

Abstract
Respiratory syncytial virus (RSV) is the leading cause of lower respiratory tract infections in infants worldwide. Despite decades of research, there is still no registered vaccine available for this major pathogen. We investigated the protective efficacy of a recombinant influenza virus, PR8/NA-F85–93, that carries the RSV CD8+ T cell epitope F85–93 in its neuraminidase stalk. F85–93-specific cytotoxic T lymphocytes (CTLs) were induced in mice after a single intranasal immunization with PR8/NA-F85-93 virus, and these CTLs provided a significant reduction in the lung viral load upon a subsequent challenge with RSV. To avoid influenza-induced morbidity, we treated mice with matrix protein 2 (M2e)-specific monoclonal antibodies before PR8/NA-F85-93 virus infection. Treatment with anti-M2e antibodies reduced the infiltration of immune cells in the lungs upon PR8/NA-F85-93 infection, whereas the formation of inducible bronchus-associated lymphoid tissue was not affected. Moreover, this treatment prevented body weight loss yet still permitted the induction of RSV F-specific T cell responses and significantly reduced RSV replication upon challenge. These results demonstrate that it is possible to take advantage of the infection-permissive protection of M2e-specific antibodies against influenza A virus to induce heterologous CD8+ T cell-mediated immunity by an influenza A virus vector expressing the RSV F85-93 epitope.
INTRODUCTION
Human respiratory syncytial virus (RSV) is the most important cause of acute lower respiratory infections in babies, especially when premature, and young children (1). Almost every child has been infected before the age of 2 years and will very likely be reinfected several times more with RSV during its further life (2). It is estimated that each year over 30 million infections with RSV result in acute lower respiratory infections (ALRI) in children younger than 5 years (3). Approximately 10% of children in this age group suffering from ALRI due to RSV require hospitalization. Moreover, it is estimated that up to 200,000 children younger than 5 years die due to complications caused by RSV, most of which occur in developing countries (3). Furthermore, severe RSV infection during infancy has been associated with an increased incidence of recurrent wheezing in later childhood (4). In the elderly, RSV causes pneumonia, bronchiolitis, and exacerbation of chronic obstructive pulmonary disease, conditions that often lead to hospitalization and excess mortality in this age group (5).
Despite the disease burden caused by RSV, no licensed RSV vaccine is currently available. The development of a safe vaccine is difficult, since natural RSV infections occur in children at very young ages and do not provide long-lasting protective immunity. The inability of natural infections to evoke protective immunity in the absence of significant antigenic drift might be attributable in part to the ability of RSV to evade the host immune response at different levels (reviewed in reference 6). The main mechanism for evasion of the host innate immune response by RSV is the inhibition of type I interferon (IFN) production and IFN-associated genes. The RSV genome encodes two nonstructural (NS) proteins, NS1 and NS2, that collaborate to suppress both the synthesis and the function of type I IFN through the transcription factors IFN regulatory factor 3 (IRF-3) and signal transducer and activator of transcription 2 (Stat-2) (7, 8). This suppression of the type I IFN response contributes to the inhibition of CD8+ and CD4+ T cell responses (9, 10).
A clinical trial with a formalin-inactivated RSV virion-based vaccine (FI-RSV) in the 1960s did not evoke protective immunity but led to enhanced disease upon infection (11). A possible explanation for this adverse response is that the FI-RSV vaccine strongly skews the immune response in an undesired allergy-like Th2 direction, which leads to enhanced infiltration of eosinophils and neutrophils into the lungs upon RSV infection, causing severe lung damage. Such a strong Th2 response blunts the CD8+ T cell response, thereby compromising viral clearance from the lungs (12). Since that fatal trial, it is generally believed that RSV vaccines that induce a strong Th2-biased immune response should be avoided.
Past attempts to produce an RSV vaccine were focused mainly on inducing neutralizing antibody responses. However, it has been suggested that an antibody response might not be sufficient for protection (as reinfections occur throughout life) and that a vaccine that elicits both an antibody and a T cell response might be more effective (13). Multiple reported studies have consistently demonstrated that fatal or severe lower respiratory tract RSV infections are characterized by high viral titers and the near absence of pulmonary infiltration of T cells or the cytokines they produce (14). Moreover, a possible role for T cells in the clearance of RSV is supported by the observation that viral clearance from the lungs occurs once a potent T cell response is induced (15, 16). Mouse studies have indicated that both CD8+ and CD4+ T cells are essential for the clearance of RSV (17, 18). In addition, CD8+ T cells have been shown to mediate protection in animals immunized with several candidate RSV vaccines, such as Mycobacterium bovis BCG-RSV (19).
Therefore, we hypothesized that priming for an RSV CD8+ T cell response might be an attractive strategy for an RSV vaccine. These primed CD8+ T cells might promote rapid clearance of the virus from the lungs and potentially prolong RSV-specific T cell memory. It has been shown that priming for an RSV KdM282–90-specific CD8+ T cell memory response abrogates the induction of an undesirable Th2 response in a model of FI-RSV-primed mice (12, 20, 21). However, Ruckwardt et al. reported that an augmented response to the dominant KdM282–90 epitope exacerbated illness upon RSV infection, whereas mutating the dominant KdM282–90 epitope enhanced the response to the subdominant DbM187–195 epitope with significantly less illness upon RSV infection (22). Moreover, CD8+ T cells specific for the subdominant KdF85–93 epitope can reduce lung eosinophilia if the total number of F85–93 epitope-specific CD8+ T cells is increased early after RSV infection (23). These results suggest that a vaccination strategy that induces an immune response to a subdominant RSV cytotoxic T lymphocyte (CTL) epitope, such as the KdF85–93 epitope, can contribute to viral clearance without exacerbating illness.
Here, we used a recombinant influenza virus as a live viral vector for mucosal delivery of the RSV KdF85–93 CTL epitope. Influenza virus is an interesting vaccine vector candidate because it induces both humoral and cellular immune responses (24) and because it can be modified by reverse genetics (25). We produced recombinant A/Puerto Rico/8/34 (PR8) influenza virus carrying the RSV F85–93 CTL epitope in the stalk of the neuraminidase and tested its protective efficacy against RSV in BALB/c mice. We showed that F85–93-specific CTLs were induced in the mice upon a single intranasal immunization with PR8/NA-F85–93 virus and that these CTLs were associated with a significant reduction in the lung viral load upon RSV challenge. We further optimized the vaccination strategy by passive administration of IgG2a monoclonal antibodies directed against the extracellular domain of influenza matrix protein 2 (M2e) to suppress morbidity associated with PR8/NA-F85–93 virus infection.
MATERIALS AND METHODS
Cell lines and viruses.
