

Abstract
The herpes simplex virus 1 (HSV-1) glycoprotein K (gK)/UL20 protein complex is incorporated into virion envelopes and cellular membranes and functions during virus entry and cell-to-cell spread. To investigate the role of gK/UL20 in the context of a highly neurovirulent virus strain, the HSV-1(McKrae) genome was cloned into a bacterial artificial chromosome plasmid (McKbac) and utilized to construct the mutant virus McK(gKΔ31-68), carrying a 37-amino-acid deletion within the gK amino terminus. The McKbac virus entered efficiently into Chinese hamster ovary (CHO) cells constitutively expressing HSV-1 human receptors, nectin-1, herpesvirus entry mediator (HVEM), or paired immunoglobulin-like type-2 receptor alpha (PILRα). In contrast, the McK(gKΔ31-68) virus failed to enter into CHO-PILRα cells, while it entered CHO cells expressing HVEM and nectin-1 more efficiently than the McKbac virus. Both McKbac and McK(gKΔ31-68) viruses entered all CHO cells expressing HSV-1 receptors via a pH-independent pathway. The HSV-1(F) gBΔ28syn mutant virus, encoding a carboxyl-terminal truncated gB, causes extensive cell fusion. Previously, we showed that the gKΔ31-68 amino acid deletion abrogated gBΔ28syn virus-induced cell fusion, indicating that the amino terminus of gK is required for gB-mediated virus-induced cell fusion (V. N. Chouljenko, A. V. Iyer, S. Chowdhury, D. V. Chouljenko, and K. G. J. Kousoulas, Virology 83:12301–12313, 2009). Surprisingly, the gKΔ31-68/gBΔ28syn virus caused extensive fusion of CHO-nectin-1 cells but limited cell fusion of CHO-PILRα cells. Coimmunoprecipitation experiments revealed that both gK and PILRα bound gB in infected cells. Collectively, these results indicate that the amino terminus of gK is functionally and physically associated with the gB-PILRα protein complex and regulates membrane fusion of the viral envelope with cellular membranes during virus entry as well as virus-induced cell-to-cell fusion.
INTRODUCTION
The herpes simplex virus 1 (HSV-1) entry mechanism is both complex and unique among enveloped viruses, involving multiple glycoproteins for attachment, binding, and membrane fusion (1). Viral glycoproteins interact with different cellular receptors to facilitate virus entry. Initial attachment of the virus to cellular membranes is mediated by interaction of glycoproteins gB and gC with glycosaminoglycan (GAG) moieties of cell surface proteoglycans (2, 3). Attachment of virions to cellular membranes facilitates subsequent binding of gD to cellular receptors, including the herpesvirus entry mediator (HVEM, also called HveA), nectin-1 (HveC), and 3-O-sulfated heparan sulfate (4–6). Apparently, gB can also bind to additional cellular receptors, including paired immunoglobulin-like type 2 receptor alpha (PILRα), nonmuscle myosin heavy chain IIA (NMHC-IIA), and myelin-associated glycoprotein (MAG) (7–9). Sequential binding of gD and then gB to their respective cellular receptors during virus entry and virus-induced cell-to-cell fusion is thought to alter gB's conformation, resulting in gB-mediated membrane fusion (1, 10–12).
HSV-1 can enter into cells by utilizing different cell-dependent pathways: (i) virus entry into Vero and HEp-2 cells is predominantly mediated via pH-independent fusion of the viral envelope with the host cell membrane (13); (ii) virus entry into HeLa and Chinese hamster ovary (CHO) cells expressing the nectin-1 gD receptor is predominantly achieved via receptor-mediated endocytosis, followed by pH-dependent fusion of the viral envelope with endocytic membranes (14); and (iii) virus entry into C10 murine melanoma cells predominantly occurs via pH-independent endocytosis (13). Recently, it has been suggested that gB-specific receptors, such as PILRα, or other cellular plasma membrane factors determine whether virions enter predominantly via fusion at the plasma membrane or via receptor-mediated endocytosis (pH dependent or pH independent), followed by fusion of the viral envelope with endosomal membranes (15).
HSV-1 gK has been shown to be involved in neurovirulence and immunomodulation (16, 17). We have shown that HSV-1 gK and UL20 functionally and physically interact, and this interaction is mandatory for their coordinated intracellular transport, cell surface expression, and functions in virion egress, virus-induced cell fusion, and virus entry (18–21). Recently, we showed that the amino terminus of gK interacts with gB and gH and can complement gB-mediated cell fusion (22, 23). Virions lacking the entire gK or the amino terminus of gK (amino acids [aa] 31 to 68) enter susceptible cells substantially slower than the wild-type virus (20, 21). Importantly, gK-null virions cannot infect neurons via their neuronal axons (24), an entry route known to involve fusion of the viral envelope with synaptic membranes of axonal neuronal termini (25). Overall, these results have strongly suggested that the gK/UL20 protein complex can regulate virus entry and virus-induced cell fusion via modulating gB and gH membrane fusion functions.
Here, we show that the gK amino terminus is necessary for efficient virus entry via the gB-specific receptor PILRα but not for entry via the gD-specific receptors nectin-1 and HVEM. Moreover, we show that PILRα forms a multiprotein complex with gB and gK but not gD. In contrast to previous reports (14), we show that HSV-1(McKrae) entry occurs via a pH-independent mechanism. These results suggest a functional and physical association of gK/UL20 with gB and gB-specific receptors, such as PILRα, during virus entry and virus-induced cell fusion.
