

Abstract
Human cytomegalovirus (HCMV) infects a variety of cell types in humans, resulting in a varied pathogenesis in the immunocompromised host. Endothelial cells (ECs) are considered an important target of HCMV infection that may contribute to viral pathogenesis. Although the viral determinants important for entry into ECs are well defined, the molecular determinants regulating postentry tropism in ECs are not known. We previously identified the UL133-UL138 locus encoded within the clinical strain-specific ULb′ region of the HCMV genome as important for the latent infection in CD34+ hematopoietic progenitor cells (HPCs). Interestingly, this locus, while dispensable for replication in fibroblasts, was required for efficient replication in ECs infected with the TB40E or fusion-inducing factor X (FIX) HCMV strains. ECs infected with a virus lacking the entire locus (UL133-UL138NULL virus) complete the immediate-early and early phases of infection but are defective for infectious progeny virus production. ECs infected with UL133-UL138NULL virus exhibited striking differences in the organization of intracellular membranes and in the assembly of mature virions relative to ECs infected with wild-type (WT) virus. In UL133-UL138NULL virus-infected ECs, Golgi stacks were disrupted, and the viral assembly compartment characteristic of HCMV infection failed to form. Further, progeny virions in UL133-UL138NULL virus-infected ECs inefficiently acquired the virion tegument and secondary envelope. These defects were specific to infection in ECs and not observed in fibroblasts infected with UL133-UL138NULL virus, suggesting an EC-specific requirement for the UL133-UL138 locus for late stages of replication. To our knowledge, the UL133-UL138 locus represents the first cell-type-dependent, postentry tropism determinant required for viral maturation.
INTRODUCTION
Cytomegaloviruses (CMV) are large, complex DNA viruses belonging to the Herpesviridae family. Human CMV (HCMV) is one of eight human herpesviruses and is ubiquitous among the population worldwide. HCMV entrenches a lifelong persistence in its host through both chronic and latent states of infection (1–3). Subclinical reactivation and low-level virus shedding events likely occur intermittently throughout the lifetime of the immunocompetent host (1). However, in individuals with compromised T cell immunity, including transplant patients, AIDS patients, and cancer patients undergoing intensive chemotherapy regimens, HCMV reactivation from latent to productive states of infection is a common cause of morbidity and mortality (4, 5). Further, HCMV persistence has been associated with increased risk of age-related pathologies including atherosclerosis, immune senescence, and frailty (6–11). The molecular mechanisms governing HCMV persistence in the infected host are largely unknown, in part, because the viral determinants and cellular reservoirs of persistence are ill defined.
HCMV infects a diverse array of cell types in humans (5, 12, 13). Among the cells infected, vascular endothelial cells (ECs) are thought to be important for viral pathogenesis in the host. By nature of the EC boundary formed between the circulation and organ tissue, ECs serve as an important interface for bidirectional virus spread (1, 14). While ECs are considered a key target of CMV infection in humans (12) and mice (15), ECs have different susceptibilities and support distinct modes of infection in vitro depending on their anatomical source (i.e., lung, dermis, or intestine) and type (micro- versus macrovascular) (16–20). This cell-type- or cell origin-dependent gradation in viral replication suggests that EC-specific determinants control infection outcomes ranging from lytic replication to “smoldering persistence” and possibly latency (17, 21). Infected ECs contribute to virus dissemination and pathogenesis, in part through the infection of circulating leukocytes or polymorphonuclear cells (1, 22–26) and by promoting monocyte recruitment and extravasation, possibly increasing vascular permeability for dissemination of infected circulating cells into tissues (27). Further, infected ECs may detach and circulate (28, 29). These modes of cell-cell-based dissemination likely define the contribution of ECs to viral spread in the host (30). Based on studies using human, murine (MCMV), or rat CMV, infection of ECs is associated with persistent proinflammatory signaling contributing to angiogenesis and vascular diseases including atherosclerosis and atherothrombosis (7, 31–38). Accordingly, HCMV is a risk factor for restenosis following angioplasty and for transplant vascular sclerosis and chronic graft rejection following solid organ allograft transplantation (39–42). Understanding the biology of HCMV infection in ECs is critical given the pivotal role of ECs in HCMV infection and pathogenesis.
As EC-specific determinants impact HCMV replication, HCMV strains also exhibit a gradation in their ability to enter and replicate in ECs (43, 44). Well-characterized viral determinants of endothelial tropism predominantly function in viral entry into ECs. Proteins encoded by UL128, UL130, and UL131A complex with glycoproteins H (gH) and L (gL) on the virion surface to mediate entry into endothelial cells, leukocytes, and dendritic cells by endocytosis (23, 45–50). This complex is not required for entry into fibroblasts; rather, HCMV enters fibroblasts by fusion mediated by the gH/gL/gO complex (51–53). HCMV strains passaged in fibroblasts accumulate mutations in the UL128-UL131A locus, resulting in lost tropism for endothelial and epithelial cells (23, 47, 49, 50, 54) and reduced transfer of virus to leukocytes (23). However, laboratory strains competent for entry into ECs (e.g., encoding wild-type [WT] or repaired UL131A) still replicate to reduced yields relative to several clinical strains (23, 49). Further, various clinical isolates of HCMV replicate with different efficiencies in ECs (19, 44, 55, 56), suggesting the existence of viral determinants specific to clinical strains that control postentry EC tropism. Few CMV-coded determinants regulating postentry EC tropism have been defined. Studies in MCMV revealed a requirement for the M45 gene to inhibit apoptosis for productive replication specifically in ECs and macrophages (57). Recently, the US16 HCMV gene was reported to be required for delivery of viral genomes and tegument proteins to the nuclei of ECs but not fibroblasts (58). Viruses containing disruptions in US16 are defective at the immediate-early (IE) stage and all subsequent stages of infection in ECs. These studies support the notion that CMV has evolved functions to specifically overcome tropism barriers to infection in unique cell types and emphasize the importance of the cellular environment in ultimately determining the success of infection.
In the present study, we investigated the requirement for the ULb′ region-encoded UL133-UL138 locus for replication in primary human lung ECs. This locus encodes four proteins, pUL133, pUL135, pUL136, and pUL138, from polycistronic transcripts (59, 60). Like the entire ULb′ region, the UL133-UL138 locus is dispensable for viral replication in fibroblasts (60). However, the locus functions to suppress virus replication in CD34+ hematopoietic progenitor cells (HPCs), contributing to the latent phenotype in vitro (60, 61). Interestingly, this locus is required for efficient viral replication in multiple types of primary human ECs (60). The defect observed in ECs was independent of the multiplicity of infection (MOI) and the UL138 latency determinant. Here, we characterize this defect and determine that the UL133-UL138 locus is required for the late stages of infection and virus maturation specifically in ECs. ECs infected with a virus lacking the UL133-UL138 locus (UL133-UL138NULL) had dramatically altered organization of intracellular membranes and failed to form the viral assembly compartment characteristic of HCMV infection. These alterations in intracellular membranes correlated with a failure to produce mature progeny virions with tegument and secondary envelope. The dissimilarities between WT and UL133-UL138NULL virus infections were unique to ECs. These studies indicate that one or more proteins encoded within the UL133-UL138 locus contribute to the reorganization of intracellular membranes and, consequently, to secondary envelopment and egress in ECs. To our knowledge, these studies point to the first EC-specific viral determinants required for virus maturation.
MATERIALS AND METHODS
Cells.
Human embryonic lung fibroblasts (MRC-5) (purchased from ATCC; Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin. Primary human microvascular lung ECs (HMVEC) (purchased from Lonza, Walkersville, MD) were cultured in EGM-2 MV BulletKit medium (Microvasular Endothelial Cell Growth Medium-2; Lonza) with 5% FBS. All cells were cultured at 37°C in 5% CO2.
Viruses.
Bacterial artificial chromosome (BAC) clones of the HCMV TB40E or fusion-inducing factor X (FIX) parental (WT) (13) strain and the UL133-UL138NULL variant express the green fluorescent protein (GFP) as a marker of infection (60). The construction of the UL133-UL138NULL variant was described previously (60, 61). Virus stocks were propagated by transfecting the viral BAC genome into MRC-5 fibroblasts and stored as described previously (61). Infectious virus yields were determined by 50% tissue culture infective dose (TCID50) on MRC-5 fibroblasts. Centrifugal enhancement was used to increase the efficiency of FIX infection in ECs. For viral growth curves, virus yields were measured by TCID50 from cell lysates harvested over a time course.