Madin-Darby canine kidney (MDCK) cells, African green monkey kidney (Vero) cells, HEp-2 cells, and HEK293T cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), nonessential amino acids, l-glutamine, sodium pyruvate, and penicillin-streptomycin at 37°C in 5% CO2. RSV-A2 (ATCC VR-1540) was propagated on HEp-2 cells and quantified by plaque titration on Vero cells. Influenza viruses were grown on MDCK cells in serum-free cell culture medium in the presence of 2 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma). The virus was pelleted from culture supernatant by overnight centrifugation at 25,000 × g.
Construction of PR8/NA-F85–93 and PR8/NA-HA518–526.
Recombinant viruses were rescued using the influenza A/Puerto Rico/8/34 virus-based reverse genetics system (25). We used fusion PCR to clone the Kd-restricted CTL epitope of the RSV fusion protein (F85–93) and the KdHA518–526 CTL epitope of influenza A virus hemagglutinin into a pHW196-NA plasmid by replacing the region encoding amino acids (aa) 65 to 71 of the neuraminidase (NA)-coding sequence with a sequence encoding 15 aa containing the CTL epitope extended with the 3 naturally flanking amino acids both C and N terminally. To generate a recombinant virus, 1 μg of each of the seven pHW plasmids (pHW191-PB2, pHW192-PB1, pHW193-PA, pHW194-HA, pHW195-NP, pHW197-M, and pHW198-NS) was transfected together with one of the NA plasmids (pHW196-NA, pHW196-NA-F85–93, or pHW196-NA-HA518–526) into a HEK293T-MDCK cell coculture using calcium phosphate coprecipitation in Opti-MEM. After 36 h, TPCK-treated trypsin (Sigma) was added to a final concentration of 2 μg/ml. After 72 h, the medium was collected. The presence of the virus in the medium was confirmed by hemagglutination of chicken red blood cells. After clonal selection by two rounds of limiting dilution, the virus was amplified on MDCK cells and the viral titer was determined by plaque assay. The presence of the epitope in the viral genome was confirmed by reverse transcription (RT)-PCR followed by sequence analysis.
Influenza virus plaque assay.
MDCK cells were seeded in complete DMEM in six-well plates at 5 × 105 cells per well 1 day before infection. The next day, the cells were washed once with serum-free medium and incubated with a 10-fold dilution series of the virus in 500 μl medium. After 1 h of incubation at 37°C, medium was removed and replaced by an overlay of 0.8% Avicel RC-591 (FMC Biopolymer) in serum-free medium with 2 μg/ml TPCK-treated trypsin (Sigma). After 72 h of incubation at 37°C, the overlay was removed, and the cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Plaques were stained with an anti-M2e IgG1 mouse monoclonal antibody (final concentration 0.4 μg/ml) followed by a secondary anti-mouse IgG horseradish peroxidase (HRP)-linked antibody. TrueBlue peroxidase substrate (KPL) was used for visualization.
RSV plaque assay.
RSV plaque assays were carried out on Vero cells, which were seeded 1 day before infection at 20,000 cells per well in a 96-well plate. Cells were infected with a 3-fold dilution series of lung homogenate or bronchoalveolar lavage (BAL) fluid in Opti-MEM. After 3 h of incubation at 37°C, the inoculum was removed and replaced with an overlay of 0.8% Avicel RC-591 (FMC Biopolymer) diluted in cell culture medium containing 2% FCS. After 4 days of incubation at 37°C, the overlay was removed, and the cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Plaques were stained using goat anti-RSV serum (AB1128; Chemicon International) followed by a secondary anti-goat IgG HRP-linked antibody and visualized by the addition of TrueBlue peroxidase substrate (KPL).
Neuraminidase activity assay.
Forty microliters of virus diluted in phosphate-buffered saline (PBS) was added to 9 μl assay buffer (1 M Na acetate, 10 mM CaCl2, 5% butanol) and 1 μl 5 mM 2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid substrate (MUNANA) (Sigma) in a 96-well plate. Fluorescence was measured every 2 min in a cytofluorometer (excitation at 360 nm, emission at 460 nm) for 3 h. The background (fluorescence measured in a sample containing 40 μl PBS combined with substrate and assay buffer) was subtracted from each measurement. Using a standard curve of free 4-methylumbelliferone, we calculated the amount of released 4-methylumbelliferone in the sample for each time point. The number of NA units in the sample was calculated by dividing the amount of free 4-methylumbelliferone by the duration (in minutes). For relative activities, the largest amount of NA units during the 3 h (correlating to the highest turnover rate of the enzyme) was divided by the largest amount of NA units in the wild-type NA sample. To confirm that equal quantities of virus were tested, the viral titer was checked by agglutination with chicken red blood cells and by plaque assay.
Immunizations and RSV challenge of mice.
Eight-week-old female BALB/c mice were housed in specified pathogen-free conditions and used in all experiments. Under mild isoflurane anesthesia the mice were immunized by intranasal administration of 5 × 103 PFU PR8/NA-F85–93 virus, 5 × 103 PFU PR8/NA-HA518–526 virus, or 1 × 103 PFU PR8 wild-type virus diluted in 50 μl PBS. Passive immunization experiments were performed by giving the mice, while under slight isoflurane anesthesia, a single intranasal (i.n.) dose of 50 μl of PBS containing 1 μg of monoclonal antibody. An IgG2a monoclonal antibody directed against M2e or an IgG1 monoclonal antibody directed against the ectodomain of the influenza B virus NB protein (NBe) were used for passive immunization before influenza A virus administration. Both antibodies were purified from hybridoma supernatants.
Challenge with 1 × 106 PFU RSV-A2 was performed on mice under slight isoflurane anesthesia by i.n. administration of 50 μl virus suspension diluted in PBS. Mice were killed on day four or five postchallenge (as indicated). Lungs were removed and homogenized in 1 ml Hanks balanced salt solution (HBSS) containing 20% sucrose, using a Heidolph RZR 2020 homogenizer. Homogenates were cleared by centrifugation (1,000 × g for 15 min at 4°C) and supernatant was used for quantitation by plaque assay.
IFN-γ enzyme-linked immunospot assay.
At various time points after immunization (mentioned in the figure legends) spleens were removed aseptically. Splenocytes were isolated and red blood cells were lysed in NH4Cl red blood cell lysis buffer. IFN-γ enzyme-linked immunospot (ELISPOT) assay was performed according to the manufacturer's instructions (U-CyTech Biosciences). Briefly, MaxiSorp 96-well plates were coated overnight with anti-IFN-γ monoclonal antibody at 4°C. The next day, plates were blocked and 3 × 105 cells were seeded per well in 100 μl culture medium (RPMI, 10% FCS, l-glutamin, and penicillin-streptomycin) supplemented with H-2d-restricted RSV-F protein-derived (KYKNAVTEL) restimulation peptide at a final concentration of 5 μg/ml. After 12 h of restimulation at 37°C, the cells were removed, the plates were washed, and the IFN-γ trapped on the plates was visualized using biotinylated polyclonal anti-IFN-γ antiserum. The spots were counted using an inverted light microscope.
