71 virions into L-SCARB2 cells and L-PSGL1 cells. (A) Immunofluorescence microscopy of EV71 and EEA1 in L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells after incubation with EV71 F particles. The cells were incubated with or without purified F particles at 37°C for 15 min. After the washes, the cells were fixed and stained with or without anti-EV71 and anti-EEA1 antibodies and detected with Alexa Fluor 488- or 568-conjugated secondary antibodies. Green and red indicate EV71 and EEA1, respectively. Blue DAPI staining indicates the location of the nuclei. Arrowheads, colocalization of EV71 with EEA1. (Inset) Enlarged view of the small square. For EV71-negative cells [EV71(−)], we showed L-SCARB2 cells as a representative of L-Empty cells, L-SCARB2 cells, and L-PSGL1 cells because we obtained essentially similar results in all of these cell types. Bars, 10 μm. (B) L-SCARB2 cells internalize EV71 in early endosomes more efficiently than L-Empty cells and L-PSGL1 cells. The ratio of EV71 puncta colocalized with EEA1 compared to EV71 puncta is shown.
Conformational alteration of EV71 by SCARB2 under acidic conditions.
After the virions are internalized by the cells, the viral genomic RNA must be released from the virion into the cytoplasm to complete the EV71 infection. We expected that SCARB2 could efficiently initiate the uncoating step. To analyze the conformational alteration of EV71 as a mimic of the uncoating step by sucrose density gradient ultracentrifugation, we prepared EV71 empty capsids by heating the purified F particles; these served as a reference (Fig. 5A). As another control, we also prepared PV A particles and empty capsids by incubating native PV virions with PVR (Fig. 5B). According to the sucrose density gradient ultracentrifugation, the EV71 and PV native virions sedimented at fractions 6 to 8 (native virion), the heat-treated EV71 sedimented at fractions 14 and 15 (empty capsid), and PV, which was treated with PVR, sedimented at fractions 10 to 12 (A particle) and fractions 14 to 16 (empty capsid, approximately 18.1% of total counts). These results indicate that the empty capsids of EV71 appeared as a peak in fractions 14 and 15 and that the A particles appeared as a peak in fractions 10 to 12.
Fig 5.

Conformational alteration of EV71 by SCARB2 under an acidic pH. (A) Characterization of EV71 F particles and heat-induced empty capsids by ultracentrifugation. Approximately 10,000 cpm of purified F particles was incubated for 15 min at 4°C or 56°C. The samples were centrifuged in 15 to 30% sucrose gradients, and the radioactivity of each fraction (0.6 ml) was measured by liquid scintillation counting. (B) Native virion, A particle, and empty capsid of PV. Approximately 10,000 cpm of the PV native virion was treated with 1 μg of PVR-His and control Fc for 1 h at 4°C and subsequently incubated for 1 h at 37°C. The samples were analyzed using 15 to 30% sucrose gradients. The number above the bracketed line indicates the proportion of fractions 14 to 16. (C) Conformational alteration of EV71 F particles via incubation with SCARB2-Fc at various pH levels. Approximately 10,000 cpm of purified F particles was treated with 1 μg of control Fc, 1 μg of SCARB2-Fc, or 1 μg of PSGL1-Fc for 60 min at 4°C and then incubated for 60 min at 37°C at a pH of 7.4, 6.0, or 4.5. The samples were analyzed using 15 to 30% sucrose gradients. The numbers above the bracketed lines indicate the proportions of fractions 12 to 15. The fractions indicated by solid bars were collected and used for analysis of VP4 and viral RNA in Fig. 6.
To analyze the conformational alteration of EV71, EV71 was incubated with 1 μg of control Fc, SCARB2-Fc, or PSGL1-Fc at 37°C at pH 7.4, 6.0, or 4.5 and subjected to sucrose density gradient ultracentrifugation (Fig. 5C). The peak of the native virion (fractions 6 to 10) did not shift when the F particles were incubated with control Fc or PSGL1-Fc at any pH, whereas a portion of the 35S-labeled virions (approximately 4.5% and 9.8% of total counts at pH 6.0 and pH 4.5, respectively) formed a shoulder peak at fractions 12 to 15 when incubated with SCARB2-Fc at an acidic pH but not under neutral conditions at pH 7.4. These data indicate that the conformational alteration of EV71 is triggered by incubation with SCARB2 under acidic conditions.
Release of VP4 and viral genomic RNA from native virions.