Virus entry.
HMVEC were plated in 12-well dishes and infected the following day at an MOI of 0.2. The infection was enhanced by centrifuging plates at 250 × g for 20 min immediately following infection. To remove unabsorbed virus, infection medium was replaced with fresh medium at 6 h postinfection (hpi). Cells were collected at 48 h postinfection and stained with propidium iodide. GFP was measured in live cells as an indicator of infection using an LSRII instrument (BD Biosciences, San Jose, CA).
Immunoblotting.
Immunoblotting was performed as described previously (61). Briefly, 15 to 20 μg of protein lysates was separated on 12% bis-Tris gels by electrophoresis and transferred to 0.45-μm-pore-size polyvinylidene difluoride (Immobilon-FL; Millipore, MA) membranes. The proteins were immunoblotted using mouse monoclonal or rabbit polyclonal antibodies directed against each protein and detected using fluorescently conjugated secondary antibodies and the Odyssey infrared imaging system (Li-Cor, NE). All antibodies used are listed in Table 1.
Table 1.
Primary antibodies used for immunofluorescence and immunoblotting
| Antigen | Antibody | Typea | Source | Dilution used for: |
|
|---|---|---|---|---|---|
| Immunofluorescenceb,d | Immunoblottingc,d | ||||
| EEA-1 | 14/EEA1 | M | BD Transduction | 1:250 | ND |
| UL44 | 10D8 | M | Virusys | ND | 1:2,500 |
| Golgi marker proteins | |||||
| giantin | Custom | M | Gifte (Abcam) | 1:250 | ND |
| GM130 | Clone 35 | M | BD Transduction | 1:100 | ND |
| EP892Y | R | Abcam | 1:250 | ND | |
| GS27 | 25 | M | BD Transduction | 1:1,100 | ND |
| β-Actin | ACTN05(C4) | R | Abcam | ND | 1:1000 |
| pp28 | 10B4-29 | M | Giftf | 1:20 | 1:50 |
| IE1/IE2 | 3H4 | M | Giftg | ND | 1:100 |
| gB | 27-156 | M | Gifth | 1:30 | ND |
| CD63 | SC-5275 | M | Santa Cruz | 1:250 | ND |
R, rabbit; M, mouse monoclonal.
Dilution in PBS supplemented with bovine serum albumin and Tween 20.
Dilution in TBS–5% milk supplemented with bovine serum albumin and Tween 20.
ND, not done.
Generous gift from Samuel Campos, University of Arizona.
Generous gift from Michael Nevels, University of Regensburg.
Generous gift from Tom Shenk, Princeton University.
Generous gift from Bill Britt, University of Alabama at Birmingham.
Transmission electron microscopy.
HMVEC or MRC-5 cells were mock infected or infected at an MOI of 4 or 2, respectively, with centrifugal enhancement. Infection medium was replaced at 24 h postinfection for HMVEC and at 6 h postinfection for MRC-5 cells with normal growth medium for each respective cell type. All cells were harvested at 5 days postinfection (dpi). Cells were fixed in 2.5% glutaraldehyde and 0.1 M PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] for 20 min. The fixed cell pellet was postfixed with osmium tetroxide in 0.1 M PIPES and dehydrated in a graded series of alcohol. Pellets were infiltrated with resin and cut into 100-nm sections. The sections were floated onto copper grids and imaged using a Phillips CM-12s transmission electron microscope. Cells were embedded and sectioned by the Arizona Research Laboratories, Arizona Health Sciences Center Core Facility.
Indirect immunofluorescence.
Indirect immunofluorescence was performed as described previously (39). Briefly, HMVEC and MRC-5 cells were seeded onto 12-mm glass coverslips in 24-well dishes 1 day prior to infection at an MOI of 2. Cells were processed for indirect immunofluorescence at 144 hpi for HMVEC and at 96 hpi for MRC-5 cells. Cells were fixed in 2% formaldehyde in phosphate-buffered saline (PBS) and permeabilized. Primary and secondary antibodies (Table 1) were incubated in PBS-Tween 20 (PBS-T) for 1 h. Poststaining, DNA was stained with 4′,6′-diamidino-2-phenylindole (DAPI) according to the manufacturer's instructions (Molecular Probes). Coverslips were mounted using Aqua Polymount according to the manufacturer's instructions (Polysciences, Inc.). Cells were imaged using a Zeiss 510 Meta Confocal Microscope as a lambda stack and unmixed using the Zeiss 510 Meta software, version 4.2, and images were processed using Zeiss software.
RESULTS
UL133-UL138 is required for efficient replication postentry in endothelial cells.
We previously determined that the ULb′-coded UL133-UL138 locus is required for efficient replication in primary human ECs such that a virus lacking this locus (UL133-UL138NULL virus) exhibited a replication defect in a variety of ECs relative to wild-type (WT) virus (60). UL133-UL138NULL virus infection results in a 5- to 1,000-fold defect in virus yields relative to the WT infection in five distinct primary EC types, including both microvascular and macrovascular cells. This defect is specific to ECs as the UL133-UL138 locus is dispensable for replication in human fibroblasts and epithelial cells (60). To further explore this phenotype, primary human microvascular ECs (HMVEC) of the lung were infected at a low MOI (0.05 TCID50 per cell), and virus yields were measured over a time course (Fig. 1A). UL133-UL138NULL virus infection resulted in an overall 1,000-fold defect relative to the WT. A similar defect was observed in HMVEC infected with the FIX strain of HCMV (Fig. 1B). This replication defect, taken together with our previous work, indicates a strong dependence on the UL133-UL138 locus in multiple endothelial cell types.
Fig 1.
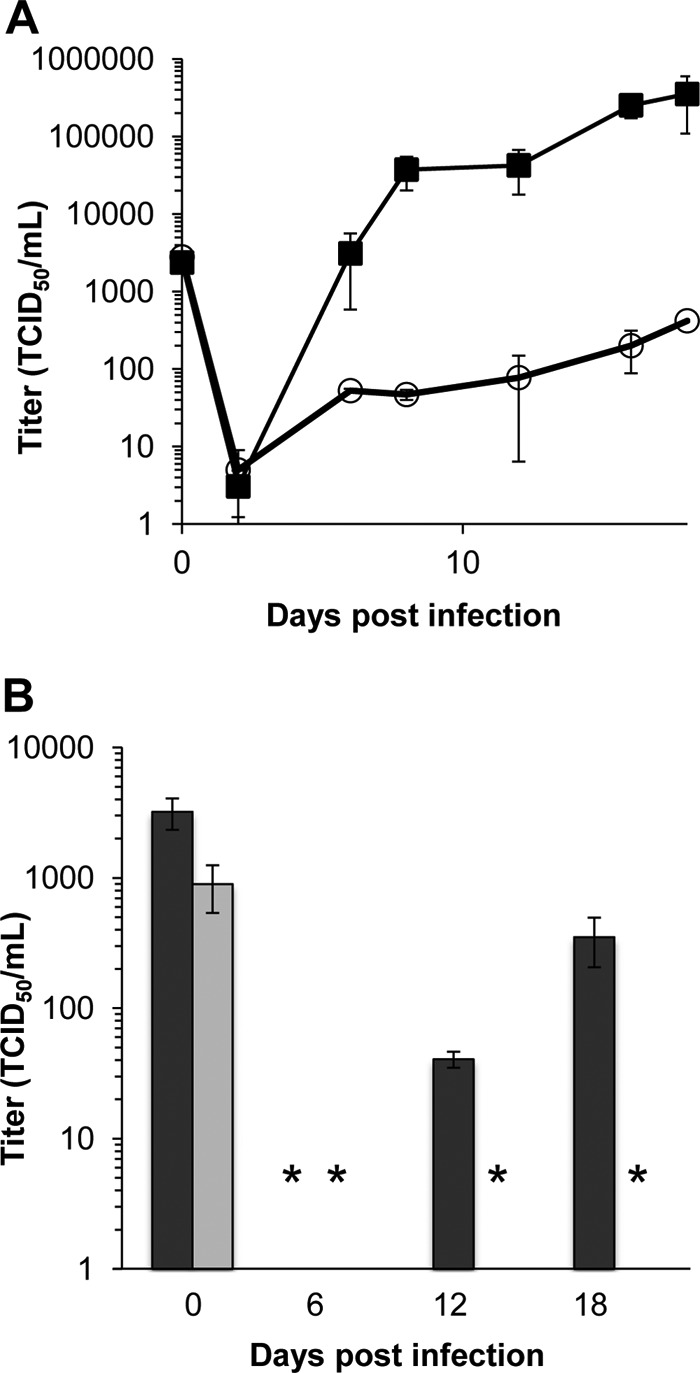
UL133-UL138 is required for viral replication in ECs. HMVEC were infected with WT or UL133-UL138NULL virus of either the TB40E (A) or FIX (B) strain at an MOI of 0.05, and virus yields were measured over a time course by TCID50 on fibroblasts. WT is represented by the filled squares (A) or dark bars (B). UL133-UL138NULL virus is represented by the open circles (A) or light bars (B). The values plotted are averages from three independent experiments, and the standard deviations are indicated by the error bars for each time point. In some cases, the error bars are too small to be seen. In panel B, the asterisks indicate values at the limit of detection.