Intracellular cytokine staining.
For intracellular cytokine staining (ICS), 2 × 106 splenocytes were seeded in 96-well suspension plates in 200 μl culture medium (RPMI, 10% FCS, l-glutamine, and penicillin-streptomycin) supplemented with restimulation peptide at a final concentration of 5 μg/ml. After 12 h of restimulation at 37°C with RSV-F protein-derived KYKNAVTEL (F85–93) or NP-derived TYQRTRALV (NP155–163) peptides, GolgiPlug (BD) was added at a final concentration of 1 μg/ml and the cells were incubated for another 4 h at 37°C. After restimulation, cells were incubated with anti-mouse CD16/CD32 antibody (BD) to avoid nonspecific staining of immune cells. Staining was performed with anti-CD8a-fluorescein isothiocyanate (FITC), anti-CD3ε-phycoerythrin (PE) (both from BD), and Live/Dead fixable Aqua dead cell stain (Molecular Probes) for 30 min. Cells were then fixed with 2% paraformaldehyde, permeabilized with Perm/Wash buffer (BD), and stained with anti-IFN-γ Alexa Fluor 647 (BD) for 30 min. IFN-γ+ CD8+ T cells were quantified on an LSRII flow cytometer (BD, San Jose, CA) and analyzed with FACSDiva software (BD).
Analysis of pulmonary cell infiltration.
Five days after influenza virus infection, mice were killed with ketamine-xylazine and the lungs were washed through the tracheas with 3 ml of HBSS + 5 mM EDTA. The first 0.5 ml was collected separately and centrifuged for 5 min at 400 × g, and the supernatant was used for viral quantification. The pelleted cells from the first BAL fluid collection were added to the rest of the BAL fluid. Cells were incubated with anti-mouse CD16/CD32 antibody (BD) to avoid nonspecific staining of immune cells, and the cells were stained with anti-CD3ε-FITC, anti-CD4-PerCP, anti-CD11c-allophycocyanin (APC), anti-CD11b-APC-Cy7 (all BD), anti-CD8a-PE-Cy7, anti-major histocompatibility complex class II (MHC-II)-eFLUOR 450 (both eBioscience), and CCR-3-PE (R&D Systems). Using FACSDiva software (BD) with a protocol similar to that described by Bogaert et al. (26), we determined bronchoalveolar lavage (BAL) fluid immune cell composition on an LSR-II flow cytometer (BD, San Jose, CA) by analyzing cellular autofluorescence and surface expression of CD3ε, CD4, CD8a, CD11b, CCR3, MHC-II, and CD11c.
Analysis of induced bronchus-associated lymphoid tissue formation.
Twenty-six days after influenza virus infection, mice were terminally anesthetized and the lungs were removed. Lungs were ground with the plunger of a syringe and passed through a 70-μm filter to produce single-cell suspensions. Red blood cells were lysed in NH4Cl red blood cell lysis buffer. They were incubated with anti-mouse CD16/CD32 antibody (BD) to avoid nonspecific staining of immune cells and stained for 30 min with anti-IgM-PerCp-Cy5.5, anti-IgD-PE, anti-B220-Alexa Fluor 700, anti-CD3ε-APC, anti-Fas-PE-Cy7, and anti-GL7-FITC (all BD) and Live/Dead fixable aqua dead cell stain (Molecular Probes). IgD− IgM− Fas+ GL7+ B cells were quantified on an LSRII flow cytometer (BD, San Jose, CA) and analyzed with FACSDiva software (BD).
RESULTS
Generation of recombinant influenza viral vector for delivery of an RSV F CTL epitope.
To evaluate whether a recombinant influenza virus harboring an RSV CTL epitope can provide protection against RSV, we used a reverse genetics system for PR8 influenza (25) to generate the following: (i) a virus containing the H-2d-restricted F85–93 CTL epitope (KYKNAVTEL) of the RSV fusion protein (PR8/NA-F85–93 virus); (ii) a virus with an H-2d-restricted CTL epitope (IYSTVASSL) of the influenza virus hemagglutinin HA518–526 (PR8/NA-HA518–526 virus). These F- and hemagglutinin (HA)-derived CTL epitopes were cloned into the neuraminidase (NA) coding sequence as replacements of part of the NA stalk by replacing amino acids (aa) 65 to 71 with a 15-aa sequence containing the CTL epitope extended with the 3 naturally flanking amino acids both C and N terminally (Fig. 1A). Both viruses were rescued and further subcloned by two rounds of limiting dilution. The presence of the epitope in the viral genome was confirmed by RT-PCR followed by sequence analysis (Fig. 1B).
Fig 1.

Characterization of recombinant PR8/NA-F85–93 and PR8/NA-HA518–526 influenza viruses. (A) Schematic representation of wild-type and mutant neuraminidases. CT, cytoplasmic tail; TM, transmembrane domain. (B) RT-PCR of an NA fragment containing the insertion site of the epitope. A band shift from 170 to 207 bp was seen when the epitope was inserted. (C) Neuraminidase activity assay on live, purified virus. Fluorescence of the cleaved MUNANA substrate was measured every 2 min during 3 h. (D) Plaque phenotypes of the PR8/NA-F85–93 and PR8/NA-HA518–526 influenza viruses do not differ from the wild-type virus plaques in an MDCK plaque assay. (E) In vitro growth kinetics. MDCK cells were infected at a multiplicity of infection of 0.001 of wild-type PR8, PR8/NA-F85–93, or PR8/NA-HA518–526 virus. Samples were taken at 0, 4, 8, 12, 24, and 48 h postinfection. The viral titer in the samples was determined by a TCID50 assay.
To test whether insertion of the epitopes affected the activity of the NAs in vitro, we performed a neuraminidase activity assay on whole purified virus (Fig. 1C). The number of virions was normalized by using equal amounts of hemagglutination units of the three viruses that were compared. Both PR8/NA-F85–93 virus and PR8/NA-HA518–526 hydrolyzed the MUNANA substrate at a lower rate than NA from the wild-type PR8 virus, suggesting a lower specific activity of both mutant NAs. The activity of NA-F85–93 was 40% of the activity of the wild-type NA, whereas the activity of NA-HA518–526 was even lower (15% of the activity of wild-type NA). In a plaque assay, the plaques of wild-type PR8, PR8/NA-F85–93, and PR8/NA-HA518–526 viruses were similar in size (Fig. 1D).
