

Abstract
Capsid (core) assembly is essential for hepatitis B virus (HBV) replication. We hypothesize that assembly kinetics and stability are tuned for optimal viral replication, not maximal assembly. Assembly effectors (AEfs) are small molecules proposed to disrupt this balance by inappropriately enhancing core assembly. Guided by the structure of an AEf-bound core, we designed a structural mimic of AEf-bound core protein, the V124W mutant. In biochemical studies, the V124W mutant recapitulated the effects of AEfs, with fast assembly kinetics and a strong protein-protein association energy. Also, the mutant was resistant to exogenous AEfs. In cell culture, the V124W mutant behaved like a potent AEf: expression of HBV carrying the V124W mutant was defective for genome replication. Critically, the V124W mutant interfered with replication of wild-type HBV in a dose-dependent manner, mimicking AEf activity. In addition, the V124W mutant was shown to adopt a more compact conformation than that of the wild type, confirming the allosteric regulation in capsid assembly. These studies show that the heteroaryldihydropyrimidine (HAP) binding pocket is a promiscuous target for inducing assembly. Suppression of viral replication by the V124W mutant suggests that mutations that fill the HAP site are not a path for HBV to escape from AEfs.
INTRODUCTION
Hepatitis B virus (HBV) chronically infects about 350 million people worldwide, resulting in ∼600,000 deaths per year (1, 2). Current treatments for HBV are unsatisfactory. The existing vaccine is preventive but not therapeutic. Interferon treatment has a low response rate and adverse effects (3). HBV reverse transcriptase inhibitors, which are nucleos(t)ide analogs, effectively suppress virus replication but are not curative, and they can select for drug-resistant mutants (3, 4), which can lead to viral rebound in long-term treatment. There is thus a tremendous unmet need for new antiviral targets.
HBV is an enveloped virus with an icosahedral, double-stranded DNA (dsDNA)-containing core (5, 6). The HBV core has a protein coat, or capsid, most often composed of 120 core protein (Cp) homodimers arranged with T=4 icosahedral symmetry. A small fraction of HBV cores have capsids with T=3 symmetry (7–10). In the cytoplasm, virion formation begins with packaging of a viral pregenomic RNA (pgRNA)-reverse transcriptase complex during capsid assembly. Inside the capsid, pgRNA is reverse transcribed into the partially double-stranded, relaxed-circular DNA (rcDNA) genome. The rcDNA-containing core can acquire an envelope and be secreted or directed to the nucleus (5).
The HBV capsid is thus an immensely complicated molecular machine. It must be able to self-assemble, constrain a dsDNA molecule with a bending energy large enough to affect capsid integrity (11), interact with its environment, and then disassemble to release its genome. Each step must be performed at the right time. To describe self-assembly, in vitro studies have focused on Cp's assembly domain (the first 149 residues of Cp, typically purified from an Escherichia coli expression system as a stable homodimer), which spontaneously assembles into capsids as a function of solution conditions (12). In vitro assembly is initiated by a nucleation event followed by a rapid elongation phase (13–15). Assembly is regulated by allosteric changes in Cp structure (presumed to control nucleation [16]), and likely by one or more chaperones (17). The dimer-dimer association that holds capsids together is based on remarkably weak hydrophobic interactions. This lack of thermodynamic stability is likely to be critical for capsid assembly and function (12). Very strong association energy can lead to trapping of partial particles and to particles with structural defects (18–20). Weak association energy could lead to particles that are too fragile to constrain HBV's dsDNA genome (11). Capsid structure and stability are also believed to affect virus intracellular trafficking and virion secretion (17, 21–23) or, as in HIV, uncoating (24).
We have shown that in vitro formation of HBV capsids from Cp can be disrupted by assembly effectors (AEfs), such as heteroaryldihydropyrimidines (HAPs) and phenylpropenamides (18, 19, 20, 25, 26). These molecules were originally discovered in cell culture-based screens for nonnucleoside HBV replication inhibitors (27–31). Their activity was later connected to HBV assembly by biochemical studies based on purified proteins (18–20, 26, 32). Their antiviral effect correlates with their ability to enhance assembly kinetics (18, 19, 26). A low-resolution (5 Å) structure of an HBV capsid cocrystallized with HAP1 suggested that the putative HAP binding site was a hydrophobic pocket at the dimer-dimer interface (25). The HAPs were also proposed to stabilize the free Cp dimer in its assembly-active conformation (16).
Here we designed a HAP-insensitive HBV Cp mutant (the V124W mutant), connected physical chemical studies with biological activities, and investigated the antiviral potential of disrupting capsid assembly. Compared to wild-type (WT) Cp, the V124W mutant assembled faster, had stronger association energy, and was highly resistant to HAPs. The V124W mutant had a HAP-like effect on wild-type capsid assembly in vitro and interfered with wild-type viral replication in cell culture.
MATERIALS AND METHODS
Cloning of Cp149-V124W and core protein purification.
The HBV Cp149-pET11c (the Swiss Protein Database accession code for full-length Cp is P03147) construct was mutated to Cp149-V124W by use of a QuikChange mutagenesis kit (Stratagene). The V124W protein was expressed in E. coli BL21(DE3) in Superior broth (Athena Enzyme Systems) with 50 μg/ml carbenicillin at 37°C overnight. V124W protein purification was performed as previously described for HBV Cp149-WT purification (33). Based on the biochemical studies in this paper, in the reassembly step, use of 50 mM NaCl instead of 500 mM NaCl to induce reassembly yielded more capsids with a slightly more active assembly activity. Protein concentration was determined using an extinction coefficient of 70,025 M−1 cm−1 per V124W mutant dimer at 280 nm, calculated by the ExPASy Proteomics Server based on the protein sequence and assuming one disulfide bond per dimer. WT dimers were purified as previously described (33). The extinction coefficient of WT dimers is 60,900 M−1 cm−1 at 280 nm. The standard assembly buffer was 50 mM HEPES, pH 7.5, at 23°C, with various NaCl concentrations. The protein stock was treated with 1% to 5% β-mercaptoethanol for 20 min before assembly.
90° light scattering.
Light scattering was monitored at 90° using a 400-nm excitation and emission wavelength with a Photon Technology International fluorometer at 23°C. WT or V124W mutant dimers (10 μM) were incubated with or without 20 μM HAP12 for 20 min prior to adding an equal volume of 2× NaCl to a final concentration of 50 mM NaCl. Each replicate was repeated 3 or 4 times independently.
Size-exclusion chromatography (SEC) and calculation of thermodynamic parameters.
The WT and V124W mutant proteins at various concentrations (WT, 40 to 80 μM; V124W mutant, 2.5 to 30 μM) were induced to assemble in 100 mM NaCl to equilibration for 72 h or longer at 23°C. We judged that 72 h of incubation was sufficient for equilibration, as longer incubations did not yield more capsid. Capsid and dimer fractions were separated in a 21-ml Superose 6 column (GE Life Sciences). ΔGcontact and the apparent dissociation constant (KD,apparent) were calculated as previously described (12). KD,apparent was the average of the equilibrium dimer concentrations, which were nearly constant.
HAP12 titration of capsid assembly.