Most known restrictions to HCMV tropism in ECs occur early in the infection process, residing at the level of viral entry (62) or delivery of the viral genome to the nucleus (58). As such, one plausible explanation for the observed defect in replication may be that a protein encoded in the UL133-UL138 locus is required for viral entry into ECs, similar to the UL128-UL131A locus. As both the WT and UL133-UL138NULL viruses used in this work have been engineered to express the green fluorescent protein (GFP) as a marker for infection, the expression of GFP can be used as an indicator of both entry and delivery of the viral genome to the nucleus. We measured the proportion of GFP-positive (GFP+) HMVEC infected with either the WT (26.6%) or UL133-UL138NULL (26.9%) virus at 48 hpi, a time preceding secondary rounds of infection (Fig. 2A). Analysis of the mean fluorescence intensities of the infected populations in four experiments indicates that the WT and UL133-UL138NULL viruses enter, deliver the genome to the nucleus, and express GFP similarly in HMVEC (Fig. 2B). Based on this result, the EC-specific replication defect exhibited by the UL133-UL138NULL virus cannot be attributed to a defect in viral entry or trafficking to the nucleus.
Fig 2.
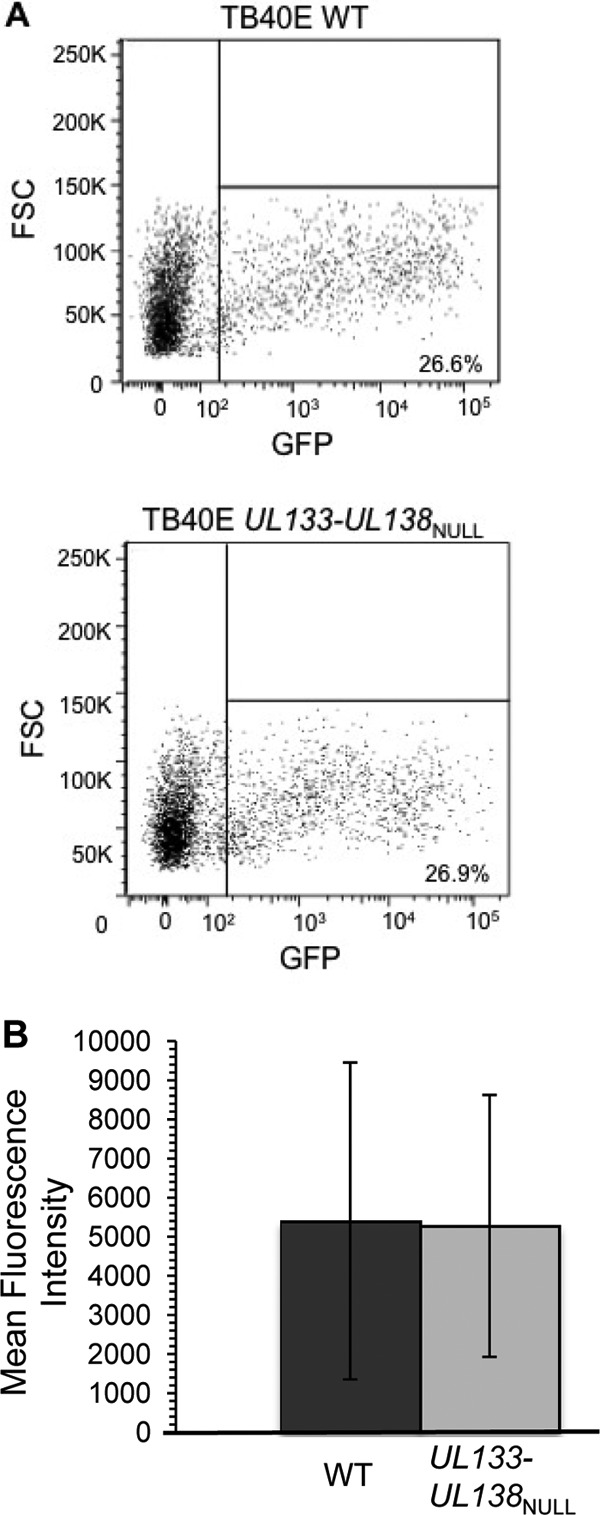
UL133-UL138 is not required for virus entry or genome transit to the nucleus in ECs. HMVEC were infected with TB40E-WT or -UL133-UL138NULL virus at an MOI of 0.02. Infected (GFP+) cells were analyzed at 48 hpi by flow cytometry. (A) Representative dot plots with the percentage of infected cells in each infection indicated. (B) The average mean fluorescence intensities for four independent experiments for each infection are shown, and the standard deviations are indicated by the error bars. FSC, forward scatter.
The UL133-UL138 locus is required for maximal late viral gene expression.
We next compared the accumulation of representative immediate-early (IE), early, and late proteins in HMVEC infected with TB40E-WT or -UL133-UL138NULL virus to define the postentry point at which viral replication is constrained in ECs. HMVEC were infected at an MOI of 2 and the IE1 72-kDa and IE2 86-kDa proteins (IE proteins), pUL44 (early protein; viral DNA polymerase processivity factor), and pp28 (late protein; prominent constituent of the tegument) were analyzed over a time course by immunoblotting (Fig. 3). IE1 and pp28 bands on immunoblots from four to six experiments were quantitated to compare the differences in protein accumulation (Fig. 3B and C). IE1 (Fig. 3B) and UL44 (data not shown) accumulated to WT levels in UL133-UL138NULL virus-infected HMVEC. pp28 levels were reduced 1.4- to 2-fold in the UL133-UL138NULL virus infection relative to levels in WT infection. However, these differences were not statistically significant (P ≥ 0.1) (Fig. 3C). These results indicate that the defect in UL133-UL138NULL virus replication is a late-phase defect.
Fig 3.