To determine the in vitro viral growth kinetics, MDCK cells were infected at a multiplicity of infection of 0.001 of wild-type PR8 virus, PR8/NA-F85–93 virus, or PR8/NA-HA518–526 virus. At various times after infection the viral titer in the supernatant was quantified by a 50% tissue culture infective dose (TCID50) assay. The growth kinetics of the PR8/NA-F85–93 virus and the PR8/NA-HA518–526 virus resembled that of the wild-type PR8 virus (Fig. 1E).
PR8/NA-F85–93 virus infection induces RSV F85–93-specific cytotoxic T lymphocytes.
We next investigated whether F85–93 epitope-specific CTLs can be retrieved from mice that have been exposed to the PR8/NA-F85–93 virus. BALB/c mice were immunized with 5 × 103 PFU of PR8/NA-F85–93 virus or, as a control, PR8/NA-HA518–526 virus, and spleens were isolated 10 days later. F85–93 peptide-specific IFN-γ ELISPOT analysis and intracellular cytokine staining revealed that F85–93 epitope-specific CTLs were induced in mice that had been exposed to the PR8/NA-F85–93 virus but not in mice that had been exposed to the PR8/NA-HA518–526 virus (Fig. 2A to C). No significant difference in response to the influenza virus-specific NP155–163 epitope was detected (Fig. 2D).
Fig 2.
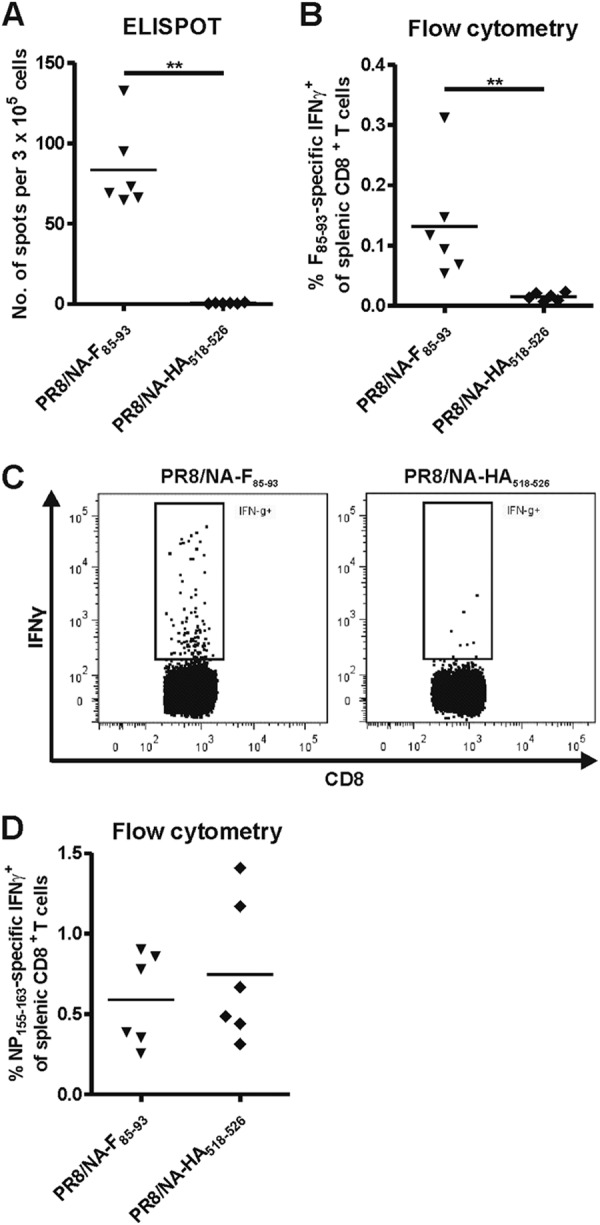
PR8/NA-F85–93 virus infection induces F85–93-specific CTLs in mice. BALB/c mice (6 per group) were infected with 5 × 103 PFU of PR8/NA-F85–93 or PR8/NA-HA518–526 virus. Spleens were isolated 10 days postinfection and stimulated with RSV F85–93 peptide (A, B, and C) or influenza virus NP155–163 peptide (D). After restimulation with RSV F85–93 peptide, IFN-γ production in splenic F85–93-specific CD8+ T cells was determined with ELISPOT assay (A) and flow cytometry (B). (C) Representative dot plots showing IFN-γ positivity in splenic CD8+ T cells after restimulation with RSV F85–93 peptide. (D) Splenocytes were restimulated with influenza virus NP155–163 peptide. The percentage of NP155–163-specific CD8+ T cells was determined by flow cytometry.
To compare the pathogenicity of PR8/NA-F85–93 and PR8/NA-HA518–526, we infected BALB/c mice with 103 or 104 PFU of wild-type PR8 virus or 102, 103, or 104 PFU of PR8/NA-F85–93 or PR8/NA-HA518–526 virus. Interestingly, both the PR8/NA-F85–93 and PR8/NA-HA518–526 viruses caused less morbidity than the wild-type PR8 virus (Fig. 3A to C). Ten-fold higher inoculum doses of PR8/NA-F85–93 and PR8/NA-HA518–526 were needed to cause morbidity resembling that caused by the wild-type PR8 virus (approximately 30% body weight loss). PR8/NA-HA518–526 was slightly more pathogenic than PR8/NA-F85–93 (Table 1). Splenocytes were isolated 14 days after infection with these different doses and cellular responses were analyzed using an F85–93 peptide-specific IFN-γ ELISPOT assay. We observed F85–93 epitope-specific CTLs in mice that had been exposed to the PR8/NA-F85–93 virus. The number of spots, reflecting the number or F epitope-specific T cells, correlated with the viral dose used for inoculation (Fig. 3D). Interestingly, the number of IFN-γ spots after infection of mice with as little as 100 PFU of PR8/NA-F85–93 was similar to that after infection with 1 × 106 PFU of RSV. This result was most likely due to the fact that the PR8/NA-F85–93 virus is able to replicate in mice, whereas for RSV, the mouse is far less permissive.
Fig 3.

PR8/NA-F85–93 virus induces F85–93-specific CTLs in a dose-dependent manner. BALB/c mice were infected with 1 × 102 (n = 4), 1 × 103 (n = 4), or 1 × 104 (n = 6) PFU of PR8/NA-F85–93 (A); 1 × 102 (n = 4), 1 × 103 (n = 4), or 1 × 104 (n = 6) PFU of PR8/NA-HA518–526 virus (B); or 1 × 103 (n = 3) or 1 × 104 (n = 3) PFU of wild-type PR8 virus (C). Body weight was recorded daily (mean ± SD; the PBS group is the same for all 3 graphs). (D) Splenocytes were isolated 14 days postinfection and ex vivo restimulated for 12 h with the F85–93 peptide. IFN-γ-secreting cells were quantified by ELISPOT assay. Control groups received PBS or 1 × 106 PFU of RSV. Bars represent the average number of spots and SD.