MATERIALS AND METHODS
Cells and plasmids.
African green monkey kidney (Vero) cells were obtained from the American Type Culture Collection (Rockville, MD) and were grown and propagated in growth medium consisting of Dulbecco's modified Eagle medium (DMEM) (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) and antibiotics (22). VK302 cells permanently expressing gK were originally obtained from David Johnson (Oregon Health Sciences University, Portland, OR) and maintained in growth medium. CHO-nectin-1(human) cells were a gift from Richard Longnecker (Northwestern University, Chicago, IL) and were propagated in Ham's F12 growth medium supplemented with 10% FBS and 200 μg/ml G418. The CHO-PILRα (human) cells were also obtained from R. Longnecker and were grown in Ham's F12 medium supplemented with 10% FBS and 700 μg/ml G418. The human CHO-HVEM cells were created by using a PiggyBac Transposon system (System Biosciences) as described earlier (26). All cells were cultured in nonselective medium prior to use in the virus entry assay. 293 PEAK TM Rapid cells (a gift from R. Longnecker) were grown in DMEM supplemented with 10% FBS and antibiotics. Plasmids expressing the soluble human PILRα-IgG Fc hybrid protein (pME18S-PILRα-Ig) and CD200-Ig Fc fusion protein were kind gifts from Hishashi Arase (Osaka University, Osaka, Japan) and Y. Kawaguchi (University of Tokyo, Tokyo, Japan), respectively. Plasmids expressing the Ig fusion proteins were constructed as described previously (8).
Viruses.
The clinical ocular, neuroinvasive HSV-1(McKrae) strain was obtained from J. M. Hill (Louisiana State University Health Sciences Center, New Orleans, LA). The McKrae genome was cloned into the bacterial artificial chromosome (BAC) vector, which also contains an excisable enhanced green fluorescent protein (EGFP) expression cassette. This BAC was used to recover the McKrae-EGFP-BAC (McKbac) virus. The recombinant mutant virus HSV-1(F) YE102-VC1 specifies gK and UL20 tagged with V5 and 3× FLAG antigenic epitopes, respectively. GFP-tagged vesicular stomatitis virus (VSV-GFP) was a kind gift from John Rose (Yale University, New Haven, CT). The mutant viruses gKΔ31-68, gBΔ28syn (deletion of 28 aa from the carboxyl terminus of gB), and gKΔ31-68/gBΔ28syn were described earlier (22).
Construction of mutant viruses.
The HSV-1 McKrae (wild-type) strain was cloned into the BAC plasmid pBeloBAC11 (NEB) essentially as described previously (27), with the following modifications. Specifically, a gene construct was created wherein the LoxP recombination site was fused together with DNA fragments of approximately 1 kb of McKrae DNA sequence, encompassing portions of the UL3(L-left)-UL4(R-right) targeted genomic region by PCR-based overlap extension. This gene construct was subsequently cloned into the pCR2.1-TOPO plasmid vector (Invitrogen, Grand Island, NY). Similarly, another gene expression cassette containing the EGFP gene flanked by FLP recombination target (FRT) sites (enabling removal of the EGFP gene cassette) was constructed and cloned into pEF6/V5-His TOPO plasmid vector (Invitrogen) under the control of the EF1α promoter. This gene cassette was excised from the vector by using EcoRI endonuclease and blunt ended by T4 polymerase. Subsequently, it was cloned into the blunt-ended BamHI site of pBeloBAC11. Cre recombinase enzyme (New England BioLabs, Ipswich, MA) was used for in vitro reaction to fuse together the L-LoxP-R ampicillin-resistant PCR 2.1-based plasmid and LoxP-EGFP chloramphenicol-resistant pBeloBAC-based plasmid. The plasmid DNA created after fusion with Cre recombinase was used to transfect Vero cells using Lipofectamine 2000 (Invitrogen). The transfected cells were infected with HSV-1(McKrae) at 6 h posttransfection. Fluorescence microscopy was used to select fluorescent viral plaques that contained the inserted McKrae genome in the BAC plasmid (McKbac). The presence of the BAC plasmid was verified by PCR-assisted DNA sequencing. The recovered McKbac virus was utilized for the construction of gK mutant viruses. The mutant viruses gKΔ31-68 (encoding gK carrying an in-frame deletion of aa 31 to 68) and gKΔ31-117 (encoding gK carrying an in-frame deletion of aa 31 to 117) were created as described earlier for the HSV-1(F) genome cloned into BAC plasmid pYEbac102 (28).
Replication kinetics, plaque morphologies of McKbac and gK mutant viruses, and electron microscopy.
Viral plaques were visualized by immunohistochemistry as we have previously described (29–32). Analysis of one-step growth kinetics was performed as described earlier (20, 22, 33). Confluent Vero cell monolayers were infected with each virus at 4°C for 1 h at a multiplicity of infection (MOI) of either 0.1 or 2. Thereafter, plates were incubated at 37°C and 5% CO2 and virus was allowed to penetrate for 1 h at 37°C. Any remaining extracellular virus was inactivated by low-pH treatment (pH 3.0), and cells were subsequently incubated at 37°C and 5% CO2. Viral titers at different times postinfection were obtained on the VK302 cell line that expresses the HSV-1(KOS) gK gene. The ultrastructural morphology of virions within infected cells was examined by transmission electron microscopy as described previously (30, 32, 34–36). All infected cells were processed at 18 h postinfection (hpi) and visualized by transmission electron microscopy.