Because a portion of the EV71 virions shifted to the fractions that sedimented at a lower rate after incubation with SCARB2 under acidic conditions, we determined whether the viral particles in fractions 12 to 15 contained VP4 and/or viral genomic RNA. For this purpose, we collected fractions 7 to 9 of the control Fc-treated EV71 and fractions 7 to 9 and 12 to 15 of SCARB2-Fc-treated EV71 (the collected fractions are indicated by solid bars in Fig. 5C). To analyze VP4, approximately 10,000 cpm of each sample was separated by SDS-PAGE and imaged with a phosphorimaging plate (Fig. 6A). VP4 together with VP1, VP2, and VP3 was clearly detected in each lane of fractions 7 to 9 of the control Fc- or SCARB2-Fc-treated EV71. VP4 was not detected in fractions 12 to 15 of SCARB2-Fc-treated EV71, whereas VP1, VP2, and VP3 were detected at levels that were similar to those in the other lanes. For the viral genomic RNA analysis, viral genomic RNA was extracted from approximately 500 cpm of each sample and quantified using real-time RT-PCR (Fig. 6B). The levels of viral genomic RNA in fractions 7 to 9 of SCARB2-Fc-treated EV71 were comparable to the levels in fractions 7 to 9 of the control Fc-treated EV71. The amount of viral genomic RNA in fractions 12 to 15 of the SCARB2-Fc-treated EV71 was significantly reduced compared with that in fractions 7 to 9 of the control Fc-treated EV71 (P = 0.00190). Taken together, these data demonstrate that the particles in fractions 12 to 15 of the SCARB2-Fc-treated EV71 are empty capsids.
Fig 6.

Release of VP4 and viral RNA. (A) Release of VP4 by treatment with SCARB2-Fc. Approximately 10,000 cpm of virus particles from fractions 7 to 9 and 12 to 15 of SCARB2-Fc-treated EV71 and from fractions 7 to 9 of control Fc-treated EV71 were collected and analyzed by SDS-PAGE and subsequent autoradiography. (B) Contents of viral RNA. The viral RNA was extracted from approximately 500 cpm of each sample. The viral RNA was quantified by quantitative RT-PCR (n = 3). The data are shown as the relative proportions of viral RNA with the SDs (n = 3). The contents of the viral RNA in fractions 7 to 9 of control Fc were set to 100%. **, P < 0.01 by Student's t test.
Time course and pH dependency of the conformational alteration of EV71 by SCARB2.
We evaluated the dependency of the conformational alteration on the incubation time and pH. First, to evaluate the time course of the conformational alteration, the native virions that bound to SCARB2-Fc were incubated at 37°C for 0, 0.5, 1, 2, or 3 h at pH 4.5 and analyzed by use of a sucrose density gradient (Fig. 7A). The empty capsid in fractions 11 to 15 was observed after incubation for 0.5 h. The proportion of these fractions increased from 0.7% (0 h) to 13.3% (0.5 h), 18.6% (1 h), 19.5% (2 h), and 21.1% (3 h) in a time-dependent manner. The proportion of virus that sedimented with fractions 11 to 15 after incubation of native virions with control Fc at 37°C for 3 h at pH 4.5 was 1.7%. These data indicate that the conformational change of EV71 is induced in a time-dependent manner. However, not all of the virions were converted into empty capsids after the longer incubation periods.
Fig 7.

Effect of incubation time, pH, and EV71 strains on the conformational alteration of EV71 in the presence of SCARB2. (A) Incubation time-dependent conformational alteration of EV71. EV71 was treated with 1 μg of SCARB2-Fc for 0, 0.5, 1, 2, and 3 h or 1 μg of control-Fc for 3 h at pH 4.5 at 37°C and analyzed by sucrose gradient centrifugation. The numbers above the bracketed lines indicate the proportions of fractions 11 to 15. (B) pH-dependent conformational alteration of EV71. [35S]methionine- and [35S]cysteine-labeled EV71 F particles were incubated with 1 μg of SCARB2-Fc at pH 7.4, 6.5, 6.0, 5.5, 5.0, and 4.5 at 37°C, and the conformational alterations were analyzed at 1 h by sucrose density gradient centrifugation. The numbers above the bracketed lines indicate the proportions of fractions 13 to 16. (C) Conformational alteration of other strains of EV71 by SCARB2. [35S]methionine- and [35S]-cysteine-labeled EV71 strains BrCr (genogroup A) and 1095 (genogroup C) were incubated with 1 μg of control Fc, 1 μg of SCARB2-Fc, and 1 μg of PSGL1-Fc at pH 4.5 at 37°C for 1 h, and the conformational alterations were analyzed by sucrose density gradient centrifugation.
Second, to evaluate the pH dependency of the conformational alteration, we incubated the native virions that bound to SCARB2-Fc at 37°C for 1 h at pH 7.4, 6.5, 6.0, 5.5, 5.0, or 4.5 and subjected them to sucrose density gradient ultracentrifugation (Fig. 7B). The conformational alteration from the native virion to the empty capsid was induced in a pH-dependent manner; empty capsid formation was rarely observed at pH 7.4 and 6.5 (approximately 1.2% and 2.6%, respectively), whereas a conformational alteration was observed at pH 6.0, 5.5, 5.0, and 4.5 (approximately 5.0, 6.1, 6.5, and 8.1%, respectively). Consistent with these data, we confirmed that infection of RD cells by EV71 was inhibited by pretreatment with NH4Cl, an endosome acidification inhibitor, as previously reported (24–26). Infection of L-PSGL1 cells with EV71 was also inhibited by the same treatment, but infection with PV was not affected (data not shown). Thus, these data suggest that the conformational alteration, an essential step in EV71 entry, requires an acidic pH below 6.0.