Table 1.
Survival of BALB/c mice infected with different doses of PR8/NA-F85–93 or PR8/NA-HA518–526 or with wild-type PR8 virus
| Infectious dose (PFU) | No. of survivors/total no. | |
|---|---|---|
| Wild-type PR8 | 103 | 3/3 |
| 104 | 0/3 | |
| PR8/NA-F85–93 | 102 | 4/4 |
| 103 | 4/4 | |
| 104 | 3/6 | |
| PR8/NA-HA518–526 | 102 | 4/4 |
| 103 | 4/4 | |
| 104 | 2/6 |
Immunization with PR8/NA-F85–93 virus reduces RSV replication in challenged mice.
Next, we evaluated the ability of the PR8/NA-F85–93 virus to protect mice against an RSV challenge. Eight-week-old BALB/c mice received a single intranasal immunization with PR8/NA-F85–93 virus, PR8/NA-HA518–526 virus, RSV, or PBS. Ten days after the immunization, the spleens of the mice were isolated and splenocytes were stimulated for 12 h with the F85–93 peptide (KYKNAVTEL) for IFN-γ ELISPOT analysis. F85–93 epitope-specific CD8+ T cells were activated in mice exposed to PR8/NA-F85–93 and to a lesser extent in RSV-exposed mice, but no F85–93 epitope-specific splenocytes were observed in the control groups (PR8/NA-HA518–526 and PBS) (Fig. 4A). Four weeks after immunization, the mice were challenged with 1 × 106 PFU of RSV-A2. Lung homogenates were prepared 4 days after infection and the RSV titer was determined by plaque assay. The mice that had received the PR8/NA-F85–93 virus as a T cell vaccine had a significantly lower RSV lung titer than the control groups (PR8/NA-HA518–526 and PBS). The median lung titers were 19 and 31 times lower in these mice than in mice immunized with PR8/NA-HA518–526 or PBS, respectively, indicating that the induced CTLs reduced RSV replication in the challenged mice (Fig. 4B). Remarkably, we repeatedly observed that mice that were vaccinated with PR8/NA-HA518–526 generally had a lower pulmonary RSV titer than PBS-treated mice. This might indicate that influenza virus induces a pathogen-independent immune response that can reduce RSV replication.
Fig 4.

Immunization with PR8/NA-F85–93 virus reduces RSV replication in challenged mice. BALB/c mice were infected with the indicated viruses or with PBS and then challenged intranasally with 1 × 106 PFU of RSV-A2. (A) Spleens were isolated 10 days after infection and restimulated ex vivo with the F85–93 peptide, and the F85–93 epitope-specific splenocytes were counted by IFN-γ ELISPOT assay. (B) Lung RSV titers were determined by plaque assay 4 days after RSV challenge. Statistical significance was determined by using a two-sided Mann-Whitney U test: **, P < 0.01.
Anti-M2e antibodies reduce infiltration of immune cells in the lungs after PR8/NA-F85–93 challenge without impairing iBALT formation.
We investigated whether we could reduce the morbidity induced by the PR8 vector but retain F85–93-specific CD8+ T cell induction and protection against RSV infection. We previously described a universal influenza A vaccine based on M2e (27) and recently reported that this vaccine prevents morbidity, but in accordance with the infection-permissive nature of M2e-based immune protection (28), it allows the induction of cross-reactive T cells upon challenge with influenza virus (29). Here, we used the passive transfer of monoclonal antibodies directed against M2e 1 day before infection with the PR8-based CTL-delivery vectors to diminish the morbidity caused by the influenza viral vector. To better understand the effect of intranasal instillation of anti-M2e antibodies on an influenza virus infection, we first characterized the immune response in the lungs of the mice by examining bronchoalveolar lavage (BAL) fluid. Mice were treated with 1 μg IgG2a anti-M2e antibodies or control antibodies directed against the ectodomain of the NB protein of influenza B virus (anti-NBe) and 24 h later they were challenged with 5 × 103 PFU of PR8/NA-F85–93 virus. Mice treated with polyclonal anti-PR8 mouse serum and mock-challenged mice (anti-NBe treated, PBS challenged) were included as controls. BAL fluid was collected 5 days after influenza virus infection. Mice treated with anti-NBe showed a strong infiltration of immune cells in the lungs (Fig. 5A). This infiltration was significantly less in anti-M2e-treated mice. Infiltration of all cell types was strongly reduced in anti-M2e-treated mice compared to anti-NBe-treated mice, except for resident alveolar macrophages (Fig. 5B and C). These effects were reflected in the viral lung titer 5 days after influenza virus infection. The viral lung titer in anti-M2e-treated mice was significantly lower than in anti-NBe-treated mice. However, anti-M2e immunity is not neutralizing in contrast to the polyclonal post-PR8 challenge serum (Fig. 5D). Infection with influenza virus is known to induce the formation of inducible bronchus-associated lymphoid tissue (iBALT). These tertiary lymphoid structures can contribute to protection by promoting T cell and B cell mediated immune responses (30). Therefore, we also investigated if iBALT is still formed after PR8/NA-F85–93 challenge in mice pretreated with anti-M2e antibodies. Germinal center formation, a prime hallmark of iBALT, was analyzed on day 26 after infection with PR8/NA-F85–93. By flow cytometry we determined the number of IgM− IgD− B cells expressing GL7 and Fas. Interestingly, iBALT formation in anti-M2e-treated mice was comparable to that in anti-NBe-treated mice (Fig. 5E). These results show that by combining the PR8/NA-F85–93 virus with anti-M2e pretreatment, we can eliminate the disadvantages of the vaccine virus (i.e., lung inflammation and substantial viral replication) without preventing the formation of iBALT.
Fig 5.