To test for resistance to HAP molecules, 10 μM dimer was incubated with HAP12 (2.5 to 60 μM) at 23°C for 20 min and then was induced to assemble at 50 mM NaCl to equilibration for 24 h. Under these conditions, 24 h was long enough to allow reactions to reach equilibrium. Capsid, abnormal structure, and dimer fractions were determined by SEC as described above. HAP12 absorbance and light scattering were subtracted in the calculation of WT aberrant structures and dimer concentrations (34).
Transmission electron microscopy (TEM).
Samples from the light-scattering experiment were diluted, applied to glow-discharged carbon copper grids, and negatively stained with either 2% uranyl acetate or 1% ammonium molybdate. Micrographs were taken at a nominal magnification of ×30,000 on a 4K × 4K charge-coupled device (CCD) camera, using a JEOL-1010 transmission electron microscope.
Cell culture and transfections.
All HBV plasmids used in cell culture experiments were derived from subtype ayw (GenBank accession no. V01460) (35). These plasmids express pgRNA under the control of the cytomegalovirus (CMV) immediate-early promoter. Plasmids WTP+C+ and V124WP+C+ express pgRNA, reverse transcriptase (P), WT or V124W core (C), and X proteins but do not express envelope proteins (36). Plasmids CWT and CV124W express only the core protein, not other viral proteins (37), and lack a functional encapsidation signal (ε). Green fluorescent protein (GFP) was expressed from a separate plasmid. Details of the construction and sequence of any plasmid will be provided upon request.
The human hepatoma cell line Huh7 was cultured pre- and posttransfection as previously described (38). Transfections were performed using calcium phosphate precipitation. For experiments using WTP+C+ and V124WP+C+, 5 μg of total plasmid mass was transfected. For V124W mutant titration experiments, 2 μg WTP+C+ and various amounts of CV124W (0 to 8 μg) were used. A filler plasmid, pCMV-Sport6, was used to keep the total plasmid mass at 10 μg. A total of 0.125 μg of the GFP expression plasmid was used in every transfection mix.
Southern blot analysis of encapsidated DNA.
Nucleic acids from capsids in a fraction of cytoplasmic lysate were isolated as described previously (38). Viral DNA was electrophoresed through a 1.25% agarose gel. DNA was denatured and neutralized in situ in 0.5 N NaOH-1.5 M NaCl and 1 M Tris-1.5 M NaCl, respectively, followed by capillary transfer to Hybond-N (GE Life Sciences) in 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). Viral DNA on the membrane was detected by use of a pool of oligonucleotides that detected minus-strand DNA. Ten picomoles of this pool was labeled at the 5′ end with [γ-32P]ATP, using T4 polynucleotide kinase (NEB), and added to the membrane in Church hybridization buffer. Hybridization was performed overnight at 48°C. Membranes were washed in Church wash buffer at room temperature. Autoradiography was performed using a Typhoon 8600 PhosphorImager (Molecular Dynamics). Levels of DNA were measured using ImageQuant 5.2 software (Molecular Dynamics). DNA levels were normalized to GFP levels as determined previously by Western blotting.
Western blot analysis of core protein and GFP.
A fraction of cytoplasmic lysate was used for 15% SDS-PAGE, followed by transfer of proteins to a polyvinylidene difluoride (PVDF-FL) membrane (Millipore) in methanol transfer buffer. Blocking was done in Li-Cor blocking buffer diluted 1:1 with 1× phosphate-buffered saline (PBS). Antibody preparations were made in the same solution, with the addition of Tween 20 to a final concentration of 0.2%. Core protein was detected using a rabbit anti-core antibody (Austral Biologicals) at a dilution of 1:500 and a goat anti-rabbit–IRDye800CW antibody (Li-Cor) at a dilution of 1:10,000. GFP was detected using a mouse anti-GFP antibody (Santa Cruz Biotechnology) at a dilution of 1:500 and a goat anti-mouse–IRDye680LT antibody (Li-Cor) at a dilution of 1:10,000. Imaging of membranes was performed on a Li-Cor Odyssey instrument. Core levels were normalized to GFP levels for quantitation. Student's t test was used to test the statistical difference of the core levels in WT and V124W mutant transfections (n = 6).
RESULTS
Design of Cp149-V124W.
The thermodynamic stability of a capsid depends on the hydrophobic interactions at the dimer-dimer interface; eliminating hydrophobic interactions was shown to weaken the HBV core protein's association energy (16, 39). HAP molecules were proposed to strengthen the association energy by filling a gap at the dimer-dimer interface, the so-called HAP pocket, thus increasing the buried hydrophobic surface (25). Residue V124 formed part of the wall of the HAP pocket but did not contribute directly to protein-protein interactions. In the HBV HAP structure, bound HAP1 made hydrophobic contacts with both walls of the pocket. V124 was in direct contact with HAP1, presumably contributing to HAP1's affinity for the pocket. To design a HAP-insensitive HBV core protein mutant, we chose to fill the putative HAP binding site with an amino acid with a large hydrophobic side chain (Fig. 1) (Protein Data Bank [PDB] accession no. 2G34).
Fig 1.