Because we used EV71 strain SK-EV006 (genogroup B), which binds to both SCARB2 and PSGL1, for the conformation alteration assays, we performed a conformation alteration assay with EV71 strain BrCr (genogroup A), which binds only to SCARB2, and strain 1095 (genogroup C), which binds to SCARB2 and PSGL1 (data not shown) (18). The purified BrCr and 1095 F particles were incubated with 1 μg of control Fc, SCARB2-Fc, or PSGL1-Fc at 37°C at pH 4.5 and subjected to a sucrose density gradient (Fig. 7C). In both cases, the major peak (fractions 5 to 8) of the native virion did not shift when the native virions were incubated with control Fc or PSGL1-Fc, whereas a portion of the native virions shifted to fractions 10 to 15 and formed a shoulder peak when incubated with SCARB2-Fc. Because the conformational alteration of three strains from genogroups A, B, and C was confirmed, we suggest that the conformational alteration of EV71 is generally mediated by SCARB2 in an acidic pH.
Uncoating of EV71 in L-SCARB2 cells.
To confirm that the same conformational alteration of EV71 occurs in vivo, we performed conformational alteration assays with virus associated with L-SCARB2 cells or L-PSGL1 cells (Fig. 8). The 35S-labeled EV71 attached to L-SCARB2 cells or L-PSGL1 cells was incubated for 0.5, 1, or 2 h at 37°C, and the conformational state of the EV71 virions was analyzed by sucrose density gradient ultracentrifugation (Fig. 8). Empty capsids formed a peak at fractions 13 to 15 when native virions were incubated with L-SCARB2 cells, and the proportion in the empty capsid fraction was increased from 5.7% (0.5 h) to 8.1% (1 h) and 17.6% (2 h) in a time-dependent manner. The distinct shift of the native virion peak (fractions 4 to 10) was not observed when the EV71 native virions were incubated with L-PSGL1 cells at any of the time points tested. These data indicate that the uncoating of EV71 occurs in L-SCARB2 cells.
Fig 8.

Conformational alteration of EV71 in L-SCARB2 cells. Purified EV71 F particles bound to L-SCARB2 cells and L-PSGL1 cells were incubated for 0.5, 1, or 2 h at 37°C. The cells were lysed with 1% Triton X-100, and these lysates were analyzed using 15 to 30% sucrose gradients. The numbers above the bracketed lines indicate the proportions of fractions 13 to 15.
DISCUSSION
For the establishment of enterovirus infection, the virus must bind to the surface of target cells, enter into the cells (this step may not be necessary in some picornaviruses), and release viral genomic RNA into the cytoplasm. The viral receptor promotes infection by playing multiple roles, including viral attachment, internalization, and uncoating. It has been independently reported that the receptors SCARB2 and PSGL1 bind to the EV71 virion and mediate EV71 infection (17, 18, 22–24, 26, 31, 63). We have demonstrated that SCARB2 supports EV71 infection in nonsusceptible cells more efficiently than PSGL1, despite the reduced binding ability of SCARB2 to the EV71 virion compared to that of PSGL1.
We first compared the virus-binding capacities of SCARB2 and PSGL1. Unexpectedly, L-PSGL1 cells could bind to more viruses than L-SCARB2 cells (Fig. 3A and B), and soluble PSGL1 could bind to more viruses than SCARB2 (Fig. 3C). The capacity of SCARB2 binding to EV71 was lower than that of PSGL1. Regions on the EV71 virion that are important for the interaction with SCARB2 and PSGL1 have been identified. The glutamine residue at position 172 in the EF loop region of VP1, which lines the wall of the canyon on the viral surface, was important for binding to SCARB2, suggesting that SCARB2 binds inside the canyon (24). Recently, structural data were collected and revealed that the canyon of EV71 is shallower than that of other enteroviruses (3, 4). It is well-known that uncoating receptors, such as PVR, CAR, and ICAM-1, have immunoglobulin-like domains and bind to the viral canyon (33). However, SCARB2 does not have immunoglobulin-like domains. We have previously demonstrated that amino acids 142 to 204 of SCARB2 are important for the interaction with EV71 (23). Thus, this region of SCARB2 may fit the shallow canyon. Regarding PSGL1, VP2 residue 149 participates in forming the puff that serves as the southern rim of the canyon (4) and is important for PSGL1-EV71 binding (18, 63). These findings suggest that SCARB2 and PSGL1 bind at different sites on the virion and that the affinity of the SCARB2-EV71 interaction is different from that of the PSGL1-EV71 interaction.