Anti-M2e antibodies reduce infiltration of immune cells in the lungs after PR8/NA-F85–93 challenge without impairing iBALT formation. Mice were immunized with anti-M2e antibodies or control anti-NBe antibodies 1 day before infection with PR8/NA-F85–93. Mice treated with polyclonal anti-PR8 mouse serum and mock-challenged mice (anti-NBe treated, PBS challenged) were included as controls. (A) The total number of cells in the BAL fluid was determined on day 5 after influenza virus infection. Bars represent average + SD. (B and C) BAL fluid cellular composition was determined by flow cytometric enumeration of eosinophils (CD3ε− CD11c− MHC-II− CD11bmed SSChi CCR3+ [med indicates medium, and hi indicates high]), neutrophils (CD3ε− CD11c− MHC-II− CD11bhi SSCmed CCR3−), resident alveolar macrophages (AM) (CD3ε− CD11c+ autofluohi CD11blo [lo indicates low]), recruited AM (CD3ε− CD11c+ autofluomed CD11b+), dendritic cells (DC) (CD3ε− CD11c+ autofluolo MHC-IIhi), CD4+ T cells (CD3ε+ CD4+), and CD8+ T cells (CD3ε+ CD8+). Bars represent the number of cells + SD. (D) Lung influenza virus titers were determined by plaque assay 5 days after the PR8/NA-F85-93 challenge. (E) We analyzed the formation of iBALT by counting the Fas+ GL7+ IgM− IgD− B cells in the lungs 26 days after challenge. Bars represent average + SD; Bdl., below detection limit. Statistical significance was determined by using a two-sided Mann-Whitney U test: **, P < 0.01.
Treatment with anti-M2e antibodies prevents PR8/NA-F85–93-induced morbidity but still allows the induction of F85–93-specific CD8+ T cell responses that reduce RSV lung viral load.
We next investigated whether anti-M2e treatment followed by a PR8/NA-F85–93 infection still allows the induction of F85–93-specific CD8+ T cell responses that are capable of reducing RSV lung viral load. Mice received a single intranasal dose of 1 μg of an anti-M2e antibody or irrelevant control anti-NBe antibody followed 24 h later by an intranasal influenza virus infection (PR8/NA-F85–93 or PR8/NA-HA518–526). Mock-immunized mice received anti-NBe antibodies followed by administration of PBS. As shown in Fig. 6A, anti-NBe treated mice displayed up to 25% weight loss after infection with PR8/NA-F85–93 or PR8/NA-HA518–526. In contrast, mice receiving anti-M2e lost very little weight after infection, and both anti-M2e-treated groups differed significantly from the corresponding groups that received anti-NBe treatment (two-way analysis of variance, P < 0.0001). Fifty days after the primary infection with the PR8 vectors, the CTL memory response in the spleen was analyzed by flow cytometry. F85–93 epitope-specific CTLs were induced in the PR8/NA-F85–93 infected groups after anti-M2e treatment or anti-NBe treatment (Fig. 6B). Although anti-M2e antibody treatment almost completely prevented body weight loss upon PR8/NA-F85–93 infection, the induction of F85–93 epitope-specific CTL responses was only partially affected. Mice were challenged with RSV 51 days after the immunization, and the viral lung titer was determined 5 days later. In anti-NBe-treated mice, the viral lung titer was reduced in the PR8/NA-F85–93-infected group relative to that in the PR8/NA-HA518–526-infected group. A significant reduction in viral lung titer was also observed in anti-M2e-treated PR8/NA-F85–93-infected mice, compared to anti-M2e-treated mice that had been vaccinated with PR8/NA-HA518–526. As in previous experiments, in all groups the lung viral titer was lower than in the PBS-immunized group. These results demonstrate that a reduction in RSV viral load can be achieved by taking advantage of the infection-permissive protection of M2e antibody pretreatment against influenza A virus to induce CD8+ T cell-mediated immunity by an influenza A virus vector expressing the RSV F85–93 epitope.
Fig 6.

Treatment with anti-M2e antibodies prevents PR8/NA-F85–93-induced morbidity but still allows the induction of F85–93-specific CD8+ T cell responses and reduced RSV lung viral load. Mice were immunized with anti-M2e antibodies or control anti-NBe antibodies 1 day before infection with PR8/NA-F85–93 or PR8/NA-HA518–526, and 7 weeks later they were challenged with 1 × 106 PFU of RSV-A2. (A) Body weight after PR8/NA-F85–93 or PR8/NA-HA518–526 infection. Graph shows average relative body weight ± SD. (B) One day before RSV challenge, splenocytes were isolated and restimulated ex vivo with F85–93 peptide for 12 h. The percentage of IFN-γ-positive CD8+ T cells was determined by flow cytometry. (C) Lung RSV titers were determined by plaque assay 5 days after RSV challenge. Statistical significance was determined by using a two-sided Mann-Whitney U test: **, P < 0.01.
DISCUSSION
We evaluated the vaccine potential of a recombinant influenza virus encoding the H-2d F85–93 CTL epitope of the RSV F protein. We demonstrate that a single intranasal immunization with this virus induces a potent F85–93 epitope-specific CD8+ T cell response in mice. RSV clearance was enhanced in PR8/NA-F85–93-immunized mice but not in control or PR8/NA-HA518–526 and PBS-immunized mice. Importantly, the enhanced clearance in PR8/NA-F85–93-immunized mice could not be explained by differences in replication efficacy of the recombinant viruses: PR8/NA-F85–93 and control PR8/NA-HA518–526 viruses replicate with similar efficacy in mice and induce similar body weight loss.
It has been reported that an enhanced illness is observed upon overcompensation of the CTL response by the immunodominant KdM282–90 epitope (22). This is due mainly to a difference between quantity and quality of the M282–90-specific CTLs: less than 50% of the M282–90-specific CTLs produce effector cytokines (IFN-γ, interleukin-2 [IL-2], and tumor necrosis factor alpha [TNF-α]) (23). In a side-by-side comparison, F85–93-specific CTLs have been shown to contain a higher frequency of cells capable of coproducing these effector cytokines, suggesting that F85–93-specific CD8+ T cells exhibit greater cytokine production capacity compared to their M282–90 counterparts. Based on these arguments we reasoned that inducing an F85–93-specific CTL response would result in at least comparable, if not greater, levels of functional CTLs compared to M282–90-specific CTLs, whereas the F85–93-specific CTL responses would not be harmful to the mice.
Remarkably, we repeatedly observed that mice vaccinated with PR8/NA-HA518–526 generally had lower pulmonary RSV titers than PBS-treated mice, indicating that influenza virus induces a pathogen-independent immune response that can partly protect against an RSV infection. It is not clear whether this effect is a result of a nonspecific innate or adaptive immune response. Similar effects have been reported for several pathogens. For example, BCG, the live attenuated vaccine against tuberculosis, induces nonspecific protection against other infections (31). In this example nonspecific innate immune responses through epigenetic reprogramming of monocytes are the basis of pathogen-independent protection (31). This innate imprinting or innate education has been defined as “the long term modification of a microenvironment, which will consequently lead to a nonspecific, but more protective, immune phenotype to a subsequent pathogen” (32). On the other hand, it has been reported that in a model of G-protein-primed mice an influenza virus infection can reduce the severity of G-protein vaccination-induced enhancement of disease (illness, lung eosinophilia, and weight loss) upon an RSV infection (33). This effect is most likely mediated by adaptive immunity, as an activation of influenza virus-specific T cells, possibly through the mechanism of bystander activation, is observed upon a secondary RSV infection. Additionally, the formation of iBALT might contribute to nonspecific protection (34) (discussed more in detail below).