Virus entry assay.
Confluent monolayers of CHO-neo (not shown), CHO-HVEM, CHO-PILRα, CHO-nectin-1, and Vero cells were infected with McKbac, McK(gKΔ31-68), and McK(gKΔ31-117) at an MOI of 1 for 1 h at 34°C. The virus inoculum was subsequently removed, and the cultures were shifted to 37°C. Eight to 12 hpi, the cells were fixed, stained with anti-ICP4 antibody (Ab) (Virusys, Inc., Taneytown, MD) and Alexa Fluor 647 goat anti-mouse IgG1 (Life Technologies, Grand Island, NY), and analyzed by flow cytometry (fluorescent-activated cell sorting [FACS]). The relative efficiency of virus entry was calculated as the percentage of cells expressing ICP4 and normalized to CHO-neo entry values. Entry experiments were performed in triplicate, and error bars represent standard errors of the means. Comparison of groups was performed by one-way analysis of variance (ANOVA) using GraphPad Prism software (version 5) (21). A P value of ≤0.05 was considered significant.
Treatments with lysosomotropic agents.
Stock solutions of ammonium chloride (1.5 M) were prepared in distilled water immediately prior to use. Monensin (75 mM; Sigma, St. Louis, MO) and bafilomycin A1 (100 mM; Sigma) were dissolved in ethanol and dimethyl sulfoxide, respectively, and stored at −20°C. Growth medium was removed from cells and replaced by medium containing inhibitors, and the plate was incubated for 1 h at 37°C. After incubation, virus was added in the presence of the inhibitor for 1 h at 34°C. Subsequently, cells were incubated again in the presence of inhibitor until they were processed for flow cytometry.
Immunohistochemistry.
Nearly confluent CHO-neo, CHO-nectin-1, and CHO-PILRα cell monolayers were infected at an MOI of 1 with mutant viruses gKΔ31-68, gBΔ28syn, and gKΔ31-68/gBΔ28syn [all mutants were constructed in the HSV-1(F) genetic background], and HSV-1(F). Fourteen hpi, monolayers were washed with Tris-buffered saline (TBS)-Ca-Mg and fixed with ice-cold 100% methanol for 10 min. Cell fusion was visualized by immunostaining with anti-HSV-1 polyclonal antibody as described previously (22).
Preparation of purified Ig fusion proteins and immunoprecipitation assay.
293 PEAK Rapid cells were transfected with the plasmid pME18S-PILRα-Ig and a control plasmid expressing human CD200-Ig Fc fusion protein with 293 Fectin (Invitrogen, Grand Island, NY). Supernatants from transfected cells were collected at 48 and 72 h after transfection and pooled. Ig fusion proteins were purified by protein A affinity chromatography and were subsequently used for immunoprecipitation. Vero cells infected with the YE102-VC1 virus were lysed with NP-40 lysis buffer (Invitrogen). Subsequently, lysates were immunoprecipitated with PILRα-Ig or CD200-Ig (control) using protein A Dynabeads (Invitrogen). Immunoprecipitates were separated on 5 to 20% polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA), which were blotted with anti-gB, anti-gD, anti-FLAG, or anti-V5 Ab (21). Ig fusion proteins used for immunoprecipitation were detected by anti-human IgG Ab (Amersham Biosciences).
RESULTS
Construction and characterization of recombinant HSV-1(McKrae) viruses.
To facilitate the construction of recombinant viruses carrying specific mutations in gK and other genes in the context of a highly neurovirulent virus strain, the HSV-1(McKrae) genome was cloned into a bacterial artificial chromosome essentially as described previously (27). Specifically, the BAC plasmid was inserted within the intergenic region between the UL3 and UL4 genes. The BAC plasmid contained a gene cassette encoding the enhanced green fluorescence protein (EGFP) under the elongation factor 1 alpha (EF1a) (Fig. 1). The cloned McKrae genome (McKbac) was subsequently utilized for the construction of recombinant viruses carrying deletions of 37 (gKΔ31-68) and 86 (gKΔ31-117) amino acids using the two-step markerless red recombination mutagenesis system (28) (Fig. 1), as previously described for the construction of similar mutant viruses in the HSV-1(F) genomic background (22).
Fig 1.

Cloning of the HSV-1(McKrae) genome into a BAC and schematic representation of mutant viruses. Shown is a linear representation of the HSV-1 genome structure, with two unique regions (long-UL and short-US), each flanked by a pair of terminal and inverted repeats (TR and IR, respectively). An expanded region of the HSV-1 genome in the intergenic region between UL3 and UL4 is shown. L and R represent DNA fragments derived from the UL3 and UL4 genes, respectively, used for recombination purposes. The BAC containing an EGFP sequence was inserted between the intergenic region of UL3 and UL4 to create a recombinant virus McKrae BAC (McKbac). The McKbac virus was utilized for the construction of gK mutant viruses gKΔ31-68 (encoding gK carrying an in-frame deletion of aa 31 to 68) and gKΔ31-117 (encoding gK carrying an in-frame deletion of aa 31 to 117). An expanded genomic region of the UL53 gene which encodes glycoprotein K (gK), flanked by UL52 and UL54 genes, is shown.