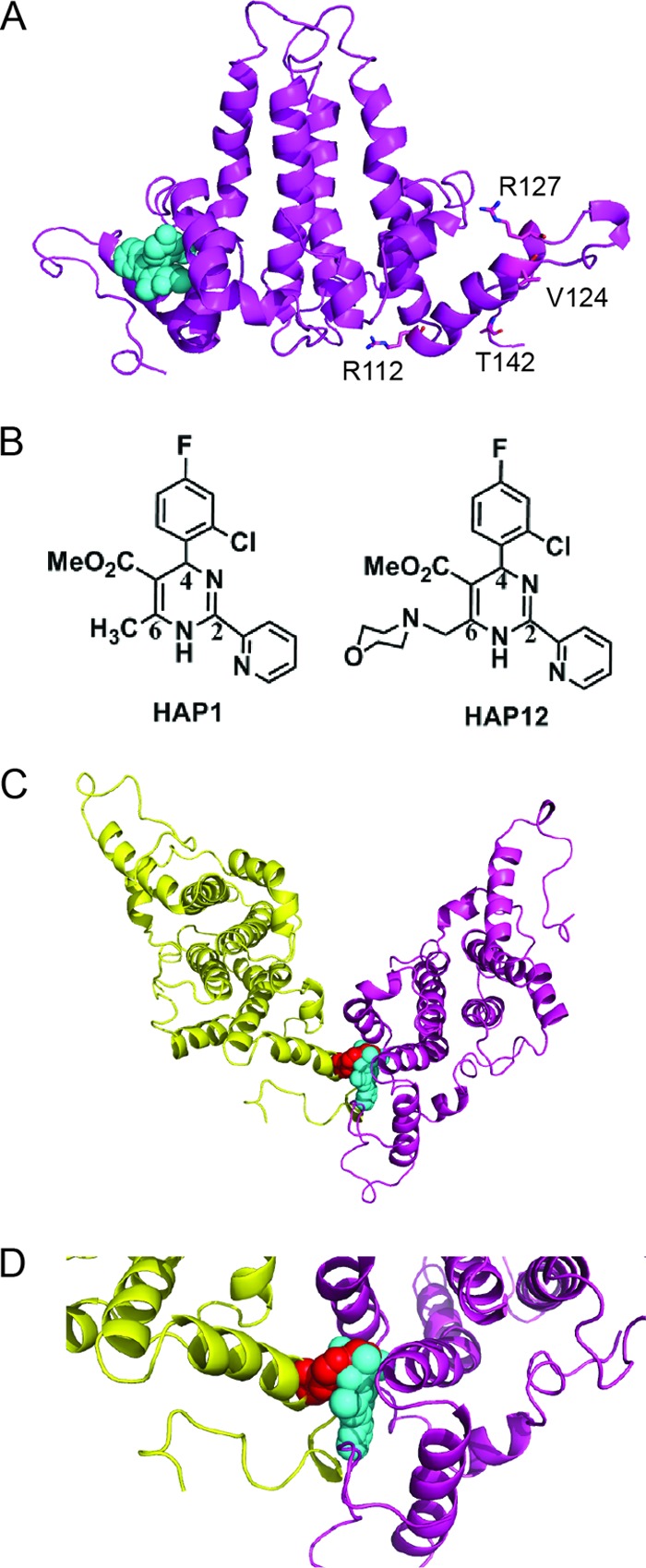
Model of Cp149-V124W dimer structure showing that the V124W mutant overlaps the HAP binding site. (A) Side view of the HBV Cp149-WT ribbon structure with bound HAP1 (cyan spheres) (PDB accession no. 2G34). Residues R112, R127, V124, and T142 are shown as sticks for reference points. (B) Structures of HAP1 and HAP12. HAP12 has an additional six-member ring linked to the methyl group at position 6. (C) Model of Cp149-V124W, generated based on Cp149-WT (PDB accession no. 2G34) with WinCoot by mutating V124 of the core protein D subunit to tryptophan. The selected rotamer accounts for 32% of the observed tryptophan rotamers. Viewed from the capsid interior, the HAP1 binding site is seen at the dimer-dimer interface. The V124W mutation (red spheres) overlaps HAP1 and partially fills the HAP binding site. The two dimers forming the site are colored yellow and magenta. (D) Close-up view of the HAP binding site at the V124W dimer-dimer interface.
Because the indole ring of tryptophan is large and relatively hydrophobic, we hypothesized that replacing V124 with tryptophan would mimic a HAP-bound wild-type core protein. In a structural model, the V124W mutant overlapped HAP1 in its predicted binding site (Fig. 1). Also, the V124W indole formed new contacts between subunits, suggesting that it would strengthen hydrophobic interactions in much the same way as a HAP molecule. Thus, the V124W mutant was predicted to resemble a HAP-bound core protein and to be insensitive to HAPs.
All in vitro studies described in this paper were based on the core protein assembly domain, Cp149, and the Cp149-V124W mutant. For our in vitro studies, the dimeric forms of these proteins are referred to as the WT for wild-type Cp149 and the V124W mutant for Cp149-V124W. In cell culture studies, the full-length core proteins, including the RNA-binding C terminus, were used.
The V124W mutant assembled faster and further than the WT in vitro.
Capsid assembly in vitro can be described in terms of the kinetics of capsid formation, thermodynamic stability of capsids, and the morphology of assembly products. We compared the assembly kinetics of the V124W mutant and the WT by using 90° light scattering. This approach is sensitive and informative, as typical HBV assembly reactions are dominated by free dimers and complete capsids, with very low concentrations of intermediates (15). We found that 10 μM V124W mutant assembled into capsids in 50 mM HEPES, pH 7.5, with 50 mM NaCl at 23°C (Fig. 2A). These conditions were chosen to highlight V124W mutant assembly; these conditions did not lead to detectable assembly of the WT (see below). In higher-ionic-strength buffers, typically used for the WT, V124W mutant assembly was faster than the dead time of our hand-mixed reactions. The difference in assembly kinetics indicated that the V124W mutant was more assembly active (i.e., able to nucleate assembly) than the WT.
Fig 2.
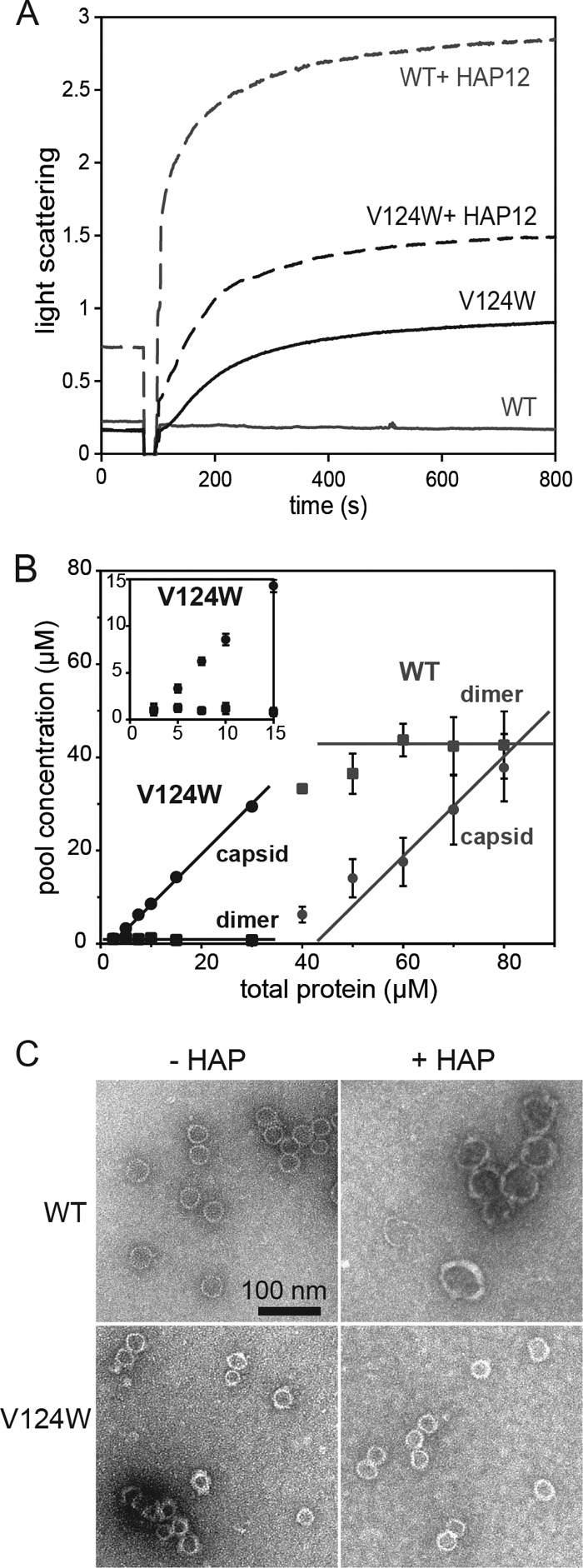
The V124W mutant is more assembly active than the WT. (A) Assembly kinetics as monitored by 90° light scattering. At 50 mM NaCl and 23°C, the WT did not assemble without HAP12. HAP12 profoundly sped up WT assembly. The V124W mutant assembled rapidly under the same conditions, but HAP12 increased V124W mutant assembly kinetics only 2-fold. Each light-scattering trace shows the average for three or four independent experiments. Light scattering is reported in relative units. (B) V124W and WT dimers at different initial concentrations were assembled and equilibrated at 100 mM NaCl, 72 h, and 23°C. Capsids and dimers were separated by SEC and quantified in terms of the dimer concentration in each pool. The pseudocritical concentration was about 43 μM for WT and about 1 μM for the V124W mutant, a 40-fold difference. The inset shows a close view of V124W mutant data at concentrations below 15 μM. Each point was repeated three times for the WT and four times for the V124W mutant. (C) Negatively stained electron micrographs of WT and V124W mutant assembly products. (Top left) 10 μM WT, 500 mM NaCl, no HAP12. (Top right) 10 μM WT, 50 mM NaCl, 20 μM HAP12. (Bottom left) 10 μM V124W mutant, 50 mM NaCl, no HAP12. (Bottom right) 10 μM V124W mutant, 50 mM NaCl, 20 μM HAP12. HBV T=4 capsids are 35 nm in diameter. T=3 capsids are 31 nm in diameter. The V124W mutant assembled into capsid-like particles in the absence and presence of HAP12. The WT assembled into aberrant structures, which were much larger than normal capsids, in the presence of HAP12. The scale bar applies to all images.