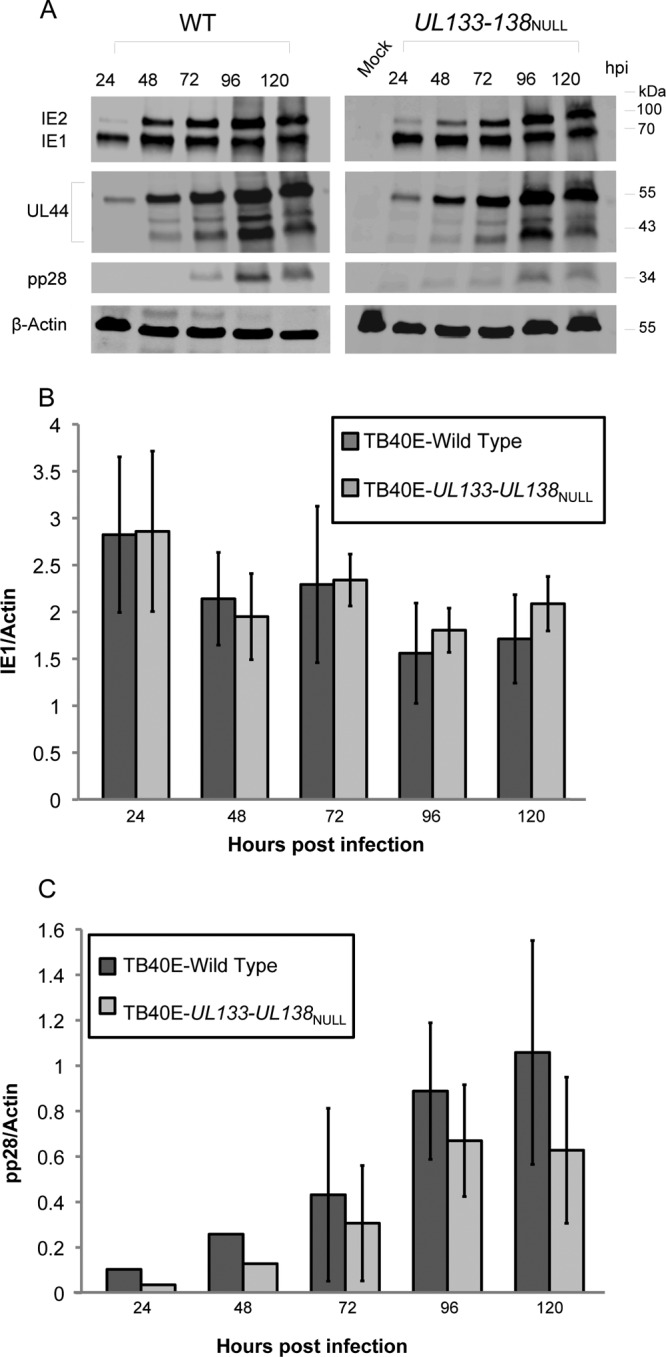
Accumulation of IE, early, and late proteins in infected HMVEC. HMVEC were infected with TB40E-WT or -UL133-UL138NULL virus at an MOI of 2. (A) Protein lysates harvested over a time course were analyzed by immunoblotting using antibodies specific to the IE1 and IE2 proteins, the UL44 early protein, and the pp28 late protein. β-Actin serves as a loading control. Quantification of average IE1 (B) and pp28 (C) protein levels (normalized to actin) in at least four independent experiments is shown. The standard deviation is indicated. Student's t test indicates that the differences between WT and UL133-UL138NULL virus at each of the time points for IE1 (P ≥ 0.2) and pp28 (P ≥ 0.1) are not significant.
UL133-UL138 locus is required for production of mature virions in ECs.
To further characterize the late-phase defect associated with the production of infectious progeny in UL133-UL138NULL virus infection (Fig. 1), we evaluated the formation of mature virions in infected ECs by transmission electron microscopy. HMVEC were infected with TB40E-WT or -UL133-UL138NULL virus at an MOI of 4 and processed for electron microscopy at 5 dpi, a time when infectious virus is produced (Fig. 1) (60). The accumulation of viral nucleocapsids appeared normal in mutant-infected HMVEC relative to the WT infection (Fig. 4A and B); both empty and genome-containing nucleocapsids were apparent with similar frequencies in each infection. Furthermore, there were no significant differences apparent between WT and mutant infections in the organization of the nucleus or viral replication centers. In stark contrast, significant differences between the WT and UL133-UL138NULL viruses emerged in the cytoplasm. The most noteworthy differences exist in (i) the integrity of the Golgi complex and cytoplasmic membranes (Fig. 4C and D), (ii) the appearance of multivesicular bodies (MVBs) (Fig. 4E and F), and (iii) the presence of mature cytoplasmic virions (Fig. 4G and H).
Fig 4.


UL133-UL138 is required for maintaining cytoplasmic membrane organization and for virion maturation. HMVEC were infected with WT or UL133-UL138NULL virus at an MOI of 4. Cells were fixed, embedded, and sectioned for transmission electron microscopy at 5 dpi. Representative micrographs are shown to illustrate the accumulation of nucleocapsids (A and B), the organization of cytoplasmic membranes (C and D), the formation of MVBs (E and F), and the maturation of cytoplasmic virions (G and H). N and C indicate nucleus and cytoplasm, respectively; DB indicates dense bodies. Open arrowheads, nucleocapsids (A and B); filled arrowheads, Golgi stacks (C) or vesicles (D); filled arrows, MVBs (E and F); open arrows, cytoplasmic virus particles (G and H). Scale bars, 2 μm (A to D) and 0.5 μm (E to H).
Golgi stacks were readily apparent in the cytoplasm of every WT virus-infected cell examined (Fig. 4C). In addition, large numbers of membrane-bound, tegument-filled dense bodies (DBs) were observed in the cytoplasm of WT virus-infected cells. In comparison, the cytoplasm of HMVEC infected with UL133-UL138NULL virus contained large electron-lucent vesicles and no apparent Golgi stacks (Fig. 4D). Further, DBs were far less abundant. These data suggest that the UL133-UL138 locus may be important for maintaining intracellular membrane organization and the formation of DBs during infection in ECs.
MVBs are late endosomes that play important roles in modulation of cell signaling, lipid and membrane protein turnover, antigen presentation, and the egress of some viruses (63). MVBs were induced by both WT and UL133-UL138NULL virus infection (Fig. 4E and F); MVBs were rarely observed in uninfected HMVEC (data not shown). The contents of the MVBs differed qualitatively between the WT and UL133-UL138NULL virus infections. In the WT infection, MVBs often contained mature virions (tegumented and enveloped), whereas this was not the case in the mutant infection (Fig. 4E). In WT infection, mature virions were present inside MVBs, while immature virions were present at the periphery of MVBs, suggesting that MVBs may play a role in the final steps of virion maturation in these cells. Further, the vast majority of membrane vesicles inside MVBs present in the WT infection were flattened or compressed. These compressed vesicles were not observed in UL133-UL138NULL virus-infected HMVEC (Fig. 4F), and the vesicles present retained a shape typically observed in MVBs.
In addition to the mature virions present in MVBs (Fig. 4E), WT infection in HMVEC produced individual enveloped, mature virions in the cytoplasm, and extracellular virions were present (Fig. 4G). In contrast, mature virions were far less abundant in the UL133-UL138NULL virus infection (Fig. 4H), reflecting diminished viral yields (Fig. 1). Instead, untegumented and unenveloped particles were commonly observed and often clustered around spherical structures in the cytoplasm. Further, these virions had a “cracked” appearance. From our electron microscopy, we conclude that UL133-UL138NULL virus infection is defective for processes in tegumentation of virus particles and secondary envelopment.
The UL133-UL138 locus is not required for virus maturation in fibroblasts.
Our result in ECs prompted analysis of TB40E UL133-UL138NULL virus-infected fibroblasts to determine if there was any alteration in mature virion formation in these cells. In stark contrast to infection in ECs, fibroblasts infected with UL133-UL138NULL virus produce WT yields of virus, suggesting that there is no defect for virus replication in the absence of the UL133-UL138 locus (60). We analyzed fibroblasts infected with WT or UL133-UL138NULL virus at an MOI of 2 at 5 dpi for the presence of mature virions by electron microscopy (Fig. 5). The cytoplasm of fibroblasts infected with UL133-UL138NULL virus contained large numbers of mature virions with both tegument and envelope similar to those of WT infection in fibroblasts (Fig. 5A and B). In addition, Golgi stacks were readily apparent in both infections, and the overall architecture of the cytoplasm did not vary. These data highlight the unique EC-specific defect associated with UL133-UL138NULL virus replication.
Fig 5.