The McKbac virus produced viral plaques on Vero cells that were similar to those of the HSV-1(McKrae) wild-type virus (not shown). The McK(gKΔ31-117) recombinant virus produced very small viral plaques containing an average of 2 to 7 cells per plaque. In contrast, the McK(gKΔ31-68) virus produced viral plaques that were much larger than those of the McK(gKΔ31-117) virus, approaching approximately half the size of the McKbac wild-type-like plaques. Infection of VK302 cells that express the HSV-1(KOS) gK gene under the gD promoter control (37) resulted in efficient complementation for plaque morphology and virus spread for the McK(gKΔ31-68) virus but not for the McK(gKΔ31-117) virus (Fig. 2I). These plaque phenotypes were consistent with similar plaque morphologies and cellular spread defects exhibited by the HSV-1(F) gKΔ31-117 and gKΔ31-68 mutant viruses with the following exceptions: (i) the McK(gKΔ31-68) virus produced partially syncytial plaques in VK302 cells (Fig. 2I, panel E), unlike the HSV-1(F) gKΔ31-68 virus characterized previously (22), and (ii) the McK(gKΔ31-117) virus was not efficiently complemented in VK302 cells (Fig. 2I, panel F), unlike the HSV-1(F) gKΔ31-117 virus (22). The apparent lack of complementation of the McK(gKΔ31-117) virus by VK302 cells was not due to potential mutations elsewhere in the viral genome, since a recombinant virus produced by rescuing the gKΔ31-117 deletion with homologous HSV-1(McKrae) gK sequence produced wild-type-like plaques (not shown). The McK(gKΔ31-68) mutant virus replicated efficiently in Vero cells, achieving viral titers of approximately 1 log less than the parental McKbac virus. In contrast, the McK(gKΔ31-117) virus replicated inefficiently, producing viral titers that were more than 3 logs less than that of the McKbac virus at both low MOI (0.1) and relatively high MOI (2) (Fig. 2II).
Fig 2.

Plaque morphology and replication kinetics of the McKbac and gK mutant viruses. (I) Plaque morphology. Vero cells were infected with each virus at an MOI of 0.001, and at 48 hpi viral plaques were fixed with methanol and stained with anti-HSV antibodies as described in Materials and Methods. Representative viral plaques of all gK mutant viruses and the McKbac virus are shown on both Vero (A to C) and VK302 (D to F) cells. (II) Replication kinetics. Vero cells were infected with each virus at either a low MOI (0.1) or a high MOI (2), and the numbers of infectious viruses produced were determined on VK302 cells at different times postinfection. Viral titers after high-MOI infection are shown in panel A, and low-MOI infections are shown in panel B. All experiments were performed in triplicate. Error bars represent standard errors of the means.
Ultrastructural characterization of McKbac and gK mutant viruses.
The ultrastructural phenotypes of all mutant viruses relative to the wild-type parental virus were investigated at 18 hpi by visually examining 50 randomly selected virus-infected Vero cells using transmission electron microscopy. The McKbac virus did not exhibit any apparent defects in cytoplasmic virion envelopment and egress. Fully enveloped virion particles were observed in both intracellular compartments as well as in extracellular spaces (Fig. 3). Similarly, mutant virus McK(gKΔ31-68) did not show any apparent defect in envelopment, and fully enveloped virion particles were excreted out of infected cells. Ultrastructural visualization of Vero cells infected with mutant virus McK(gKΔ31-117) showed severe defects in virion envelopment and egress, characterized by the presence of numerous unenveloped capsids in the cytoplasm. These results are in agreement with the ultrastructural morphologies of mutant viruses carrying the same gK mutations in the HSV-1(F) genetic background (22).
Fig 3.

Ultrastructural morphology of McKbac and gK mutant viruses. Electron micrographs of Vero cells infected at an MOI of 2 with McKbac (A), McK(gKΔ31-68) (B), and McK(gKΔ31-117) (C) and processed for electron microscopy at 18 hpi are shown. Nucleus (n), cytoplasm (c), and extracellular space (e) are marked. Arrows indicate virion particles.
Characterization of McKbac and gK mutant viral entry into Vero and CHO cells expressing different HSV-1 cellular receptors.
We have shown previously that HSV-1(F) recombinant viruses gKΔ31-68 and gKΔ31-117 exhibit slower kinetics of entry into Vero cells than their parental wild-type virus (21). To ascertain the relative efficiencies of virus entry into cells expressing different viral receptors, viruses were allowed to enter into cells for an hour at 34°C, and virus entry was detected by monitoring the level of ICP4 protein expression at 8 to 12 hpi as described previously (21) (also see Materials and Methods). Both gK mutant viruses entered into Vero cells, albeit with decreased efficiencies compared to the McKbac virus (Fig. 4A). These entry defects were particularly pronounced in PILRα-expressing CHO cells, in which both gK mutant viruses entered up to 8-fold less efficiently than the McKbac virus (Fig. 4B). In contrast, all viruses entered efficiently into both CHO-nectin-1 and CHO-HVEM cells, while gK mutant viruses appeared to enter more efficiently than the McKbac virus (Fig. 4C and D).