Capsid stability and the association energy of dimer-dimer interactions can readily be determined from their pseudocritical concentrations (the concentrations of free dimers at equilibrium, equivalent to the apparent dissociation constant [KD,apparent]) (12). We compared equilibrated WT and V124W mutant assembly in 100 mM NaCl at 23°C by SEC. SEC separates solutes based on their Stokes radii. At all ionic strengths tested, much higher concentrations of the WT than the V124W mutant were required for detectable assembly. An assembly isotherm, a plot of the equilibrium concentrations of dimers and capsids (in terms of the concentration of dimers in each pool) at constant temperature, showed that when the total protein concentration was above the pseudocritical concentration, the concentration of free dimers was constant (Fig. 2B). The constant free dimer concentration is diagnostic for a capsid assembly reaction at equilibrium (40). To obtain a robust capsid assembly reaction, the total protein concentration must exceed the pseudocritical concentration. The KD,apparent for the WT was 43.28 ± 4.97 μM, and the KD,apparent for the V124W mutant was 0.99 ± 0.42 μM, a 40-fold difference (Table 1). At 150 mM NaCl, where the WT has a KD,apparent of 14 μM (12), the value for the V124W mutant was too low to measure accurately.
Table 1.
Thermodynamics of HBV Cp149-WT and Cp149-V124W assembly at 100 mM NaCl and 23°C
| Parameter | Valuea |
|
|---|---|---|
| WT | V124W mutant | |
| ΔGcontact (kcal/mol) | −2.74 ± 0.04 | −3.84 ± 0.14 |
| KD,apparent (μM) | 43.28 ± 4.97 | 0.99 ± 0.42 |
Values shown are means ± standard deviations based on 10 to 24 independent measurements (n = 16 for WT ΔGcontact, n = 24 for V124W mutant ΔGcontact, n = 10 for WT KD,apparent, and n = 24 for V124W mutant KD,apparent).
From the amount of free dimer (KD,apparent), we calculated the pairwise dimer-dimer association energy (ΔGcontact) (12, 14) (Table 1). V124W dimer-dimer interactions were more stable than those of the WT by −1.1 kcal/mol, similar to the stabilizing effect of HAPs and phenylpropenamides (18, 19). Considering the weak interaction driving WT HBV capsid assembly (−3.87 kcal/mol at 37°C and 150 mM NaCl [18]), a 1.1-kcal/mol increase in the association energy, multiplied by the 240 contacts in a T=4 capsid, is substantial. Strong association energy can trap a misassembled V124W mutant, leading to defects and aberrant noncapsid polymers. We used low ionic strength and 23°C instead of physiological ionic strength and temperature in our studies, specifically to minimize such kinetic traps in our studies.
To ascertain assembly product morphology, we examined V124W assembly products by TEM (Fig. 2C). Though the V124W mutant was shown to mimic the HAP-bound WT with fast kinetics and strong association energy, we observed only capsid-like particles in V124W mutant assembly reactions. No aberrant structures were observed for V124W mutant assembly at 50 mM NaCl. As such, the V124W mutant more closely mimicked the behavior of the phenylpropenamides. Phenylpropenamides belong to another class of AEfs, which speed up assembly kinetics but do not induce formation of aberrant structures. In cell culture, phenylpropenamides inhibit pgRNA packaging, probably by disrupting the timing of assembly (19, 41).
Comparison of the V124W mutant based on assembly kinetics and SEC results yielded an important observation. The fast kinetics of the V124W mutant met with our prediction that the V124W mutant would behave like the HAP-bound or phenylpropenamide-bound WT (Fig. 2A) (19, 20). The mechanism of assembly acceleration by HAPs and phenylpropenamides led to the hypothesis that small-molecule AEfs transiently stabilize an assembly-active conformation of the core protein. We observed evidence of such a conformation of the V124W mutant by SEC (Fig. 3B; see below). WT dimers eluted at 18 ml on a 21-ml Superose 6 column; V124W dimers eluted at 18.5 ml, reproducibly later. The slower migration of V124W dimers indicated that the mutant had a smaller Stokes radius and was more compact than the WT, i.e., it had a different conformation or a shift in the constellation of available conformations.
Fig 3.