The discrepancy between the infection efficiency and binding suggests that SCARB2 plays additional essential roles in the internalization and/or induction of viral uncoating, the step after viral attachment. We first examined whether EV71 was internalized via a SCARB2- or PSGL1-dependent pathway (Fig. 4). We observed internalized viruses in both L-SCARB2 cells and L-PSGL1 cells. In some instances, the EV71 puncta colocalized with the endosome marker. The amount of virus associated with endosomes in L-SCARB2 cells is greater than that in L-PSGL1 cells. SCARB2 is normally present in lysosomes and shuttles via vesicular transport to the plasma membrane through endosomes (19). It is likely that SCARB2 captures EV71 when SCARB2 is present at the plasma membrane. SCARB2 on the cell surface is transported to lysosomes by clathrin-mediated endocytosis (19). EV71 infects RD cells via the SCARB2-dependent pathway (22, 26). EV71 infection of RD cells was inhibited by knocking down the expression of the clathrin heavy peptide (CLTC) by treatment with chlorpromazine, an inhibitor of clathrin-mediated endocytosis, and by the expression of a dominant-negative mutant, EPS15 (25). The inhibition of EV71 infection by a depletion of clathrin-mediated endocytosis was similarly observed in mouse 3T3 cells that expressed human SCARB2 (26). These reports, together with our data, suggest that SCARB2 plays a role in internalizing the viral particles and delivering them to the endosome using the physiological pathway of SCARB2. Our data showed that at least a portion of the viruses that were internalized via the SCARB2 pathway was delivered to the endosome, where the viruses were exposed to an acidic pH. It is therefore likely that the EV71 puncta that colocalized with the endosomal marker represent the viruses that are involved in a productive stage of infection. However, the precise mechanisms of EV71 entry via SCARB2 should be studied more in detail.
Next, we performed a conformational alteration assay with 35S-labeled EV71 to determine whether SCARB2 or PSGL1 induces the uncoating process (Fig. 5). A particles and empty capsids are considered to be important intermediates and end products, respectively, during the uncoating process. Therefore, detecting these particles is important in order to confirm the role of these two receptors in the uncoating process. The empty capsids were generated by incubating the native virions with SCARB2 but not with PSGL1 both in vitro and in vivo. Recently, the crystal structure of the EV71 E particle was reported with high resolution (3, 4). The E particle of EV71 has properties similar to those of the PV and HRV empty capsids, as determined by cryo-electron microscopy and X-ray diffraction (64, 65). Therefore, it is possible that the empty capsid of EV71 also has exits for viral genomic RNA near the 2-fold axis. We demonstrated that the conformation of the EV71 virion was altered by incubation with soluble SCARB2 under acidic conditions (Fig. 5C). Consistent with these results, previous studies have found that EV71 entry via the SCARB2-dependent pathway was inhibited by endosome acidification inhibitors (24–26). Soluble PSGL1 did not induce a conformational change under any tested physiological pH. Taken together, these data demonstrate that SCARB2 functions as an uncoating receptor during EV71 entry, and this property is a major reason that L-SCARB2 cells support an efficient EV71 infection compared with the efficiency of infection for L-PSGL1 cells. Because SCARB2 shuttles between lysosomes and the plasma membrane (19), the EV71-SCARB2 complex that is formed at the plasma membrane may enter the endosomal compartment, in which the complex is then exposed to an acidic pH. A viral conformation alteration occurred when the pH was less than 6.0 in the cell-free system (Fig. 7B), suggesting that the uncoating events begin in the early endosomes.
To date, several patterns of uncoating processes have been reported for picornaviruses. One pattern is the conformational alteration of PV, CVB, and the major-group HRVs, which is facilitated by PVR, CAR, and ICAM-1, respectively (Fig. 9A) (34, 46, 48). The native virions of PV, CVB and the major-group HRVs treated with respective receptors become A particles after releasing VP4 at neutral pH. The A particle becomes an empty capsid after releasing viral RNA. In the case of HRV14 and HRV16, the alteration is dependent on ICAM-1 and low pH (53). Another pattern is the conformational alteration of the minor-group HRVs, which is dependent on an acidic pH (Fig. 9B) (34, 50). VP4 and viral RNA are sequentially released from the native virion under an acidic environment because both A particles and empty capsids were detected. In addition, the other pattern of conformational alteration in aphthovirus also depends on an acidic pH (Fig. 9B) (34, 51). Under acidic conditions, the native virion dissociates into an empty capsid and then into pentameric subunits for viral RNA release. In this study, we revealed that the conformational alteration of EV71 is different from these patterns and requires both SCARB2 and an acidic pH (Fig. 9C). Additionally, we observed the formation of empty capsids but not A particles (Fig. 5 and 6). Therefore, either the native EV71 virion is converted directly into an empty capsid or the A particles were not detected because they have a short half-life. Chen et al. performed a conformational alteration assay by monitoring the shift of the viral RNA peak using purified EV71 and soluble receptors (24). In contrast to our findings, they observed the formation of an RNA peak that sedimented at a lower rate than the native virion (24). Chen et al. claimed that the shifted viral RNA peak was representative of A particles; however, they did not demonstrate whether VP4 existed in putative A-particle fractions or whether empty capsids were formed (24). In either case, the mode of EV71 conformational alteration is a novel pattern among picornaviruses because both SCARB2 and an acidic pH are essential for conformational alteration.