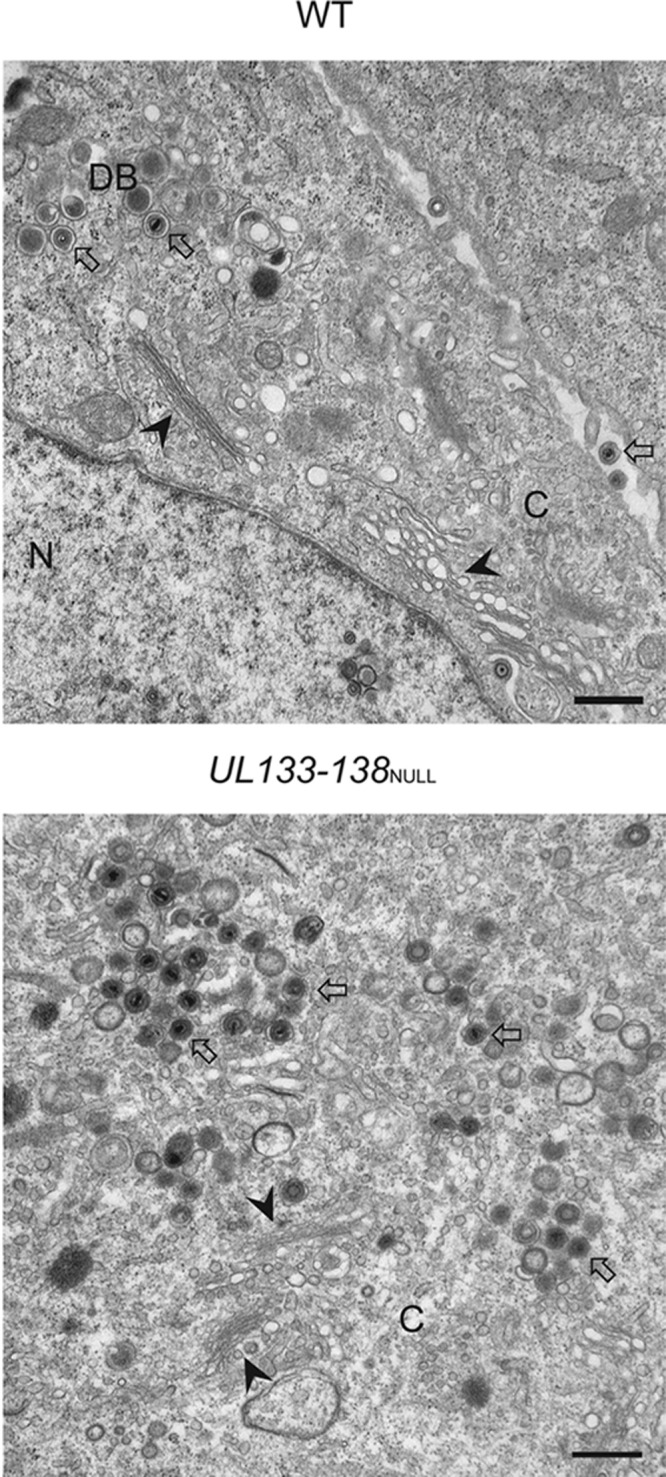
UL133-UL138 is dispensable for mature virion formation in infected fibroblasts. MRC-5 fibroblasts were infected with WT (top panel) or UL133-UL138NULL (bottom panel) virus at an MOI of 2. At 5 dpi, cells were fixed, embedded, and sectioned for transmission electron microscopy. Representative micrographs are shown to illustrate the presence of Golgi stacks and mature virions in each infection. N and C indicate nucleus and cytoplasm, respectively; DB indicates dense bodies. Filled arrowheads, Golgi stacks; open arrows, virions. Scale bar, 0.5 μm.
The assembly compartment is poorly formed in ECs infected with TB40E-UL133-UL138NULL virus.
In fibroblasts, HCMV infection results in a dramatic reorganization of cellular cytoplasmic membranes to form a juxtanuclear compartment commonly referred to as the assembly compartment (AC) (1, 64–70). Due to the altered morphology of the Golgi compartment and the lack of mature virions in ECs infected with UL133-UL138NULL virus, we examined the formation of the AC. ECs were infected with TB40E-WT or -UL133-UL138NULL virus at an MOI of 2. We examined the distribution of the late viral protein, pp28, and the cis-medial Golgi marker, GM130, in infected ECs by indirect immunofluorescence at 144 hpi (Fig. 6A and B). pp28 is known to localize to the AC and is an abundant tegument protein in the mature virion. GM130, marking Golgi membranes, is dramatically reorganized to the peripheral zone of the AC (66, 67, 69). In both WT and UL133-UL138NULL virus infections, the characteristic Golgi stack-staining of GM130 was disrupted. In WT infection of HMVEC, the characteristic ring-like structure stained by GM130 indicated the formation of ACs, similar to our results described in fibroblasts (66, 67). In the WT infection, pp28 exhibited remarkable overlap in its staining with GM130, characteristic of the formation of late-phase ACs. In contrast, GM130 and pp28 staining was dispersed throughout the cytoplasm of ECs infected with UL133-UL138NULL virus. While there was some overlap in GM130 and pp28 staining in UL133-UL138NULL virus-infected HMVEC, colocalization was reduced compared to that of the WT infection. Low-magnification images illustrate the range of differences in staining observed in a field of ECs infected with either the WT or UL133-UL138NULL virus (Fig. 6B). In the analysis of pp28 staining in more than 100 cells, a focal AC was observed in 85% of WT-infected ECs but in only 34% of UL133-UL138NULL virus-infected ACs. These results indicate that ACs form inefficiently in ECs infected with UL133-UL138NULL virus.
Fig 6.

UL133-UL138NULL virus-infected ECs fail to form assembly compartments. HMVEC were infected with WT or UL133-UL138NULL virus at an MOI of 2 or mock infected. At 144 h postinfection, cells were processed for indirect immunofluorescence using antibodies specific to the GM130 Golgi marker or pp28 viral protein. The nuclei are indicated by DAPI staining. Localization was visualized by confocal microscopy using a Zeiss 510 Meta Confocal microscope. Panel B contains low-magnification fields of cells to demonstrate the range of AC morphology in each infection. Magnification, ×60 (A) and ×40 (B).
To further investigate the role of the UL133-UL138 locus in the formation of ACs, we analyzed cellular Golgi markers (giantin and GS27), the early endosomal marker (EEA1), the MVB marker (CD63), viral pp28, and viral glycoprotein B (gB) in both MRC-5 fibroblasts (Fig. 7) and HMVEC (Fig. 8). Each of these protein markers localizes to the AC in WT infection in fibroblasts (66). MRC-5 cells or HMVEC infected with TB40E-WT or -UL133-UL138NULL virus at an MOI of 2 were processed for indirect immunofluorescence at 96 hpi in fibroblasts (Fig. 7) and at 144 hpi in HMVEC (Fig. 8). ECs were processed at a later time postinfection due to slower kinetics of infection in these cells. Infected cells were identified by GFP fluorescence (data not shown). gB and pp28 staining was evaluated as a marker of ACs.
Fig 7.

Assembly compartment formation in fibroblasts is similar for WT and UL133-UL138NULL virus infection. MRC-5 fibroblasts were infected with WT or UL133-UL138NULL virus at an MOI of 2 or mock infected. At 96 hpi, cells were processed for indirect immunofluorescence using antibodies specific to giantin (A), GS27 (B), EEA1 (C), CD63 (D), pp28 (E), or gB (F). Infection of the cells imaged was determined by GFP fluorescence (data not shown). The nuclei are indicated by DAPI staining. Localization was visualized by confocal microscopy using a Zeiss 510 Meta Confocal microscope. Magnification, ×60.
Fig 8.

Analysis of cellular membrane organization and viral assembly compartment markers in WT and UL133-UL138NULL virus infection in ECs. HMVEC were infected with WT or UL133-UL138NULL virus at an MOI of 2 or mock infected. At 144 hpi, cells were processed for indirect immunofluorescence using antibodies specific to giantin (A), GS27 (B), EEA1 (C), CD63 (D), pp28 (E), or gB (F). Infection of the cells imaged was determined by GFP fluorescence (data not shown). The nuclei are indicated by DAPI staining. Localization was visualized by confocal microscopy using a Zeiss 510 Meta Confocal microscope. Magnification, ×60.