Fig 4.

Comparison of entry efficiencies of McKbac and gK mutant viruses. (A) Entry into Vero cells. (B) Entry into CHO-PILRα cells. (C) Entry into CHO-HVEM cells. (D) Entry into CHO-nectin-1 cells. All cells were infected with McKbac and the gK mutant viruses gKΔ31-68 and gKΔ31-117 at an MOI of 1. At 8 to 12 hpi, the cells were stained with anti-ICP4 antibody and analyzed by flow cytometry to determine the percentage of infected cells. Comparison of groups was performed by one-way ANOVA (see Materials and Methods). P ≤ 0.05 is considered significant. *, 0.01 < P ≤ 0.05; **, 0.001 <P ≤ 0.01; ***, P ≤ 0.001.
Effects of lysosomotropic agents on entry of McKbac and gK mutant viruses into different cell types.
It has been reported that HSV-1 enters into HeLa and CHO-nectin-1 cells via pH-dependent endocytosis (14). Lysomotropic inhibitors NH4Cl, monensin, and bafilomycin inhibit the acidification of endosomes. The effect of these inhibitors on virus entry was investigated and compared to virus entry of vesicular stomatitis virus (VSV), which is known to enter via pH-sensitive endocytosis (38). NH4Cl and monensin inhibited VSV entry into CHO-nectin-1 cells in a dose-dependent manner (Fig. 5C and D). However, the two inhibitors inhibited neither McKbac nor gK mutant virus entry (Fig. 5A and B). Similarly, NH4Cl, monensin, and bafilomycin did not inhibit McKbac virus entry into CHO-PILRα cells (Fig. 5E, F, and G).
Fig 5.

Effect of lysosomotropic agents on McKbac and gK mutant virus entry. Cells (CHO-nectin-1) were pretreated with different concentrations of ammonium chloride (A and C) and monensin (B and D) and infected with McKbac and McKbac gK mutants (A and B) or VSV (C and D) for 1 h at 34°C in the presence of the inhibitor. One hour after infection, the inoculum was removed and the cells were incubated in the presence of each inhibitor at 37°C until the cells were processed. The percentages of infected cells were determined after staining with anti-ICP4 antibody and analyzed by flow cytometry. Six hours after VSV infection, the percentage of GFP-positive cells were measured by flow cytometry. CHO-PILRα cells were pretreated with different concentrations of ammonium chloride (E), monensin (F), and bafilomycin (G), infected with McKbac, and processed for flow cytometry as described above.
Effects of gK mutations on the ability of gB to cause virus-induced cell fusion in CHO cells expressing gB- or gD-specific receptors.
To investigate the relationship between gB-induced cell fusion and specific viral receptors, the previously characterized set of viruses, HSV-1(F)gBΔ28syn, HSV-1(F)gKΔ31-68, and HSV-1(F)gKΔ31-68/gBΔ28syn, was utilized (22). Cells expressing different viral receptors were infected with these viruses, and the relative extent of cell-to-cell fusion was compared to that of the HSV-1(F) wild-type virus (Fig. 6). The gBΔ28syn virus caused extensive fusion in Vero (22), CHO-nectin-1, CHO-HVEM (not shown), and CHO-PILRα cells but not in CHO-neo cells. In contrast, gKΔ31-68/gBΔ28syn caused extensive cell fusion in both CHO-HVEM (not shown) and CHO-nectin-1 cells, limited fusion in CHO-PILRα cells (Fig. 6), and no fusion in Vero cells (reference 22 and data not shown).
Fig 6.

Visualization of relative extent of virus-induced cell fusion in CHO cells expressing different cellular receptors. CHO-nectin-1, CHO-PILRα, and CHO-neo cells were infected with HSV-1(F), gKΔ31-68, gBΔ28syn, and gKΔ31-68/gBΔ28syn. Fourteen hpi the cells were fixed with methanol and stained with anti-HSV antibody.
PILRα interacts with gB, gK, and UL20p in virus-infected cells.
gB interacts with the amino terminus of PILRα (8). Moreover, we have shown that the amino terminus of gK, as a free peptide of 82 amino acids, interacted with the amino terminus of gB and complemented gB-mediated cell fusion (23). To investigate whether PILRα interacted simultaneously with both gB and gK, the purified PILRα-Ig fusion protein was mixed with cellular extracts obtained from infected cells and immunoprecipitated using magnetic beads coated with protein A (see Materials and Methods). Immunoprecipitates were subsequently tested for the presence of gB, gD, gK, and UL20 using specific antibodies for each protein in Western immunoblotting. Detection of gK and UL20 was accomplished after infection of Vero cells with the YE102-VC1 virus, which expresses gK tagged with the V5 epitope and UL20 tagged with the 3× FLAG epitope (21). Virus-infected cell lysates were immunoprecipitated with soluble PILRα (Fig. 7A, C, E, G, and I) and control IgG fusion protein CD200 IgG (Fig. 7B, D, F, H, and J). PILRα-Ig fusion protein immunoprecipitated gB (Fig. 7C, lane 2), UL20p (Fig. 7E, lane 2), and glycoprotein K (Fig. 7G, lane 2) but not gD (Fig. 7A, lane 2). The control CD200 IgG fusion protein did not react with any of the viral proteins (Fig. 7).