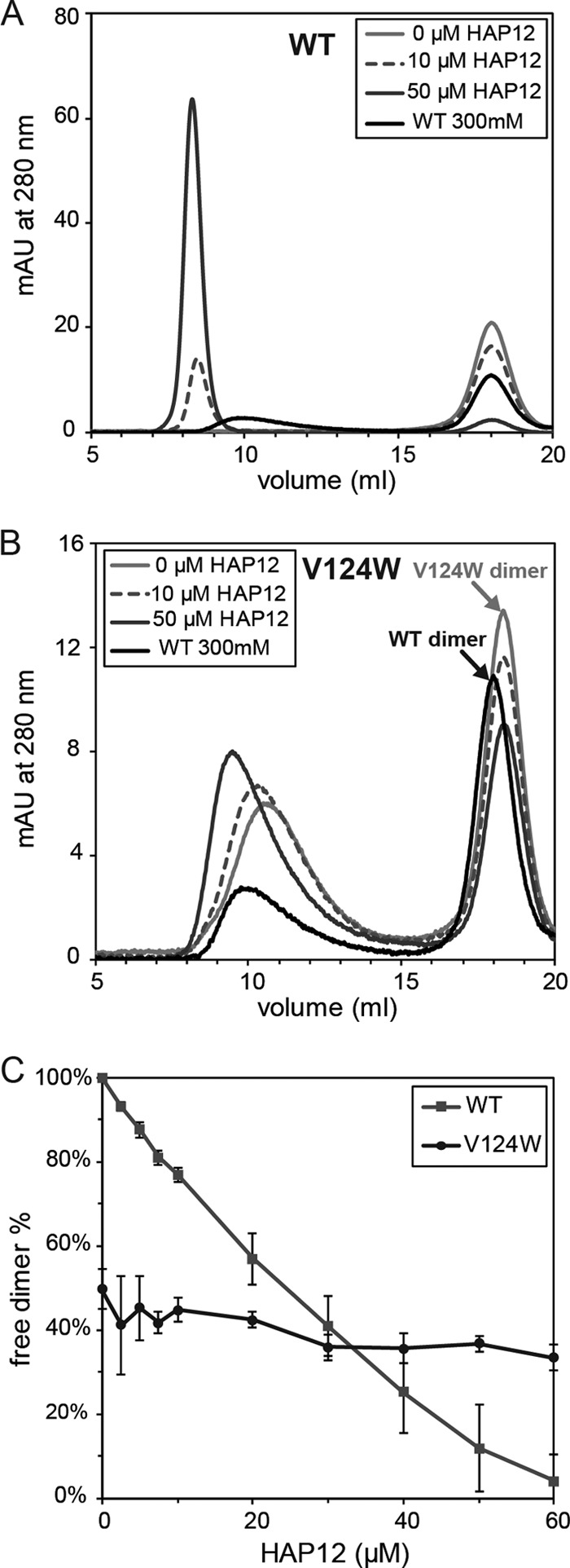
Effects of HAP12 on V124W mutant and WT assembly. (A) Representative chromatograms of 10 μM WT assembly with HAP12. A control chromatogram of 10 μM WT assembly at 300 mM NaCl without HAP12 is shown by the black lines in both panels A and B to indicate the WT capsid and dimer positions. The void volume (V0) for the 21-ml Superose 6 column was 7 ml. WT capsid eluted at 10 ml, WT dimers eluted at 18 ml, and small molecules, including HAP12 and β-mercaptoethanol, eluted after 20 ml. With increasing HAP12 concentrations, the WT dimer peak decreased and an assembled dimer peak appeared, increased in peak area, and shifted progressively to the void volume. Peak height is in milli-absorbance units. (B) Representative chromatograms of 10 μM V124W mutant assembly with HAP12. With increasing HAP12 concentrations, the V124W capsid peak eluted earlier, but not into the void volume, unlike the case for the WT. V124W dimers eluted later than WT dimers. (C) HAP12 titration on WT and V124W mutant assembly. WT assembly was enhanced from no assembly without HAP12 to 95% assembly with 60 μM HAP12. The extent of V124W mutant assembly was changed only from 50% to 65% at 60 μM HAP12. The extent of assembly is indicated by the decreasing dimer fraction.
V124W mutant assembly has dramatically reduced HAP sensitivity.
We studied the effect of HAP molecules on V124W mutant assembly kinetics, thermodynamics, and product morphology to determine the sensitivity of the V124W mutant to HAP-induced assembly effects. HAP12 has previously been described as a potent effector of HBV capsid assembly (18). HAP12 increases the rate of light-scattering change by as much as 5,000-fold and decreases the pseudocritical concentration by as much as 500-fold (18). HAP12 also induces formation of large aberrant structures in capsid assembly reactions.
We studied the effect of HAP12 on V124W mutant assembly kinetics by 90° light scattering. In the presence of excess HAP12 (two HAP12 molecules per dimer), WT assembly was activated and very fast (Fig. 2A). The absolute change in assembly kinetics was impossible to calculate under these conditions, as the WT did not assemble in the absence of HAP12. The strong light-scattering signal was due to the formation of large aberrant structures, which were confirmed by TEM (Fig. 2C). Under the same conditions, HAP12 increased the rate of V124W mutant assembly only 2-fold (Fig. 2A). Thus, HAP12 could interact with the V124W mutant, but it either had a weaker interaction than it had with the WT or had an effect on the interaction that was much weaker.
HAP molecules increase the thermodynamic stability of dimer-dimer interactions, resulting in increased assembled protein and decreased free dimers as observed by SEC (Fig. 3). In this experimental setup, WT capsids eluted at 10 ml and WT dimers eluted at 18 ml. Without HAP12, there was no assembly of 10 μM WT protein in 50 mM NaCl at 23°C (Fig. 3A). With increasing HAP12, WT assembly increased and resulted in formation of progressively larger noncapsid polymers. By TEM, WT assembly with HAP12 resulted in structures with diameters ranging from about 40 nm to >80 nm instead of the normal, 35-nm capsids (Fig. 2C). The absorbance at 280 nm of the capsid and overlapping noncapsid polymer peaks includes protein absorbance, HAP12 absorbance, and light scattering. At the highest HAP12 concentration, the light-scattering signal, which can only be estimated because of the size and irregular shape of the particles (34), accounted for approximately 9% of the total signal, while the HAP12 absorbance was about 16% of the total signal (about 1.5 HAPs/dimer); thus, HAP12 binds the capsid with a high affinity. Under these conditions, HAP12 drove the WT from no assembly without HAP12 to about 95% assembly at 60 μM HAP12 (Fig. 3C).
Under the same conditions where the WT did not assemble into capsids and showed extreme sensitivity to HAP12, 50% of the V124W mutant assembled into capsids, and the extent of assembly was comparatively HAP insensitive (Fig. 3B). The V124W capsid eluted later than the WT capsid. As the HAP12 concentration increased, the V124W capsid eluted earlier. However, by TEM, we did not observe aberrant structures such as those seen for WT assembly with HAP12 (Fig. 2C). We were unable to detect evidence of bound HAP12 in the V124W capsid peak based on absorbance at 280 nm, in contrast to the case for WT assembly. Increasing HAP12 concentrations only slightly affected V124W mutant assembly thermodynamics, decreasing the fraction of free dimers from 50% to 35% at 60 μM HAP12 (Fig. 3C). The HAP12 titration results indicated that the binding affinity of HAP12 for the V124W mutant was extremely weak, which correlated with a relatively weak effect on assembly. To summarize, V124W mutant assembly appeared to be largely insensitive to the effects of HAPs, in contrast to WT assembly.
The V124W mutant coassembled with the WT in vitro.
Because the V124W mutant was shown to mimic the HAP-treated WT in vitro in terms of kinetics and thermodynamics, we hypothesized that the V124W mutant and HAPs would have similar effects on WT assembly both in vitro and in vivo. Alternatively, it was possible that the V124W mutant would assemble only with itself and would not coassemble with the WT or affect WT assembly.
We studied the coassembly kinetics by 90° light scattering under conditions where the V124W mutant and the WT individually assembled, although they required different concentrations. At 100 mM NaCl and 23°C, the pseudocritical concentration was 1 μM for the V124W mutant and 43 μM for the WT (Table 1). Under these conditions, 10 μM WT and 1 μM V124W mutant did not assemble into detectable amounts of capsids on their own (Fig. 4A). When 1 μM V124W mutant and 10 μM WT were mixed, we observed a substantial time-dependent increase in light scattering, indicating protein self-assembly (Fig. 4A). Since the V124W mutant was more assembly active, we reasoned that it played a prominent role in nucleating the assembly reaction and that both V124W and WT dimers participated in elongation. However, because of the difficulty in evaluating assembly products by light scattering, we examined coassembly reactions by SEC.