Fig 9.
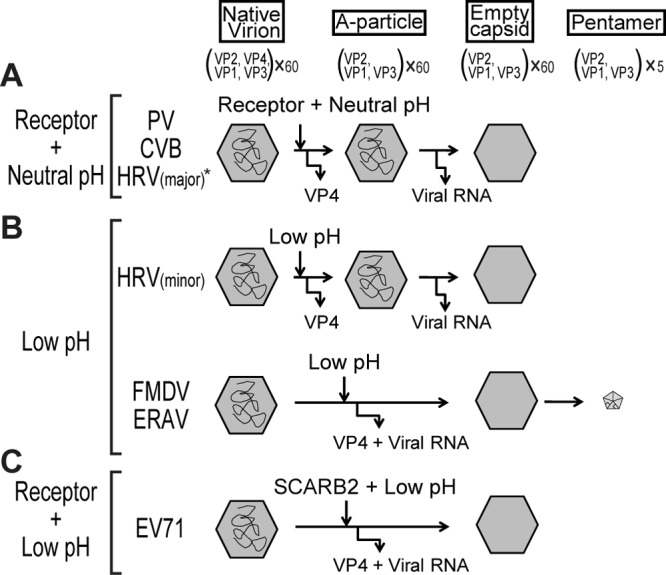
Schematic diagrams of the conformational alterations of picornaviruses. (A) Receptor-dependent conformational alteration at a neutral pH pattern. The conformational alteration of PV, CVB, and the major-group HRVs is triggered by their respective receptors at a neutral pH. The native virion becomes an A particle when VP4 is released from the native virion. The A particle becomes an empty capsid when the viral RNA is released from the A particle. *, HRV14 and HRV16 require both receptor and low pH. (B) Low-pH-dependent conformational alteration pattern. The conformational alteration is triggered by an acidic environment. VP4 and the viral RNA of the minor-group HRVs are released sequentially in a pattern similar to that in panel A. The conformational alteration of FMDV and ERAV is also triggered by an acidic pH. The native virion releases both VP4 and viral RNA and changes into empty capsid and then into pentamers. In the case of FMDV uncoating, the existence of an empty capsid has not been proven. (C) Receptor-dependent conformational alteration at a low pH pattern. The conformational alteration of EV71 is triggered by the receptor SCARB2 under an acidic pH. VP4 and viral RNA are released from the native virion, and the native virion directly becomes an empty capsid.
We have demonstrated that PSGL1 binds to a large amount of EV71 (a PB strain) but does not induce conformational alterations. This situation is similar to the DAF in CVB receptors. DAF binds to several strains of CVB and does not induce conformational alterations (33). Because DAF functions only as a binding factor, an additional molecule, CAR, is needed to establish an efficient infection through the DAF-dependent pathway. However, in polarized epithelial cells, CAR is exclusively located in the tight junctions and is not directly accessible. Thus, DAF-mediated signals permit the virus to move across the apical cell surface to the tight junction, where they can interact with CAR (47). The virus captured by PSGL1 may be similarly uncoated by SCARB2 in human cells that expressed both receptors or by unknown molecules if SCARB2 is absent.
The data suggest that SCARB2 is an authentic receptor for EV71. Only EV71 PB strains have adapted to use PSGL1 as a receptor. The evolutionary significance of using PSGL1 as a receptor is not presently clear. PSGL1 might contribute by broadening the target cells in vivo. It may also act as more than an attachment site on the cell surface. The infection of lymphocytes may trigger the induction of proinflammatory cytokines and participate in pathogenesis because individuals with severe EV71-associated encephalitis and neurogenic pulmonary edema have exhibited high levels of proinflammatory cytokines (66, 67). Further analyses are needed to evaluate the role of PSGL1 as a receptor for EV71.
In summary, we compared SCARB2 and PSGL1 as EV71 receptors. These two receptors demonstrated different abilities in viral attachment, internalization, and uncoating. SCARB2 plays all three roles, whereas PSGL1 does not have the ability to initiate EV71 uncoating. Therefore, PSGL1 alone is not sufficient to enhance EV71 infection in mice (32). Our preliminary results suggest that the transgenic expression of SCARB2 is sufficient to cause neurological diseases in mice. The mouse model that expresses human SCARB2 will greatly contribute to the understanding of EV71 neuropathogenicity in vivo and the development of a vaccine and/or antiviral drugs.
ACKNOWLEDGMENTS
We thank H. Shimizu (NIDD of Japan) for helpful discussions.