Fig 7.
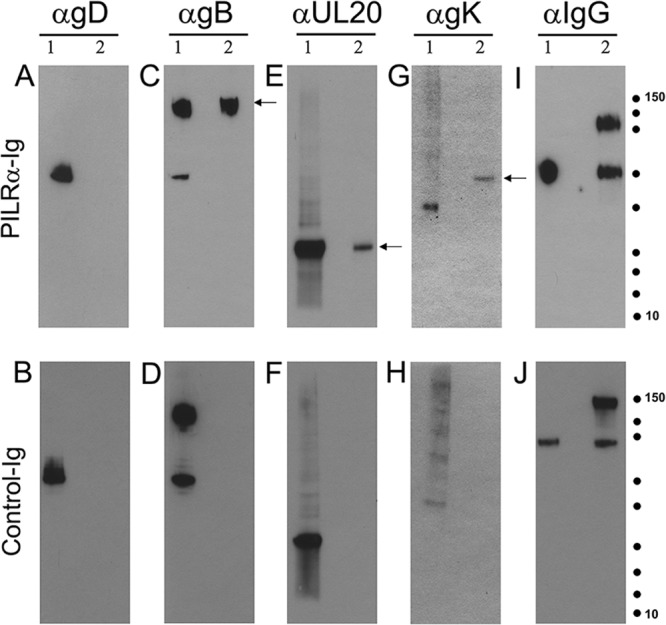
Soluble PILRα interacts with gB, gK, and UL20p in virus-infected cells. Western blot analysis of immunoprecipitates of PILRα ligand from HSV-1-infected cells is shown. Vero cells were infected with a double-tagged HSV-1 virus (YE102-VC1). (A to H) Infected lysates were immunoprecipitated with PILRα-Ig or CD200-Ig (control) and the immunoprecipitates were separated by SDS-PAGE, followed by blotting with anti-gB, anti-gD, anti-V5 (gK), and anti-FLAG (UL20) Ab. (I and J) The Ig fusion proteins used for immunoprecipitation were detected by anti-human IgG Ab. Molecular mass standards are shown as dots (150, 100, 75, 50, 37,25, 20, 15, and 10 kDa; Precision Plus protein standards; Bio-Rad). Lane 1, lysate from infected cells; lane 2, lysates immunoprecipitated with PILRα-Ig or CD200-Ig. Arrows indicate protein species of interest.
DISCUSSION
Glycoprotein K and UL20p are highly conserved among alphaherpesviruses, suggesting that they serve important functions that have been evolutionarily conserved (39). Both HSV-1 proteins have been intimately associated with membrane fusion phenomena, since mutations in either gK or UL20p cause extensive virus-induced cell fusion (31, 37, 40–45). Moreover, both gK and UL20p are structural components of the virion particle and function during virus entry (20, 21). Recent evidence suggests that gK and UL20p modulate these membrane fusion phenomena by direct interactions with viral gB (22, 23). Here, we have utilized CHO cells expressing individual viral receptors to demonstrate that gK is functionally and physically associated with the gB-PILRα interacting protein complex.
The McKrae viral strain was isolated from an ocular clinical sample of a patient suffering from acute infection (46). This viral strain is particularly virulent in mice and rabbits compared to other laboratory and clinical strains (47). Initial investigations suggested that the McKrae viral strain is able to utilize the PILRα receptor more efficiently than other viral strains (26). Therefore, the McKrae genome was cloned into a BAC plasmid to facilitate the construction of additional recombinant viruses with defined mutations in gK and UL20p. The McKbac virus appeared to have characteristics similar to those of the McKrae wild-type virus with regard to viral growth and plaque formation. Moreover, initial results suggest that insertion of the BAC plasmid did not substantially affect the neurovirulent characteristics of the parental McKrae virus in mice infected via the ocular route (not shown).
Recombinant McKbac viruses containing the gKΔ31-68 and gKΔ31-117 mutations appeared to replicate in a manner similar to that previously reported for identical mutations in the HSV-1(F) pYEbac102 genetic background. Plaque phenotypes, replication kinetics, and ultrastructure morphologies were generally similar irrespective of the viral genetic background (22), with the following differences. The McK(gKΔ31-68) virus caused cell fusion in the gK-complementing cell line VK302, while the HSV-1(F) gKΔ31-68 virus did not (22). Moreover, the HSV-1(F) gKΔ31-117 virus was efficiently complemented by VK302 cells (22), while the McK(gKΔ31-117) virus was not. Recently, we sequenced all major HSV-1(McKrae) genes encoding membrane proteins and glycoproteins (gB, gC, gD, gH, gL, gK, and UL20p) involved in membrane fusion and compared their primary structures to those of other viral strains. These results showed that the McKrae and F viral strains have identical gK and UL20 amino acid sequences; however, there are specific amino acid differences between their respective gB glycoproteins, predominantly located in their amino-terminal portions. Similarly, there are two amino acid differences between the gK expressed in VK302 cells (KOS genetic origin) and the gK amino acid sequences encoded by the McKrae and F strains at amino acid positions 101 (KOS-Thr to McKrae-Met) and 309 (KOS-Met to McKrae-Val) (26, 42). Therefore, these gB and gK amino acid differences may alter the known interactions of gK with gB (22, 23), resulting in the observed differential complementation of McK(gKΔ31-117) and F(gKΔ31-117) mutant viruses in VK302 cells.