In both WT and UL133-UL138NULL virus-infected fibroblasts, giantin (Fig. 7A), GS27 (Fig. 7B), EEA1 (Fig. 7C), and CD63 (Fig. 7D) were reorganized by infection to virus-induced ACs as previously described (66, 67). The characteristic ring-like structure of the AC was apparent for each of the markers staining cellular cytoplasmic structures. The localization of pp28 and gB to assembly compartments in UL133-UL138NULL virus-infected fibroblasts was similar to that of the WT infection although in some cases the staining for these proteins was slightly more diffuse relative to that in the WT infection. These results suggest that the formation of ACs in fibroblasts does not require the UL133-UL138 locus.
Similar to WT infection in fibroblasts, cytoplasmic membrane markers were reorganized to form ACs in WT-infected ECs (Fig. 8A to D). Further, pp28 and gB staining was indicative of ACs in the WT infection (Fig. 8E and F). Consistent with the data shown in Fig. 6, HMVEC infected with UL133-UL138NULL virus displayed poorly formed ACs, and all cellular membrane markers were more diffusely localized (Fig. 8A to D) relative to WT infection in fibroblasts. This diffuse distribution of membrane markers is consistent with the loss of Golgi stacks and increased numbers of large cytoplasmic vesicles observed by electron microscopy (Fig. 4). Therefore, our data indicate a role for the UL133-UL138 proteins in efficient formation of the AC for virus assembly.
DISCUSSION
The endothelium represents an important target for HCMV infection as it contributes to viral dissemination and pathogenesis. HCMV determinants important for postentry endothelial tropism have remained elusive and represent an important point of control. Our work reveals a novel EC-specific function of the UL133-UL138 locus encoded within the ULb′ region of the HCMV genome in maintaining intracellular membrane organization and in the formation of the AC for mature virus production. These results identify the first virus-coded determinants important for late stages of viral infection in ECs. The EC-specific function of the UL133-UL138 locus was revealed by a specific defect in UL133-UL138NULL virus replication in a variety of EC types but not in fibroblasts or epithelial cells (Fig. 1) (60). There was no defect in entry or in immediate-early and early gene expression and less than a 2-fold defect in late gene expression in ECs infected with UL133-UL138NULL virus relative to the wild-type infection (Fig. 2 and 3). Strikingly, virions formed during UL133-UL138NULL virus infection in ECs lacked tegument and secondary envelopes (Fig. 4). The defect in virion maturation observed in UL133-UL138NULL virus-infected cells may be secondary to a defect in intracellular membrane organization and the failure to form an AC for virus maturation (Fig. 6 and 8).
Herpesviruses acquire their tegument and a host-derived envelope during a complex process of egress involving a variety of membrane-bound cellular compartments (71–74). HCMV is unique among the herpesviruses in that it induces the formation of a juxtanuclear virus factory, or AC, comprised of secretory/trafficking membranes, including those of the Golgi complex, early endosomes, lysosomes, and MVBs (1, 64–70). Many late viral proteins that are components of the mature virion, including the viral glycoproteins (i.e., gB), pp28, and pp150, localize to the AC (75). As such, the AC is thought to facilitate the acquisition of tegument and secondary envelopes in the maturation of HCMV particles during the late, postnuclear stages of infection. While the AC has been described predominantly in fibroblasts, the conventional cell culture model for HCMV research, ACs are formed in HCMV-infected ECs (Fig. 6 and 8). Cellular secretory markers such as GM130, giantin, GS27K, EEA1, and CD63 localize to the AC in WT HCMV-infected ECs (Fig. 6 and 8) in a pattern and organization similar to those described in fibroblasts (66, 67, 69). Strikingly, the UL133-UL138 locus was required for the formation of the AC in ECs but not in fibroblasts. The cell-type-specific requirement for the UL133-UL138 locus in the formation of the AC likely reflects specific interactions between UL133-UL138 proteins and cellular factors that may function in membrane transport including, but not limited to, the endosomal sorting complex required for transport (ESCRT) machinery.
Host vesicles and membrane-bound organelles reorganized to become the AC are important for HCMV envelopment (71–73). MVBs (CD63-positive) and their possible role in constitutive or alternative pathways of virus maturation are of particular interest (76). A role for MVBs in HCMV egress is supported by the observations that MVBs were induced by infection, that they reorganized into the AC late in infection, as indicated by CD63 staining (Fig. 8) (67), and that they often contained mature virions in WT-infected ECs (Fig. 4E). Consistent with a possible role of the UL133-UL138 locus in mediating intracellular membrane organization, striking differences were observed in MVBs induced by the WT and UL133-UL138NULL virus infections in ECs. Vesicles contained within MVB were often flattened or compressed in ECs infected with WT but not UL133-UL138NULL virus (Fig. 4E and F). The observation of flattened vesicles within MVBs in WT virus-infected ECs taken together with the lack of Golgi stacks in UL133-UL138NULL virus-infected cells suggests a possible role for UL133-UL138 proteins in “stacking” membranes.
In fibroblasts, MVBs and ESCRT machinery play an important role in HCMV egress (76, 77). MVBs and ESCRT machinery are associated with a number of cellular processes that may impact outcomes of infection, including signaling, membrane and lipid turnover, and cellular proliferation and polarization (78). In addition to HCMV, MVBs and ESCRT machinery have been shown to play a role in the assembly and egress of numerous viruses, including Marburg virus (79), retroviruses (80–83), hepatitis B virus (84), herpes simplex virus 1 (HSV-1) (85–88), Epstein-Barr virus (89), and human herpesvirus 6 (77, 90). Blocking MVB formation by targeting either Vps4 or Vps24 of the ESCRT-III complex during HSV infection results in a dramatic reduction in the yield of cell-associated and cell-free virus, depending on the cell type infected (85, 86, 88). In HIV infection, the virus buds directly into MVBs in monocytic cells, whereas MVBs are recruited to the plasma membrane to become a site of budding in lymphocytic cells (80). These studies, among others, underscore the central importance of MVBs to virus envelopment and egress. Indeed, MVBs may play different roles in egress in a cell-type- and infection-dependent manner. Further work is required to determine the role of MVBs and the ESCRT machinery in HCMV egress from ECs and how the UL133-UL138 proteins may contribute to this process through EC-specific interactions.
While it is not fully understood how the HCMV virion acquires its tegument during egress, the tegument is critical for at least three important functions. These include the packaging and structural stability of nucleocapsids, mediating interactions necessary for final envelopment and egress, as well as playing a vital role in initiating the second round of infection (75). While the UL133-UL138 proteins localize to Golgi membranes (60, 61), these proteins are not thought to be packaged in progeny virions (61). However, the UL133-UL138 locus appears to contribute to the acquisition of the virus tegument. Further work is required to understand if the failure of virions to acquire tegument is a direct function of these proteins or a consequence of the failure to reorganize intracellular membranes into an AC. Viruses lacking the HCMV gene, UL71, exhibit a similar phenotype to what we have observed during infection with UL133-UL138NULL virus (76). Infection with a UL71 mutant virus alters MVB morphology, mislocalizes pp28, pp150, and gB, alters the morphology of the AC, and impairs secondary envelopment. Interestingly, the UL71 mutant virus phenotype is apparent only in fibroblasts and not ECs. Taken together with our study, these results suggest that HCMV has evolved similar but distinct mechanisms for viral egress in different cell types.
Our study defines a novel EC-specific role for the UL133-UL138 locus in mediating intracellular membrane reorganization for egressing virus to acquire both tegument and secondary envelope. Ongoing work is focused on determining how the phenotypes observed in ECs segregate between the four genes encoded within the locus. Preliminary studies indicate that at least two of the UL133-UL138 proteins are required for the EC-specific functions we have ascribed to the UL133-UL138 locus in this study (F. Bughio and F. Goodrum, unpublished results). We previously demonstrated a role for the UL133-UL138 locus, specifically pUL133 and pUL138, in promoting a latent infection in CD34+ hematopoietic progenitor cells (HPCs). It is not yet clear how these new functions of the UL133-UL138 locus in ECs may contribute to infection in HPCs. Defining the mechanisms by which the individual proteins encoded by the UL133-UL138 locus contribute to the replication in ECs and latency in CD34+ HPCs is a primary focus of our ongoing research. The relationship of these four proteins to one another and their role in viral replication and persistence will undoubtedly prove complex. The existence of viral genes important for viral replication in ECs opens up new avenues for targeting virus replication to ameliorate vascular-related pathologies associated with HCMV infection, including atherosclerosis, restenosis, and chronic graft rejection.