This work was supported in part by a Grant-in-Aid for Scientific Research (B) (23390116) and a Grant-in-Aid for Scientific Research (C) (23590557) from the Japan Society for the Promotion of Science and in part by a Grant-in-Aid for Research on Emerging and Re-Emerging Infectious Diseases from the Ministry of Health, Labor and Welfare, Japan.
Footnotes
Published ahead of print 9 January 2013
REFERENCES
- 1. Racaniello V. 2007. Picornaviridae: the viruses and their replication, p 795–838 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Rossmann MG, Johnson JE. 1989. Icosahedral RNA virus structure. Annu. Rev. Biochem. 58:533–573 [DOI] [PubMed] [Google Scholar]
- 3. Wang X, Peng W, Ren J, Hu Z, Xu J, Lou Z, Li X, Yin W, Shen X, Porta C, Walter TS, Evans G, Axford D, Owen R, Rowlands DJ, Wang J, Stuart DI, Fry EE, Rao Z. 2012. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 19:424–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Plevka P, Perera R, Cardosa J, Kuhn RJ, Rossmann MG. 2012. Crystal structure of human enterovirus 71. Science 336:1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rossmann MG, He Y, Kuhn RJ. 2002. Picornavirus-receptor interactions. Trends Microbiol. 10:324–331 [DOI] [PubMed] [Google Scholar]
- 6. Schmidt NJ, Lennette EH, Ho HH. 1974. An apparently new enterovirus isolated from patients with disease of the central nervous system. J. Infect. Dis. 129:304–309 [DOI] [PubMed] [Google Scholar]
- 7. Blomberg J, Lycke E, Ahlfors K, Johnsson T, Wolontis S, von Zeipel G. 1974. New enterovirus type associated with epidemic of aseptic meningitis and-or hand, foot, and mouth disease. Lancet ii:112. [DOI] [PubMed] [Google Scholar]
- 8. Hagiwara A, Tagaya I, Yoneyama T. 1978. Epidemic of hand, foot and mouth disease associated with enterovirus 71 infection. Intervirology 9:60–63 [DOI] [PubMed] [Google Scholar]
- 9. McMinn PC. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol. Rev. 26:91–107 [DOI] [PubMed] [Google Scholar]
- 10. Fujimoto T, Chikahira M, Yoshida S, Ebira H, Hasegawa A, Totsuka A, Nishio O. 2002. Outbreak of central nervous system disease associated with hand, foot, and mouth disease in Japan during the summer of 2000: detection and molecular epidemiology of enterovirus 71. Microbiol. Immunol. 46:621–627 [DOI] [PubMed] [Google Scholar]
- 11. Chan LG, Parashar UD, Lye MS, Ong FG, Zaki SR, Alexander JP, Ho KK, Han LL, Pallansch MA, Suleiman AB, Jegathesan M, Anderson LJ. 2000. Deaths of children during an outbreak of hand, foot, and mouth disease in Sarawak, Malaysia: clinical and pathological characteristics of the disease. For the Outbreak Study Group. Clin. Infect. Dis. 31:678–683 [DOI] [PubMed] [Google Scholar]
- 12. Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, Wang JR, Shih SR. 1999. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N. Engl. J. Med. 341:929–935 [DOI] [PubMed] [Google Scholar]
- 13. Ahmad K. 2000. Hand, foot, and mouth disease outbreak reported in Singapore. Lancet 356:1338. [DOI] [PubMed] [Google Scholar]
- 14. Qiu J. 2008. Enterovirus 71 infection: a new threat to global public health? Lancet Neurol. 7:868–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan JJ, Wang JR, Liu CC, Yang HB, Su IJ. 2000. An outbreak of enterovirus 71 infection in Taiwan 1998: a comprehensive pathological, virological, and molecular study on a case of fulminant encephalitis. J. Clin. Virol. 17:13–22 [DOI] [PubMed] [Google Scholar]
- 16. Yang F, Ren L, Xiong Z, Li J, Xiao Y, Zhao R, He Y, Bu G, Zhou S, Wang J, Qi J. 2009. Enterovirus 71 outbreak in the People's Republic of China in 2008. J. Clin. Microbiol. 47:2351–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, Koike S. 2009. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 15:798–801 [DOI] [PubMed] [Google Scholar]
- 18. Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. 2009. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat. Med. 15:794–797 [DOI] [PubMed] [Google Scholar]
- 19. Eskelinen EL, Tanaka Y, Saftig P. 2003. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13:137–145 [DOI] [PubMed] [Google Scholar]
- 20. Reczek D, Schwake M, Schroder J, Hughes H, Blanz J, Jin X, Brondyk W, Van Patten S, Edmunds T, Saftig P. 2007. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131:770–783 [DOI] [PubMed] [Google Scholar]
- 21. Blanz J, Groth J, Zachos C, Wehling C, Saftig P, Schwake M. 2010. Disease-causing mutations within the lysosomal integral membrane protein type 2 (LIMP-2) reveal the nature of binding to its ligand beta-glucocerebrosidase. Hum. Mol. Genet. 19:563–572 [DOI] [PubMed] [Google Scholar]