Fig 4.
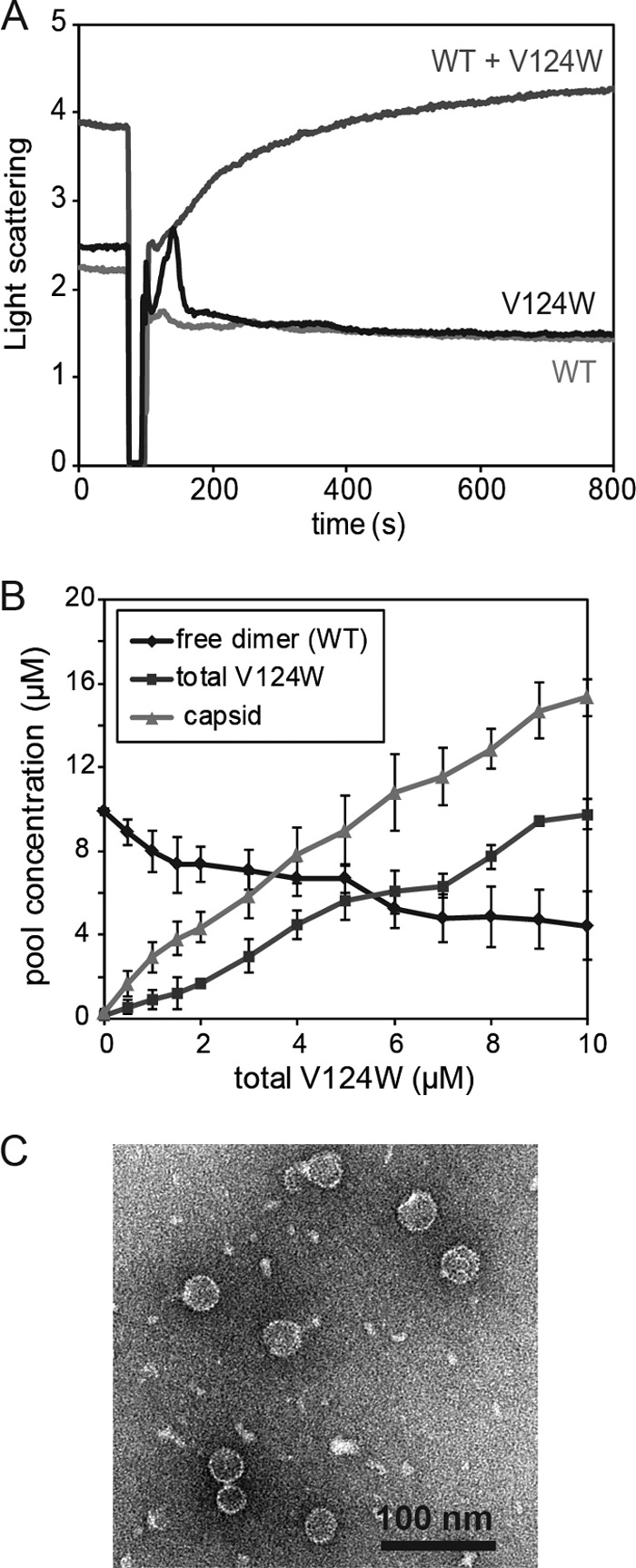
WT and V124W dimers coassemble into capsids in vitro. (A) Coassembly kinetics of 10 μM WT and 1 μM V124W mutant at 100 mM NaCl and 23°C was monitored by 90° light scattering. The proteins alone (10 μM WT or 1 μM V124W mutant) did not assemble under these conditions. However, if the two proteins were mixed together, time-dependent coassembly kinetics was observed. Each light-scattering trace shows the average for four independent experimental results. (B) Coassembly thermodynamics at 100 mM NaCl and 23°C. In the absence of the V124W mutant, 10 μM WT dimers did not assemble. With increasing concentrations of the V124W mutant, the free WT dimer concentration decreased and the yield of capsids increased. The total capsid concentration exceeded the total concentration of the V124W mutant, indicating coassembly. Each point shows the average for three to five independent experimental results. (C) Negatively stained electron micrographs of 10 μM WT and 5 μM V124W mutant coassembly products. No aberrant structures were observed.
With homogenous protein, we expected that at equilibrium, essentially all dimers above the KD,apparent would participate in capsid formation. In coassembly, where the more active V124W dimers were varied against a constant concentration of WT dimers, we anticipated that the amount of the WT incorporated would be proportional to the amount of the V124W mutant. When the amount of the V124W mutant was limiting, we anticipated maximum incorporation of the WT.
To define the thermodynamics of coassembly, different concentrations of the V124W mutant mixed with 10 μM WT (final concentration) were induced to assemble at 100 mM NaCl and 23°C, and the equilibrated reaction mixtures were examined by SEC. Based on the elution profiles, noting the different elution positions of V124W and WT dimers, no V124W dimers were detected at equilibrium. With increasing V124W mutant concentrations, the total capsid concentration increased and WT dimers decreased, indicating coassembly (Fig. 4B). Typically, about 40% of the protein in the capsid fraction was WT dimers, although different protein preparations showed slightly different activities. TEM showed that the coassembled capsids looked identical to WT capsids and that there were no large aberrant structures (Fig. 4C).
The V124W mutant dominantly interfered with virus replication in cell culture.
Our studies demonstrated that the V124W mutant mimicked a HAP-bound core protein in vitro, in terms of fast kinetics and strong association energy. We have proposed that a mechanism of antiviral activity of HAPs is their ability to inappropriately induce assembly (19, 20). We hypothesized that the V124W mutant would have HAP-like effects on viral replication in cell culture.
We examined the ability of the V124W mutant containing capsids to support DNA synthesis. We transfected Huh7 cells with expression plasmids encoding pgRNA, P protein (or reverse transcriptase), and X protein, as well as either full-length WT or V124W core protein (plasmids WTP+C+ and V124WP+C+). Southern blotting was used to evaluate the synthesis of intracellular HBV DNA. Expression of plasmid WTP+C+ produced the typical constellation of replicative intermediates: rcDNA, double-stranded linear DNA (dlDNA), single-stranded DNA (ssDNA), and a broad swath of partially double-stranded intermediates (Fig. 5A) (42). However, expression of plasmid V124WP+C+ led to nearly undetectable levels of replicative intermediates (Fig. 5).
Fig 5.