Viral glycoproteins gB, gD, gH, and gL play important roles in mediating fusion of the viral envelope with cellular membranes and fusion of adjacent infected cells (1, 10). Historically, gK and UL20 have been intimately associated with virus-induced cell fusion because mutations in either gK or UL20 cause extensive cell fusion in a variety of cell types. A largely unexplained phenomenon is that gK and UL20 are absolutely required for virus-induced cell fusion even in the presence of highly fusogenic mutations in gB (22). Moreover, gK syncytial mutations cause extensive cell fusion of many cell types, unlike mutations in gB and UL20 that do not (48). In addition, mutants lacking gK or lacking a portion of the amino terminus of gK cause delayed entry kinetics in Vero cells, suggesting that gK and UL20 function similarly during cell-to-cell fusion and virus envelope-to-cell fusion. In support of these findings, we show here for the first time that gK functions in both virus entry and virus-induced cell fusion in conjunction with the PILRα receptor. Specifically, gK mutants were unable to enter into CHO-PILRα cells, while they efficiently entered CHO-nectin-1 and CHO-HVEM cells. This functional association between gK and the gB-specific receptor PILRα was also demonstrated by the inability of the gKΔ31-68/gBΔ28Syn virus to cause extensive fusion of CHO-PILRα cells, while both CHO-nectin-1 and CHO-HVEM cells were efficiently fused. Similarly, the previously reported inability of the gKΔ31-68/gBΔ28syn virus to cause fusion in Vero cells (22) may be due to other gB-specific receptors, since Vero cells do not express PILRα (S. Chowdhury and K. G. Kousoulas, unpublished data). Vero cells are known to express the nonmuscle myosin heavy chain IIA (NMHC-IIA) gB-specific receptor (7), which may function in a manner similar to that of PILRα, requiring gK for optimum gB-mediated membrane fusion.
HSV-1 virions can enter cells by fusion at the plasma membrane or by fusion with endosomal membranes in acidic or neutral endosomes after endocytosis (14). Previous work has indicated that HSV-1 enters via a pH-dependent endocytic pathway into CHO-nectin-1 cells (14). However, testing a variety of lysosomotropic inhibitors that prevent acidification of endosomes revealed that the McKbac virus entered into all cells in the presence of increasing concentrations of inhibitors, i.e., through a pH-independent entry mechanism. In contrast with our results, it has been suggested that HSV-1 enters into CHO-nectin-1 cells via a pH-dependent mechanism (14). We have found that the viability of cells treated with the various inhibitors should be carefully monitored to ensure that cell death does not affect virus entry experiments. Clearly, HSV-1 McKbac and gK mutant viruses enter into all cells via a pH-independent mechanism, unlike VSV. Additional studies are required to discern whether the different receptors influence virus entry via either fusion at the plasma membrane or fusion with endosomal membranes.
The amino terminus of gK was shown to physically and functionally interact with the amino terminus of gB (23). Here, we show that soluble PILRα was able to precipitate gB, gK, and UL20, suggesting that PILRα, gB, gK, and UL20 form a functional protein complex in infected cells. Based on the fact that PILRα binds to the amino terminus of gB, it is possible that gB binds both PILRα and the gK/UL20 protein complex independently of each other, utilizing distinct gB domains. PILRα may also bind gK/UL20 in the presence or absence of gB. These possibilities will be discerned in future experiments. The observed functional and physical association between gK/UL20 and the gB-specific receptor PILRα and potentially other gB-specific receptors, such as NMHCIIA, suggest that gK/UL20 is required for optimum regulation of the fusogenic properties of gB. Finally, our recent findings that gK-null virions do not infect neuronal axons (24, 49) suggest the potential involvement of gB-specific receptors in neuronal infections.
ACKNOWLEDGMENTS
This work was supported in part by NIH NIAID grant AI43000 to K.G.K. We acknowledge financial support by the LSU School of Veterinary Medicine to BIOMMED.
We acknowledge the technical assistance of Yulia Sokolova with electron microscopy and helpful discussions with Jason Walker and other BIOMMED staff.