To prevent influenza virus-induced morbidity, we further optimized our vaccination strategy by passive administration of IgG2a, monoclonal antibody directed against M2e, to control morbidity associated with PR8/NA-F85–93 virus infection. In contrast to vaccination with conventional HA-based influenza vaccines, M2e-based vaccination with M2e-VLPs (virus-like particles) is not sterilizing (28, 29). Immunity induced by M2e-VLPs allows limited virus replication, and hence viral antigen processing and presentation to the host immune system, which leads to the induction of a functional influenza virus-specific T cell response (29). In agreement with previous results, we observed that passively transferred anti-M2e monoclonal antibodies protected the mice against weight loss following PR8/NA-F85–93 infection and allowed the induction of an F85–93 epitope-specific CTL response, which correlated with reduced RSV lung viral load upon a subsequent RSV infection. Additionally, anti-M2e antibody treatment was associated with reduced infiltration of immune cells into the lungs after a PR8/NA-F85–93 infection. However, the formation of iBALT, a hallmark of influenza infection, was not affected. These tertiary lymphoid structures might contribute to protection, as they can initiate a localized immune response consisting of B and T cells (30, 34). A recent study demonstrated that iBALT induced by protein cage nanoparticles protects against multiple respiratory viruses, at least until 35 days after immunization with the nanoparticles (34). This report is the first to describe nonspecific protection evoked by iBALT. In our study, we confirmed the presence of iBALT 26 days after PR8/NA-F85–93 virus infection; however, we did not investigate whether the presence of iBALT persists until the moment of RSV infection, which is 51 days after the influenza A virus infection. If iBALT is still present upon RSV infection, this might contribute to the nonspecific protection observed upon PR8/NA-HA518–526 infection.
The body weight loss caused by the influenza vaccine vector might be prevented or reduced by administering the virus as a live attenuated influenza vaccine (LAIV). Since 2003, LAIV (FluMist) has been approved in the United States as an influenza vaccine for healthy persons aged 2 to 49 years (35). In Europe, the European Medicines Agency has approved Fluenz only for healthy individuals aged 2 to 18 years (36). Compared to an inactivated influenza vaccine, LAIV has the advantage of inducing an immune response that more closely resembles the immune response induced by natural infection (37). Besides a local mucosal immune response, the LAIV also induces a systemic cellular immune response (38). An alternative strategy for producing live attenuated influenza virus was recently described by Mueller and colleagues (39). Using a computer algorithm, Mueller and colleagues codon pair deoptimized the A/PR/8/34 PB1, NP and HA genes, while the wild-type protein sequence was not affected. This led to an in vivo attenuation of the virus in mice due to a less-than-optimal arrangement of codon pairs. Such deoptimized live attenuated viruses are highly unlikely to revert to their original virulence because that would involve hundreds of nucleotide mutations (39).
To adapt the vaccination strategy for humans, one or several human CTL epitopes have to be introduced into the influenza virus. It has been shown that up to 58 aa can be incorporated into the stalk of the neuraminidase (40). In this way, several human epitopes can be included in the NA to generate a vaccine that covers most human HLA types. Alternatively, large fragments can be introduced in the NS1/NS2 gene fragment. Manicassamy et al. reported the production of an influenza virus containing green fluorescent protein in the NS segment. This virus has the additional advantage of being attenuated in vivo (41).
In conclusion, we describe a novel vaccine approach against RSV that is based on the induction of a CTL immune response. Our results demonstrate that it is possible to take advantage of the infection-permissive protection of M2e-specific antibodies against influenza A virus to induce CD8+ T cell-mediated immunity by an influenza A virus vector expressing the RSV F85–93 epitope.
ACKNOWLEDGMENTS
We thank Anouk Smet and Tine Ysenbaert for excellent technical support and Amin Bredan for editing the manuscript. We thank St. Jude Children's Research Hospital for providing us the A/PR/8/34-based eight-plasmid system for generating recombinant influenza A viruses.
This work was supported by Agentschap voor Innovatie door Wetenschap en Techniek (IWT) grants 81050 and 83050 (to S.D.B.), by SBO grant 110038 (to K.R.), and by the Fonds Wetenschappelijk Onderzoek Vlaanderen (FWO-Vlaanderen) (to B.S and K.S.). The flow cytometer core facility at DMBR is supported by a Methusalem grant (BOF09/01M00709) from Ghent University.
Footnotes
Published ahead of print 9 January 2013
REFERENCES
- 1. Centers for Disease Control and Prevention 2010. Respiratory syncytial virus infection (RSV). Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/rsv/about/index.html [Google Scholar]
- 2. Glezen WP, Taber LH, Frank AL, Kasel JA. 1986. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 140:543–546 [DOI] [PubMed] [Google Scholar]
- 3. Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bacharier LB, Cohen R, Schweiger T, Yin-Declue H, Christie C, Zheng J, Schechtman KB, Strunk RC, Castro M. 2012. Determinants of asthma after severe respiratory syncytial virus bronchiolitis. J. Allergy Clin. Immunol. 130:91–100 e103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, Kjellman B. 2005. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care Med. 171:137–141 [DOI] [PubMed] [Google Scholar]
- 6. van Drunen Littel-van den Hurk S, Watkiss ER. 2012. Pathogenesis of respiratory syncytial virus. Curr. Opin. Virol. 2:300–305 [DOI] [PubMed] [Google Scholar]
- 7. Spann KM, Tran KC, Collins PL. 2005. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J. Virol. 79:5353–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Swedan S, Musiyenko A, Barik S. 2009. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J. Virol. 83:9682–9693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Munir S, Hillyer P, Le Nouen C, Buchholz UJ, Rabin RL, Collins PL, Bukreyev A. 2011. Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog. 7:e1001336 doi:10.1371/journal.ppat.1001336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kotelkin A, Belyakov IM, Yang L, Berzofsky JA, Collins PL, Bukreyev A. 2006. The NS2 protein of human respiratory syncytial virus suppresses the cytotoxic T-cell response as a consequence of suppressing the type I interferon response. J. Virol. 80:5958–5967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89:422–434 [DOI] [PubMed] [Google Scholar]
- 12. Olson MR, Varga SM. 2007. CD8 T cells inhibit respiratory syncytial virus (RSV) vaccine-enhanced disease. J. Immunol. 179:5415–5424 [DOI] [PubMed] [Google Scholar]