- 22. Yamayoshi S, Iizuka S, Yamashita T, Minagawa H, Mizuta K, Okamoto M, Nishimura H, Sanjoh K, Katsushima N, Itagaki T, Nagai Y, Fujii K, Koike S. 2012. Human SCARB2-dependent infection by coxsackievirus A7, A14, and A16 and enterovirus 71. J. Virol. 86:5686–5696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamayoshi S, Koike S. 2011. Identification of a human SCARB2 region that is important for enterovirus 71 binding and infection. J. Virol. 85:4937–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen P, Song Z, Qi Y, Feng X, Xu N, Sun Y, Wu X, Yao X, Mao Q, Li X, Dong W, Wan X, Huang N, Shen X, Liang Z, Li W. 2012. Molecular determinants of enterovirus 71 viral entry: cleft around GLN-172 on VP1 protein interacts with variable region on scavenge receptor B 2. J. Biol. Chem. 287:6406–6420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hussain KM, Leong KL, Ng MM, Chu JJ. 2011. The essential role of clathrin-mediated endocytosis in the infectious entry of human enterovirus 71. J. Biol. Chem. 286:309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin YW, Lin HY, Tsou YL, Chitra E, Hsiao KN, Shao HY, Liu CC, Sia C, Chong P, Chow YH. 2012. Human SCARB2-mediated entry and endocytosis of EV71. PLoS One 7:e30507 doi:10.1371/journal.pone.0030507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sako D, Chang XJ, Barone KM, Vachino G, White HM, Shaw G, Veldman GM, Bean KM, Ahern TJ, Furie B, Cumming DA, Larsen GR. 1993. Expression cloning of a functional glycoprotein ligand for P-selectin. Cell 75:1179–1186 [DOI] [PubMed] [Google Scholar]
- 28. Laszik Z, Jansen PJ, Cummings RD, Tedder TF, McEver RP, Moore KL. 1996. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood 88:3010–3021 [PubMed] [Google Scholar]
- 29. Somers WS, Tang J, Shaw GD, Camphausen RT. 2000. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell 103:467–479 [DOI] [PubMed] [Google Scholar]
- 30. Ley K, Kansas GS. 2004. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 4:325–335 [DOI] [PubMed] [Google Scholar]
- 31. Nishimura Y, Wakita T, Shimizu H. 2010. Tyrosine sulfation of the amino terminus of PSGL-1 is critical for enterovirus 71 infection. PLoS Pathog. 6:e1001174 doi:10.1371/journal.ppat.1001174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu J, Dong W, Quan X, Ma C, Qin C, Zhang L. 2012. Transgenic expression of human P-selectin glycoprotein ligand-1 is not sufficient for enterovirus 71 infection in mice. Arch. Virol. 157:539–543 [DOI] [PubMed] [Google Scholar]
- 33. Bergelson JM. 2010. Receptors, p 73–86 In Ehrenfeld E, Domingo E, Roos RP. (ed), The picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 34. Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. 2010. Picornaviruses. Curr. Top. Microbiol. Immunol. 343:43–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mendelsohn CL, Wimmer E, Racaniello VR. 1989. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56:855–865 [DOI] [PubMed] [Google Scholar]
- 36. Koike S, Horie H, Ise I, Okitsu A, Yoshida M, Iizuka N, Takeuchi K, Takegami T, Nomoto A. 1990. The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 9:3217–3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. 1989. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell 56:849–853 [DOI] [PubMed] [Google Scholar]
- 38. Tomassini JE, Graham D, DeWitt CM, Lineberger DW, Rodkey JA, Colonno RJ. 1989. cDNA cloning reveals that the major group rhinovirus receptor on HeLa cells is intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. U. S. A. 86:4907–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW, Kamarck ME, McClelland A. 1989. The major human rhinovirus receptor is ICAM-1. Cell 56:839–847 [DOI] [PubMed] [Google Scholar]
- 40. Duque H, Baxt B. 2003. Foot-and-mouth disease virus receptors: comparison of bovine alpha(V) integrin utilization by type A and O viruses. J. Virol. 77:2500–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, Blaas D. 1994. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. U. S. A. 91:1839–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323 [DOI] [PubMed] [Google Scholar]
- 43. Tomko RP, Xu R, Philipson L. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. U. S. A. 94:3352–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. 1995. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 69:3873–3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shafren DR, Williams DT, Barry RD. 1997. A decay-accelerating factor-binding strain of coxsackievirus B3 requires the coxsackievirus-adenovirus receptor protein to mediate lytic infection of rhabdomyosarcoma cells. J. Virol. 71:9844–9848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Milstone AM, Petrella J, Sanchez MD, Mahmud M, Whitbeck JC, Bergelson JM. 2005. Interaction with coxsackievirus and adenovirus receptor, but not with decay-accelerating factor (DAF), induces A-particle formation in a DAF-binding coxsackievirus B3 isolate. J. Virol. 79:655–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131 [DOI] [PubMed] [Google Scholar]