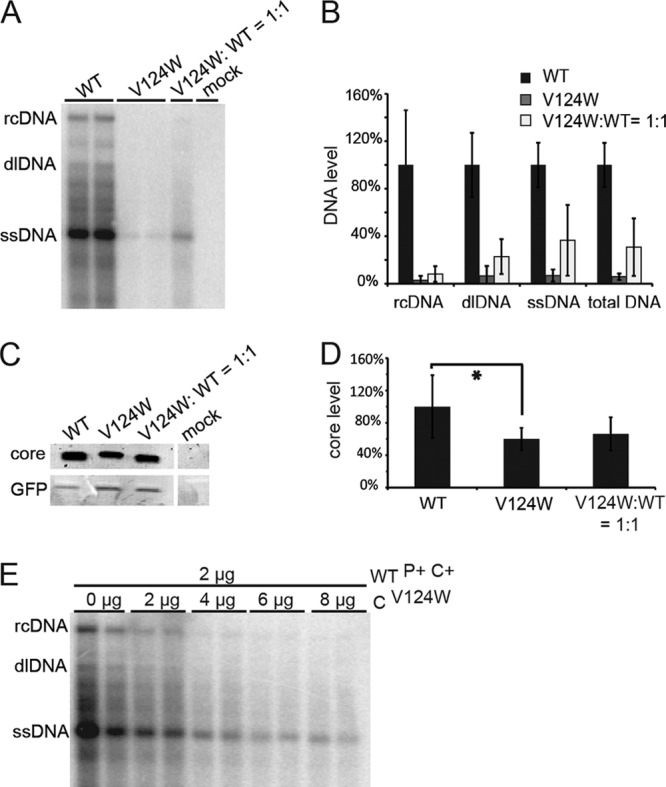
The V124W mutant has a dominant-negative effect on HBV replication in cell culture. (A) Southern blot showing that V124W mutant expression profoundly suppressed HBV DNA synthesis. Duplicate WT and V124W lanes are shown. (B) Quantification of Southern blot data from three independent transfections. WT yields of rcDNA, dlDNA, ssDNA, and total DNA were normalized to 100%. Yields of DNA in transfections exclusively with the V124W mutant were 3 to 7% of WT levels. Cotransfections (1:1) of the WT and the V124W mutant yielded 8 to 37% of the DNA in WT transfections. (C) Western blot of core protein in WT, V124W mutant, and cotransfection experiments. Mock transfection did not show any detectable proteins with a molecular weight corresponding to that of the HBV core protein. (D) Quantification of Western blot data from the same three independent transfections. The V124W core protein yield was statistically less than the WT yield (as indicated by an asterisk). However, there was no statistical difference between the core protein levels for 1:1 cotransfection and WT transfection alone. (E) V124W mutant titration of WT expression in Huh7 cells. With increasing amounts of the CV124W expression plasmid, WT DNA synthesis was progressively suppressed. Each cotransfection experiment is shown in duplicate.
According to Deres et al., HAP treatment caused depletion of the WT core protein in HepG2.2.15 cells (27). To examine if suppression of DNA synthesis by the V124W mutant had a similar basis, we examined V124W core protein levels by Western blotting (Fig. 5C). The V124W core protein level was 60% of the WT core protein level (Fig. 5D) (Student's two-tailed t test; P = 0.0178; n = 6). The observed effect of the V124W mutant on DNA replication was greater than its effect on core protein concentration, which indicated that the V124W mutant's suppression of DNA synthesis was attributable mainly to factors other than reduction of cytoplasmic core protein.
Our earlier analysis (Fig. 4) showed that the WT and the V124W mutant could coassemble into capsids in vitro. We hypothesized that the V124W mutant would dominantly interfere with viral DNA synthesis when coexpressed with the WT in cell culture. We examined the effect of cotransfection of both HBV expression plasmids (plasmids WTP+C+ and V124WP+C+). If the V124W core protein did not have a dominant-negative effect on the WT capsid's capacity to support rcDNA synthesis, we hypothesized that a 1:1 coexpression of WTP+C+ and V124WP+C+ plasmids would result in levels of viral DNA that were near 50% of WT levels. Instead, a 1:1 coexpression of the WTP+C+ and V124WP+C+ plasmids resulted in a profound suppression of viral DNA synthesis: 8% of rcDNA and 31% of total DNA compared to those with the WT alone (Fig. 5B). These values are much lower than predicted.
When core protein levels in the 1:1 coexpression experiments were evaluated, they were not statistically different from levels of WT core protein expressed alone (Fig. 5D), further indicating that reduction in the core protein level was not the main reason for suppression of DNA synthesis. To further corroborate these findings, we initiated DNA synthesis from plasmid WTP+C+ and expressed the V124W core from a second plasmid, CV124W, which expressed only the V124W core protein, with no other viral components. We found that with increasing V124W mutant expression, WT DNA synthesis was progressively suppressed (Fig. 5E). In total, our results demonstrate that the V124W mutant has a dominant-negative effect on WT core protein function for pgRNA packaging and/or genome replication.
DISCUSSION
Based on a 5-Å structure of an HBV capsid cocrystallized with HAP1, we designed a core protein mutant that was predicted to mimic the activity of HAPs. Our goals were to confirm the putative HAP binding site, to test if filling the HAP binding site could alter HAP sensitivity, and to study the mechanism of HAP activity. In our study, we mutated core protein residue 124 from V to W, which is predicted to fill the HAP site and increase the buried hydrophobic surface. The V124W core protein showed enhanced assembly kinetics, strong association energy, and HAP resistance, and in cell culture, it dominantly interfered with HBV DNA synthesis.
We also found evidence supporting the hypothesis that HBV capsid assembly is allosterically regulated. In allostery-regulated assembly, the free dimer undergoes a conformational change to become competent to nucleate and participate in assembly. We hypothesized that HAP molecules increased assembly kinetics by favoring the assembly-active state (16). We found that the V124W mutant, which assembled very rapidly compared to the WT, adopted a more compact conformation as shown by SEC elution (Fig. 3B). SEC separates solutes based on their Stokes radii; the slower elution of the V124W mutant indicated conformational differences between WT and V124W dimers. Previous work has shown that WT dimers in a capsid have a more compact conformation than that of assembly-incompetent dimers (16, 39), which elute earlier than WT dimers in SEC (see the inset in Fig. 4 of reference 39). Together, these data support the hypothesis that HBV core protein dimers undergo allosteric changes associated with assembly and that the V124W mutant and HAPs favor the assembly-active state.
As shown in the electron micrographs, the V124W mutant did not assemble into large aberrant structures, unlike aberrant HAP-induced WT assembly products. Similarly, phenylpropenamides induce fast assembly but do not affect the global capsid structure (19). At a low HAP-to-dimer stoichiometry, normal capsids are observed. Indeed, using pure BAY41-4109, it has been shown that more than one HAP per two dimers is necessary to generate aberrant structures (26). The tertiary and quaternary structures of capsids may be changed slightly due to the V124W mutation; however, this cannot be observed in negatively stained micrographs.
In the HAP12 titration, we observed that V124W capsids eluted later than WT capsids and that, with increasing HAP12 concentrations, the V124W capsids eluted earlier. It is possible that HAP12 altered the dynamics of the assembled capsids. HBV capsids are highly dynamic, opening and closing on a scale of seconds (43), which could certainly affect the elution volume. V124W mutant dynamics and its response to HAPs have not yet been investigated, and other explanations cannot be excluded. We note that with <50 μM HAP12, V124W capsids did not appear as large aberrant structures: they eluted after the SEC void volume (Fig. 3B) and migrated as a well-demarcated band in sucrose gradients (data not shown). We observed capsid-like particles and broken capsids by TEM, but not large aberrant structures for V124W mutant assembly with 60 μM HAP12 (data not shown).