- 13. Graham BS. 2011. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 239:149–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Welliver TP, Garofalo RP, Hosakote Y, Hintz KH, Avendano L, Sanchez K, Velozo L, Jafri H, Chavez-Bueno S, Ogra PL, McKinney L, Reed JL, Welliver RC., Sr 2007. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J. Infect. Dis. 195:1126–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heidema J, Lukens MV, van Maren WW, van Dijk ME, Otten HG, van Vught AJ, van der Werff DB, van Gestel SJ, Semple MG, Smyth RL, Kimpen JL, van Bleek GM. 2007. CD8+ T cell responses in bronchoalveolar lavage fluid and peripheral blood mononuclear cells of infants with severe primary respiratory syncytial virus infections. J. Immunol. 179:8410–8417 [DOI] [PubMed] [Google Scholar]
- 16. Lukens MV, van de Pol AC, Coenjaerts FE, Jansen NJ, Kamp VM, Kimpen JL, Rossen JW, Ulfman LH, Tacke CE, Viveen MC, Koenderman L, Wolfs TF, van Bleek GM. 2010. A systemic neutrophil response precedes robust CD8(+) T-cell activation during natural respiratory syncytial virus infection in infants. J. Virol. 84:2374–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cannon MJ, Openshaw PJ, Askonas BA. 1988. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J. Exp. Med. 168:1163–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Graham BS, Bunton LA, Wright PF, Karzon DT. 1991. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Invest. 88:1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bueno SM, Gonzalez PA, Cautivo KM, Mora JE, Leiva ED, Tobar HE, Fennelly GJ, Eugenin EA, Jacobs WR, Jr, Riedel CA, Kalergis AM. 2008. Protective T cell immunity against respiratory syncytial virus is efficiently induced by recombinant BCG. Proc. Natl. Acad. Sci. U. S. A. 105:20822–20827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Srikiatkhachorn A, Braciale TJ. 1997. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J. Exp. Med. 186:421–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stevens WW, Sun J, Castillo JP, Braciale TJ. 2009. Pulmonary eosinophilia is attenuated by early responding CD8(+) memory T cells in a murine model of RSV vaccine-enhanced disease. Viral Immunol. 22:243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruckwardt TJ, Luongo C, Malloy AM, Liu J, Chen M, Collins PL, Graham BS. 2010. Responses against a subdominant CD8+ T cell epitope protect against immunopathology caused by a dominant epitope. J. Immunol. 185:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olson MR, Hartwig SM, Varga SM. 2008. The number of respiratory syncytial virus (RSV)-specific memory CD8 T cells in the lung is critical for their ability to inhibit RSV vaccine-enhanced pulmonary eosinophilia. J. Immunol. 181:7958–7968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kreijtz JH, Fouchier RA, Rimmelzwaan GF. 2011. Immune responses to influenza virus infection. Virus Res. 162:19–30 [DOI] [PubMed] [Google Scholar]
- 25. Hoffmann E, Krauss S, Perez D, Webby R, Webster RG. 2002. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 20:3165–3170 [DOI] [PubMed] [Google Scholar]
- 26. Bogaert P, Naessens T, De Koker S, Hennuy B, Hacha J, Smet M, Cataldo D, Di Valentin E, Piette J, Tournoy KG, Grooten J. 2011. Inflammatory signatures for eosinophilic vs. neutrophilic allergic pulmonary inflammation reveal critical regulatory checkpoints. Am. J. Physiol. Lung Cell. Mol. Physiol. 300:L679–L690 [DOI] [PubMed] [Google Scholar]
- 27. Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM, Fiers W. 1999. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat. Med. 5:1157–1163 [DOI] [PubMed] [Google Scholar]
- 28. El Bakkouri K, Descamps F, De Filette M, Smet A, Festjens E, Birkett A, Van Rooijen N, Verbeek S, Fiers W, Saelens X. 2011. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 186:1022–1031 [DOI] [PubMed] [Google Scholar]
- 29. Schotsaert M, Ysenbaert T, Neyt K, Ibanez LI, Bogaert P, Schepens B, Lambrecht BN, Fiers W, Saelens X. 18 July 2012, posting date Natural and long-lasting cellular immune responses against influenza in the M2e-immune host. Mucosal Immunol. (Epub ahead of print.) doi:10.1038/mi.2012.1069 [DOI] [PubMed] [Google Scholar]
- 30. Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, Woodland DL, Lund FE, Randall TD. 2004. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat. Med. 10:927–934 [DOI] [PubMed] [Google Scholar]
- 31. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, Jacobs C, van Loenhout J, de Jong D, Stunnenberg HG, Xavier RJ, van der Meer JW, van Crevel R, Netea MG. 2012. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. U. S. A. 109:17537–17542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goulding J, Snelgrove R, Saldana J, Didierlaurent A, Cavanagh M, Gwyer E, Wales J, Wissinger EL, Hussell T. 2007. Respiratory infections: do we ever recover? Proc. Am. Thorac. Soc. 4:618–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walzl G, Tafuro S, Moss P, Openshaw PJ, Hussell T. 2000. Influenza virus lung infection protects from respiratory syncytial virus-induced immunopathology. J. Exp. Med. 192:1317–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wiley JA, Richert LE, Swain SD, Harmsen A, Barnard DL, Randall TD, Jutila M, Douglas T, Broomell C, Young M. 2009. Inducible bronchus-associated lymphoid tissue elicited by a protein cage nanoparticle enhances protection in mice against diverse respiratory viruses. PLoS One 4:e7142 doi:10.1371/journal.pone.0007142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Centers for Disease Control and Prevention 2012. Influenza vaccine, live, intranasal; what you need to know. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/vaccines/pubs/vis/downloads/vis-flulive.pdf [Google Scholar]
- 36. European Medicines Agency 2012. Assessment report: FLUENZ. European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001101/WC500103711.pdf [Google Scholar]
- 37. Gasparini R, Amicizia D, Lai PL, Panatto D. 2011. Live attenuated influenza vaccine—a review. J. Prev. Med. Hyg. 52:95–101 [PubMed] [Google Scholar]
- 38. Cox RJ, Brokstad KA, Ogra P. 2004. Influenza virus: immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand. J. Immunol. 59:1–15 [DOI] [PubMed] [Google Scholar]
- 39. Mueller S, Coleman JR, Papamichail D, Ward CB, Nimnual A, Futcher B, Skiena S, Wimmer E. 2010. Live attenuated influenza virus vaccines by computer-aided rational design. Nat. Biotechnol. 28:723–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Castrucci MR, Hou S, Doherty PC, Kawaoka Y. 1994. Protection against lethal lymphocytic choriomeningitis virus (LCMV) infection by immunization of mice with an influenza virus containing an LCMV epitope recognized by cytotoxic T lymphocytes. J. Virol. 68:3486–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. U. S. A. 107:11531–11536 [DOI] [PMC free article] [PubMed] [Google Scholar]