Footnotes
Published ahead of print 9 January 2012
REFERENCES
- 1. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65:1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Investig. 108:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620 [DOI] [PubMed] [Google Scholar]
- 5. Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436 [DOI] [PubMed] [Google Scholar]
- 6. Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22 [DOI] [PubMed] [Google Scholar]
- 7. Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862 [DOI] [PubMed] [Google Scholar]
- 8. Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 107:866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 13. Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J. Virol. 83:4520–4527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J. Virol. 81:12962–12972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Osorio Y, Mott KR, Jabbar AM, Moreno A, Foster TP, Kousoulas KG, Ghiasi H. 2007. Epitope mapping of HSV-1 glycoprotein K (gK) reveals a T cell epitope located within the signal domain of gK. Virus Res. 128:71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Foster TP, Chouljenko VN, Kousoulas KG. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 82:6310–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foster TP, Melancon JM, Olivier TL, Kousoulas KG. 2004. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 78:13262–13277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Foster TP, Rybachuk GV, Kousoulas KG. 2001. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J. Virol. 75:12431–12438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jambunathan N, Chowdhury S, Subramanian R, Chouljenko VN, Walker JD, Kousoulas KG. 2011. Site-specific proteolytic cleavage of the amino terminus of herpes simplex virus glycoprotein K on virion particles inhibits virus entry. J. Virol. 85:12910–12918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chouljenko VN, Iyer AV, Chowdhury S, Chouljenko DV, Kousoulas KG. 2009. The amino terminus of herpes simplex virus type 1 glycoprotein K (gK) modulates gB-mediated virus-induced cell fusion and virion egress. J. Virol. 83:12301–12313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J. Virol. 84:8596–8606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. David AT, Baghian A, Foster TP, Chouljenko VN, Kousoulas KG. 2008. The herpes simplex virus type 1 (HSV-1) glycoprotein K(gK) is essential for viral corneal spread and neuroinvasiveness. Curr. Eye Res. 33:455–467 [DOI] [PubMed] [Google Scholar]
- 25. Aggarwal A, Miranda-Saksena M, Boadle RA, Kelly BJ, Diefenbach RJ, Alam W, Cunningham AL. 2012. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J. Virol. 86:6123–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chowdhury S, Naderi M, Chouljenko VN, Walker JD, Kousoulas KG. 2012. Amino acid differences in glycoproteins B (gB), C (gC), H (gH) and L(gL) are associated with enhanced herpes simplex virus type-1 (McKrae) entry via the paired immunoglobulin-like type-2 receptor. Virol. J. 9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 29. Fulmer PA, Melancon JM, Baines JD, Kousoulas KG. 2007. UL20 protein functions precede and are required for the UL11 functions of herpes simplex virus type 1 cytoplasmic virion envelopment. J. Virol. 81:3097–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee HC, Chouljenko VN, Chouljenko DV, Boudreaux MJ, Kousoulas KG. 2009. The herpes simplex virus type 1 glycoprotein D (gD) cytoplasmic terminus and full-length gE are not essential and do not function in a redundant manner for cytoplasmic virion envelopment and egress. J. Virol. 83:6115–6124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Melancon JM, Foster TP, Kousoulas KG. 2004. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J. Virol. 78:7329–7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Melancon JM, Luna RE, Foster TP, Kousoulas KG. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 79:299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Foster TP, Alvarez X, Kousoulas KG. 2003. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 77:499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foster TP, Kousoulas KG. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 73:8457–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foster TP, Melancon JM, Baines JD, Kousoulas KG. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 78:5347–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jayachandra S, Baghian A, Kousoulas KG. 1997. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 71:5012–5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hutchinson L, Goldsmith K, Snoddy D, Ghosh H, Graham FL, Johnson DC. 1992. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J. Virol. 66:5603–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Le Blanc I, Luyet PP, Pons V, Ferguson C, Emans N, Petiot A, Mayran N, Demaurex N, Faure J, Sadoul R, Parton RG, Gruenberg J. 2005. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 7:653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dietz P, Klupp BG, Fuchs W, Kollner B, Weiland E, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein K requires the UL20 gene product for processing. J. Virol. 74:5083–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baines JD, Ward PL, Campadelli-Fiume G, Roizman B. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 65:6414–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bond VC, Person S. 1984. Fine structure physical map locations of alterations that affect cell fusion in herpes simplex virus type 1. Virology 132:368–376 [DOI] [PubMed] [Google Scholar]
- 42. Debroy C, Pederson N, Person S. 1985. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 145:36–48 [DOI] [PubMed] [Google Scholar]
- 43. Pogue-Geile KL, Lee GT, Shapira SK, Spear PG. 1984. Fine mapping of mutations in the fusion-inducing MP strain of herpes simplex virus type 1. Virology 136:100–109 [DOI] [PubMed] [Google Scholar]
- 44. Pogue-Geile KL, Spear PG. 1987. The single base pair substitution responsible for the syn phenotype of herpes simplex virus type 1, strain MP. Virology 157:67–74 [DOI] [PubMed] [Google Scholar]
- 45. Ruyechan WT, Morse LS, Knipe DM, Roizman B. 1979. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J. Virol. 29:677–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kaufman HE, Ellison ED, Waltman SR. 1969. Double-stranded RNA, an interferon inducer, in herpes simplex keratitis. Am. J. Ophthalmol. 68:486–491 [DOI] [PubMed] [Google Scholar]
- 47. Halford WP, Balliet JW, Gebhardt BM. 2004. Re-evaluating natural resistance to herpes simplex virus type 1. J. Virol. 78:10086–10095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bzik DJ, Person S. 1981. Dependence of herpes simplex virus type 1-induced cell fusion on cell type. Virology 110:35–42 [DOI] [PubMed] [Google Scholar]
- 49. David AT, Saied A, Charles A, Subramanian R, Chouljenko VN, Kousoulas KG. 2012. A herpes simplex virus 1 (McKrae) mutant lacking the glycoprotein K gene is unable to infect via neuronal axons and egress from neuronal cell bodies. mBio 3:e00144–00112 doi:10.1128/mBio.00144-12 [DOI] [PMC free article] [PubMed] [Google Scholar]