ACKNOWLEDGMENTS
We thank William Day of the Arizona Research Laboratories and Patricia Jansma of the Molecular and Cellular Biology Imaging Facility for their expertise and assistance with transmission electron and confocal microscopy, respectively. We acknowledge Paula Campbell and the AZCC/ARL Division of Biotechnology Cytometry Core Facility for expertise in flow cytometry. We gratefully acknowledge the gift of antibodies from Thomas Shenk, William Britt, Jay Nelson, Michael Nevels, and Samuel Campos. We thank Jean Wilson for helpful discussions. We acknowledge M. Umashankar, K. Caviness, and P. Zagallo for critical reading of the manuscript.
This work was supported by Public Health Service grant AI079059 from the National Institute of Allergy and Infectious Diseases and by a Cancer Center Support grant (CA020374). F.G. is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts.
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, or The Pew Charitable Trusts.
Footnotes
Published ahead of print 2 January 2013
REFERENCES
- 1. Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470 [DOI] [PubMed] [Google Scholar]
- 2. Goodrum F, Caviness K, Zagallo P. 2012. Human cytomegalovirus persistence. Cell. Microbiol. 14:644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jarvis MA, Nelson JA. 2007. HCMV: molecular basis of persistence and latency, p 780–794 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 4. Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J. Clin. Invest. 121:1673–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 6. Caposio P, Orloff SL, Streblow DN. 2011. The role of cytomegalovirus in angiogenesis. Virus Res. 157:204–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horvath R, Cerny J, Benedik J, Jr, Hokl J, Jelinkova I, Benedik J. 2000. The possible role of human cytomegalovirus (HCMV) in the origin of atherosclerosis. J. Clin. Virol. 16:17–24 [DOI] [PubMed] [Google Scholar]
- 8. Pawelec G, Derhovanessian E. 2011. Role of CMV in immune senescence. Virus Res. 157:175–179 [DOI] [PubMed] [Google Scholar]
- 9. Pourgheysari B, Khan N, Best D, Bruton R, Nayak L, Moss PA. 2007. The cytomegalovirus-specific CD4+ T-cell response expands with age and markedly alters the CD4+ T-cell repertoire. J. Virol. 81:7759–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vasto S, Colonna-Romano G, Larbi A, Wikby A, Caruso C, Pawelec G. 2007. Role of persistent CMV infection in configuring T cell immunity in the elderly. Immun. Ageing 4:2 doi:10.1186/1742-4933-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang GC, Kao WH, Murakami P, Xue QL, Chiou RB, Detrick B, McDyer JF, Semba RD, Casolaro V, Walston JD, Fried LP. 2010. Cytomegalovirus infection and the risk of mortality and frailty in older women: a prospective observational cohort study. Am. J. Epidemiol. 171:1144–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sinzger C, Grefte A, Plachter B, Gouw AS, The TH, Jahn G. 1995. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76:741–750 [DOI] [PubMed] [Google Scholar]
- 13. Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368 [DOI] [PubMed] [Google Scholar]
- 14. Adler B, Sinzger C. 2009. Endothelial cells in human cytomegalovirus infection: one host cell out of many or a crucial target for virus spread? Thromb. Haemost. 102:1057–1063 [DOI] [PubMed] [Google Scholar]
- 15. Koffron AJ, Hummel M, Patterson BK, Yan S, Kaufman DB, Fryer JP, Stuart FP, Abecassis MI. 1998. Cellular localization of latent murine cytomegalovirus. J. Virol. 72:95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fish KN, Soderberg-Naucler C, Mills LK, Stenglein S, Nelson JA. 1998. Human cytomegalovirus persistently infects aortic endothelial cells. J. Virol. 72:5661–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jarvis MA, Nelson JA. 2007. Human cytomegalovirus tropism for endothelial cells: not all endothelial cells are created equal. J. Virol. 81:2095–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kahl M, Siegel-Axel D, Stenglein S, Jahn G, Sinzger C. 2000. Efficient lytic infection of human arterial endothelial cells by human cytomegalovirus strains. J. Virol. 74:7628–7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sinzger C, Kahl M, Laib K, Klingel K, Rieger P, Plachter B, Jahn G. 2000. Tropism of human cytomegalovirus for endothelial cells is determined by a post-entry step dependent on efficient translocation to the nucleus. J. Gen. Virol. 81:3021–3035 [DOI] [PubMed] [Google Scholar]
- 20. Sinzger C, Knapp J, Plachter B, Schmidt K, Jahn G. 1997. Quantification of replication of clinical cytomegalovirus isolates in cultured endothelial cells and fibroblasts by a focus expansion assay. J. Virol. Methods 63:103–112 [DOI] [PubMed] [Google Scholar]
- 21. Jarvis M, Nelson J. 2002. Human cytomegalovirus persistence and latency in endothelial cells and macrophages. Curr. Opin. Microbiol. 5:403–407 [DOI] [PubMed] [Google Scholar]
- 22. Grundy JE, Lawson KM, MacCormac LP, Fletcher JM, Yong KL. 1998. Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil-endothelial cell contact and during neutrophil transendothelial migration. J. Infect. Dis. 177:1465–1474 [DOI] [PubMed] [Google Scholar]
- 23. Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 78:10023–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Revello MG, Gerna G. 2010. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev. Med. Virol. 20:136–155 [DOI] [PubMed] [Google Scholar]
- 25. Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 325:63–82 [DOI] [PubMed] [Google Scholar]
- 26. Waldman WJ, Knight DA, Huang EH, Sedmak DD. 1995. Bidirectional transmission of infectious cytomegalovirus between monocytes and vascular endothelial cells: an in vitro model. J. Infect. Dis. 171:263–272 [DOI] [PubMed] [Google Scholar]
- 27. Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. 2006. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J. Virol. 80:11539–11555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grefte A, Blom N, van der Giessen M, van Son W, The TH. 1993. Ultrastructural analysis of circulating cytomegalic cells in patients with active cytomegalovirus infection: evidence for virus production and endothelial origin. J. Infect. Dis. 168:1110–1118 [DOI] [PubMed] [Google Scholar]
- 29. Grefte A, Harmsen MC, van der Giessen M, Knollema S, van Son WJ, The TH. 1994. Presence of human cytomegalovirus (HCMV) immediate early mRNA but not ppUL83 (lower matrix protein pp65) mRNA in polymorphonuclear and mononuclear leukocytes during active HCMV infection. J. Gen. Virol. 75:1989–1998 [DOI] [PubMed] [Google Scholar]
- 30. Sacher T, Andrassy J, Kalnins A, Dolken L, Jordan S, Podlech J, Ruzsics Z, Jauch KW, Reddehase MJ, Koszinowski UH. 2011. Shedding light on the elusive role of endothelial cells in cytomegalovirus dissemination. PLoS Pathog. 7:e1002366 doi:10.1371/journal.ppat.1002366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bentz GL, Yurochko AD. 2008. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and β1 and β3 integrins. Proc. Natl. Acad. Sci. U. S. A. 105:5531–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Botto S, Streblow DN, DeFilippis V, White L, Kreklywich CN, Smith PP, Caposio P. 2011. IL-6 in human cytomegalovirus secretome promotes angiogenesis and survival of endothelial cells through the stimulation of survivin. Blood 117:352–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hsich E, Zhou YF, Paigen B, Johnson TM, Burnett MS, Epstein SE. 2001. Cytomegalovirus infection increases development of atherosclerosis in apolipoprotein-E knockout mice. Atherosclerosis 156:23–28 [DOI] [PubMed] [Google Scholar]
- 34. Koskinen PK, Nieminen MS, Krogerus LA, Lemstrom KB, Mattila SP, Hayry PJ, Lautenschlager IT. 1993. Cytomegalovirus infection and accelerated cardiac allograft vasculopathy in human cardiac allografts. J. Heart Lung Transplant. 12:724–729 [PubMed] [Google Scholar]
- 35. Popovic M, Smiljanic K, Dobutovic B, Syrovets T, Simmet T, Isenovic ER. 2012. Human cytomegalovirus infection and atherothrombosis. J. Thromb. Thrombolysis 33:160–172 [DOI] [PubMed] [Google Scholar]
- 36. Streblow D N, Orloff SL, Nelson JA. 2001. Do pathogens accelerate atherosclerosis? J. Nutr. 131:2798S–2804S [DOI] [PubMed] [Google Scholar]
- 37. Vliegen I, Stassen F, Grauls G, Blok R, Bruggeman C. 2002. MCMV infection increases early T-lymphocyte influx in atherosclerotic lesions in apoE knockout mice. J. Clin. Virol. 25(Suppl. 2):S159–S171 [DOI] [PubMed] [Google Scholar]