- 48. Arita M, Koike S, Aoki J, Horie H, Nomoto A. 1998. Interaction of poliovirus with its purified receptor and conformational alteration in the virion. J. Virol. 72:3578–3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fricks CE, Hogle JM. 1990. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prchla E, Kuechler E, Blaas D, Fuchs R. 1994. Uncoating of human rhinovirus serotype 2 from late endosomes. J. Virol. 68:3713–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Curry S, Abrams CC, Fry E, Crowther JC, Belsham GJ, Stuart DI, King AM. 1995. Viral RNA modulates the acid sensitivity of foot-and-mouth disease virus capsids. J. Virol. 69:430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tuthill TJ, Harlos K, Walter TS, Knowles NJ, Groppelli E, Rowlands DJ, Stuart DI, Fry EE. 2009. Equine rhinitis A virus and its low pH empty particle: clues towards an aphthovirus entry mechanism? PLoS Pathog. 5:e1000620 doi:10.1371/journal.ppat.1000620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nurani G, Lindqvist B, Casasnovas JM. 2003. Receptor priming of major group human rhinoviruses for uncoating and entry at mild low-pH environments. J. Virol. 77:11985–11991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nagata N, Shimizu H, Ami Y, Tano Y, Harashima A, Suzaki Y, Sato Y, Miyamura T, Sata T, Iwasaki T. 2002. Pyramidal and extrapyramidal involvement in experimental infection of cynomolgus monkeys with enterovirus 71. J. Med. Virol. 67:207–216 [DOI] [PubMed] [Google Scholar]
- 55. Shiroki K, Kato H, Koike S, Odaka T, Nomoto A. 1993. Temperature-sensitive mouse cell factors for strand-specific initiation of poliovirus RNA synthesis. J. Virol. 67:3989–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hatakeyama S, Sakai-Tagawa Y, Kiso M, Goto H, Kawakami C, Mitamura K, Sugaya N, Suzuki Y, Kawaoka Y. 2005. Enhanced expression of an alpha2,6-linked sialic acid on MDCK cells improves isolation of human influenza viruses and evaluation of their sensitivity to a neuraminidase inhibitor. J. Clin. Microbiol. 43:4139–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamayoshi S, Noda T, Ebihara H, Goto H, Morikawa Y, Lukashevich IS, Neumann G, Feldmann H, Kawaoka Y. 2008. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe 3:168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. 27:493–499 [Google Scholar]
- 59. Arita M, Takebe Y, Wakita T, Shimizu H. 2010. A bifunctional anti-enterovirus compound that inhibits replication and the early stage of enterovirus 71 infection. J. Gen. Virol. 91:2734–2744 [DOI] [PubMed] [Google Scholar]
- 60. Dierssen U, Rehren F, Henke-Gendo C, Harste G, Heim A. 2008. Rapid routine detection of enterovirus RNA in cerebrospinal fluid by a one-step real-time RT-PCR assay. J. Clin. Virol. 42:58–64 [DOI] [PubMed] [Google Scholar]
- 61. Kaplan G, Freistadt MS, Racaniello VR. 1990. Neutralization of poliovirus by cell receptors expressed in insect cells. J. Virol. 64:4697–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu CC, Guo MS, Lin FH, Hsiao KN, Chang KH, Chou AH, Wang YC, Chen YC, Yang CS, Chong PC. 2011. Purification and characterization of enterovirus 71 viral particles produced from Vero cells grown in a serum-free microcarrier bioreactor system. PLoS One 6:e20005 doi:10.1371/journal.pone.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miyamura K, Nishimura Y, Abo M, Wakita T, Shimizu H. 2011. Adaptive mutations in the genomes of enterovirus 71 strains following infection of mouse cells expressing human P-selectin glycoprotein ligand-1. J. Gen. Virol. 92:287–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garriga D, Pickl-Herk A, Luque D, Wruss J, Caston JR, Blaas D, Verdaguer N. 2012. Insights into minor group rhinovirus uncoating: the X-ray structure of the HRV2 empty capsid. PLoS Pathog. 8:e1002473 doi:10.1371/journal.ppat.1002473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bostina M, Levy H, Filman DJ, Hogle JM. 2011. Poliovirus RNA is released from the capsid near a twofold symmetry axis. J. Virol. 85:776–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lin TY, Hsia SH, Huang YC, Wu CT, Chang LY. 2003. Proinflammatory cytokine reactions in enterovirus 71 infections of the central nervous system. Clin. Infect. Dis. 36:269–274 [DOI] [PubMed] [Google Scholar]
- 67. Wang SM, Lei HY, Huang KJ, Wu JM, Wang JR, Yu CK, Su IJ, Liu CC. 2003. Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J. Infect. Dis. 188:564–570 [DOI] [PubMed] [Google Scholar]