- 38. Zhou YF, Leon MB, Waclawiw MA, Popma JJ, Yu ZX, Finkel T, Epstein SE. 1996. Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N. Engl. J. Med. 335:624–630 [DOI] [PubMed] [Google Scholar]
- 39. Almond P. S., Matas A., Gillingham K., Dunn D. L., Payne W. D., Gores P., Gruessner R., Najarian J. S. 1993. Risk factors for chronic rejection in renal allograft recipients. Transplantation 55:752–757 [DOI] [PubMed] [Google Scholar]
- 40. Melnick JL, Adam E, DeBakey ME. 1996. Cytomegalovirus and atherosclerosis. Arch. Immunol. Ther. Exp. (Warsz.). 44:297–302 [PubMed] [Google Scholar]
- 41. Speir E, Huang ES, Modali R, Leon MB, Shawl F, Finkel T, Epstein SE. 1995. Interaction of human cytomegalovirus with p53: possible role in coronary restenosis. Scand. J. Infect. Dis. Suppl. 99:78–81 [PubMed] [Google Scholar]
- 42. Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE. 1994. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 265:391–394 [DOI] [PubMed] [Google Scholar]
- 43. Bolovan-Fritts CA, Wiedeman JA. 2002. Mapping the viral genetic determinants of endothelial cell tropism in human cytomegalovirus. J. clinical virology: the official publication of the Pan American Society for Clinical Virology 25(Suppl. 2):S97–S109 [DOI] [PubMed] [Google Scholar]
- 44. Sinzger C, Schmidt K, Knapp J, Kahl M, Beck R, Waldman J, Hebart H, Einsele H, Jahn G. 1999. Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J. Gen. Virol. 80:2867–2877 [DOI] [PubMed] [Google Scholar]
- 45. Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J. Gen. Virol. 87:2451–2460 [DOI] [PubMed] [Google Scholar]
- 46. Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. U. S. A. 105:14118–14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scrivano L, Sinzger C, Nitschko H, Koszinowski UH, Adler B. 2011. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 7:e1001256 doi:10.1371/journal.ppat.1001256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 79:10330–10338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Compton T, Nepomuceno RR, Nowlin DM. 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387–395 [DOI] [PubMed] [Google Scholar]
- 52. Huber MT, Compton T. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 72:8191–8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li L, Nelson JA, Britt WJ. 1997. Glycoprotein H-related complexes of human cytomegalovirus: identification of a third protein in the gCIII complex. J. Virol. 71:3090–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dargan DJ, Douglas E, Cunningham C, Jamieson F, Stanton RJ, Baluchova K, McSharry BP, Tomasec P, Emery VC, Percivalle E, Sarasini A, Gerna G, Wilkinson GW, Davison AJ. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 91:1535–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bolovan-Fritts C, Wiedeman JA. 2001. Human cytomegalovirus strain Toledo lacks a virus-encoded tropism factor required for infection of aortic endothelial cells. J. Infect. Dis. 184:1252–1261 [DOI] [PubMed] [Google Scholar]
- 56. Slobbe-van Drunen ME, Hendrickx AT, Vossen RC, Speel EJ, van Dam-Mieras MC, Bruggeman CA. 1998. Nuclear import as a barrier to infection of human umbilical vein endothelial cells by human cytomegalovirus strain AD169. Virus Res. 56:149–156 [DOI] [PubMed] [Google Scholar]
- 57. Brune W, Menard C, Heesemann J, Koszinowski UH. 2001. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291:303–305 [DOI] [PubMed] [Google Scholar]
- 58. Bronzini M, Luganini A, Dell'Oste V, De Andrea M, Landolfo S, Gribaudo G. 2012. The US16 gene of human cytomegalovirus is required for efficient viral infection of endothelial and epithelial cells. J. Virol. 86:6875–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grainger L, Cicchini L, Rak M, Petrucelli A, Fitzgerald KD, Semler BL, Goodrum F. 2010. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J. Virol. 84:9472–9486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog. 7:e1002444 doi:10.1371/journal.ppat.1002444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi-localized latency determinant encoded by human cytomegalovirus. J. Virol. 83:5615–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Isaacson MK, Juckem LK, Compton T. 2008. Virus entry and innate immune activation. Curr. Top. Microbiol. Immunol. 325:85–100 [DOI] [PubMed] [Google Scholar]
- 63. Katzmann DJ, Odorizzi G, Emr SD. 2002. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3:893–905 [DOI] [PubMed] [Google Scholar]
- 64. Britt B. 2007. Maturation and egress, p 311–323 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 65. Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J. Virol. 83:11421–11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 85:5864–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Homman-Loudiyi M, Hultenby K, Britt W, Soderberg-Naucler C. 2003. Envelopment of human cytomegalovirus occurs by budding into Golgi-derived vacuole compartments positive for gB, Rab 3, trans-Golgi network 46, and mannosidase II. J. Virol. 77:3191–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sanchez V, Sztul E, Britt WJ. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-Golgi-intermediate compartment. J. Virol. 74:3842–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cepeda V, Esteban M, Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell. Microbiol. 12:386–404 [DOI] [PubMed] [Google Scholar]
- 72. Henaff D, Radtke K, Lippe R. 2012. Herpesviruses exploit several host compartments for envelopment. Traffic 13:1443–1449 [DOI] [PubMed] [Google Scholar]
- 73. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 74. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 75. Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 20:392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T, von Einem J. 2011. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J. Virol. 85:3821–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tandon R, AuCoin DP, Mocarski ES. 2009. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 83:10797–10807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Slagsvold T, Pattni K, Malerod L, Stenmark H. 2006. Endosomal and non-endosomal functions of ESCRT proteins. Trends Cell Biol. 16:317–326 [DOI] [PubMed] [Google Scholar]
- 79. Kolesnikova L, Berghofer B, Bamberg S, Becker S. 2004. Multivesicular bodies as a platform for formation of the Marburg virus envelope. J. Virol. 78:12277–12287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pelchen-Matthews A, Kramer B, Marsh M. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, Mothes W. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4:785–801 [DOI] [PubMed] [Google Scholar]
- 82. Stuchell MD, Garrus JE, Muller B, Stray KM, Ghaffarian S, McKinnon R, Krausslich HG, Morham SG, Sundquist WI. 2004. The human endosomal sorting complex required for transport (ESCRT-I) and its role in HIV-1 budding. J. Biol. Chem. 279:36059–36071 [DOI] [PubMed] [Google Scholar]
- 83. Usami Y, Popov S, Popova E, Gottlinger HG. 2008. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a Nedd4-like ubiquitin ligase. J. Virol. 82:4898–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Watanabe T, Sorensen EM, Naito A, Schott M, Kim S, Ahlquist P. 2007. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. U. S. A. 104:10205–10210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Calistri A, Sette P, Salata C, Cancellotti E, Forghieri C, Comin A, Gottlinger H, Campadelli-Fiume G, Palu G, Parolin C. 2007. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 81:11468–11478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Crump CM, Yates C, Minson T. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 81:7380–7387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, Mettenleiter TC. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pawliczek T, Crump CM. 2009. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J. Virol. 83:11254–11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, Chang CW, Tsai CH, Liu FT, Chen MR. 2012. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr virus. PLoS Pathog. 8:e1002904 doi:10.1371/journal.ppat.1002904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mori Y, Koike M, Moriishi E, Kawabata A, Tang H, Oyaizu H, Uchiyama Y, Yamanishi K. 2008. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 9:1728–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]