AEfs such as HAPs and phenylpropenamides have been suggested as antiviral agents (18, 19, 27, 44). A caveat to their use as therapy would be the emergence of resistant mutants. We have demonstrated that V124W dimers are HAP resistant. HAP12 only modestly affected V124W mutant assembly kinetics and thermodynamics. Unfortunately, from the virus's perspective, the V124W mutant acts like a WT-assembly effector complex (Fig. 2). In cell culture, the phenotype of the V124W mutant is similar to the effects of HAP treatment (Fig. 5) (27). Resistance to HAP12 conferred by the V124W mutant is destructive to HBV infection and is unlikely to be a pathway to viral escape of HAPs, phenylpropenamides, or other AEfs that bind the HAP pocket.
Coassembly studies showed that the V124W mutant enhanced WT assembly similarly to the effect of HAP molecules. HAPs induce assembly by transiently stabilizing the assembly-active state of WT dimers and strengthening dimer-dimer interactions by increasing the buried surface area (16, 25). The V124W mutant also appeared to favor the assembly-active conformation, while the increased buried surface due to the tryptophan enhanced dimer association.
Similar to the HAP-like behavior in the coassembly studies, cell culture studies showed that the V124W mutant inhibited viral genome replication in a dominant-negative manner. To understand this, we must consider capsid formation in a WT infection. Core proteins assemble around the pgRNA-reverse transcriptase complex to form an RNA-filled capsid (6). In the capsid, pgRNA is reverse transcribed to minus-strand DNA, and then plus-strand DNA is synthesized. We propose three possible mechanisms for the dominant-negative effect of the V124W mutant (Fig. 6). (i) The V124W mutant might nucleate assembly in the absence of the pgRNA-reverse transcriptase complex to form empty particles or package nonviral RNAs (an effect seen with phenylpropenamides [19, 41]). (ii) The V124W mutant might assemble into kinetically trapped intermediates instead of complete capsids. These complexes might not support normal DNA synthesis, or viral DNA may have been digested during the isolation of cytoplasmic capsid DNA. (iii) The V124W mutant might assemble into pgRNA-filled capsids; however, the physical chemistry of the V124W capsid, with its unusually strong association energy, might adversely affect DNA synthesis. The biochemical coassembly experiments showed that the V124W mutant interacted with the WT, and thus coexpression in cells can have a dominant-negative effect analogous to the antiviral effects of HAPs.
Fig 6.
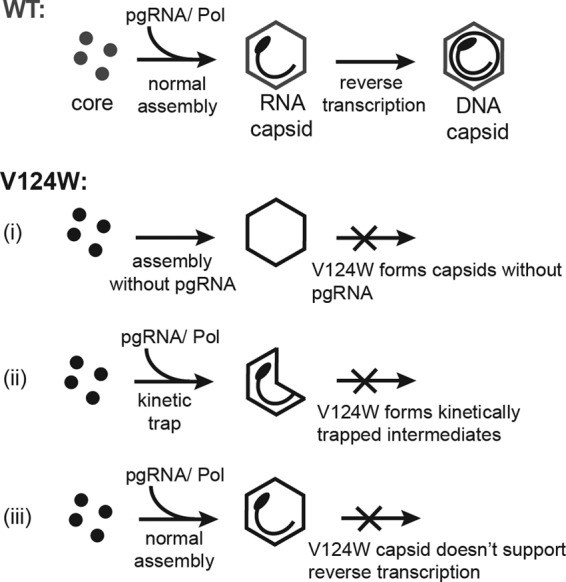
Proposed mechanisms of suppression of HBV replication by the V124W mutant. In WT capsid assembly, core proteins assemble around the pgRNA-reverse transcriptase complex to form an RNA-filled capsid, in which pgRNA can be reverse transcribed to rcDNA. For V124W capsid assembly, we propose three possible explanations for the observed suppression in DNA synthesis. (i) The V124W mutant might nucleate assembly in the absence of the pgRNA-reverse transcriptase complex to form empty particles or package nonviral RNAs. (ii) The V124W mutant may assemble into kinetically trapped intermediates which do not support reverse transcription, or it may produce viral DNA which can be digested during intracellular capsid DNA isolation. (iii) The V124W mutant may assemble into pgRNA-filled capsids; however, the physical chemistry of the V124W capsid, with its unusually strong association energy, may adversely affect reverse transcription.
HBV DNA synthesis in the presence of the V124W mutant did not correlate with the level of expression of core protein. V124W mutant expression led to a >90% reduction in the yield of total DNA, though V124W protein accumulation was decreased by only 40%. When the V124W mutant and the WT were coexpressed at a 1:1 ratio, the total core protein accumulation was not affected by the V124W mutant, although the DNA synthesis was profoundly suppressed. When increasing amounts of CV124W plasmid were cotransfected with WTP+C+ plasmid, we observed a HAP-like dose dependence for suppression of DNA synthesis. While this was not explored in this report, there could be many reasons for the decrease of V124W core protein, including decreased protein synthesis or the protein degradation associated with HAP molecules (27). These will be evaluated in future studies.
We designed a novel HBV core protein that dominantly interferes with HBV replication. When this project was started, we had only a 5-Å structure for guidance. Based on HAP resistance, we confirmed the putative HAP binding site, accentuating the importance of the dimer-dimer interface for regulating assembly. We demonstrated that assembly effectors have tremendous potential as antiviral drugs. Because the development of assembly effectors is in its infancy, this study is the first to examine resistance to their action. We observed that resistance to HAPs through mutations that block the HAP site do not benefit virus replication, suggesting that AEfs may have a high barrier to resistance as antiviral drugs. Additional studies of mutations that affect the HAP binding pocket will provide new and exciting opportunities to study capsid assembly. The use of the HBV capsid-HAP structure to design these mutations is an example of the power of chemical biology.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01-AI077688 and R01-AI067417 to A.Z. and P01-CA022443 to D.D.L.
A.Z. reports a potential conflict of interest related to an interest in a company based on assembly effectors.
Footnotes
Published ahead of print 2 January 2013
REFERENCES
- 1. Colvin HM, Mitchell AE. (ed). 2010. Hepatitis and liver cancer: a national strategy for prevention and control of hepatitis B and C. National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- 2. WHO 2004. WHO position on the use of hepatitis B vaccines. Wkly. Epidemiol. Rec. 28:255–263 [Google Scholar]
- 3. Tillmann HL. 2007. Antiviral therapy and resistance with hepatitis B virus infection. World J. Gastroenterol. 13:125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kwon H, Lok AS. 2011. Hepatitis B therapy. Nat. Rev. Gastroenterol. Hepatol. 8:275–284 [DOI] [PubMed] [Google Scholar]
- 5. Ganem D, Prince AM. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118–1129 [DOI] [PubMed] [Google Scholar]
- 6. Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235–249 [DOI] [PubMed] [Google Scholar]
- 7. Crowther RA, Kiselev NA, Bottcher B, Berriman JA, Borisova GP, Ose V, Pumpens P. 1994. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 77:943–950 [DOI] [PubMed] [Google Scholar]
- 8. Kenney JM, von Bonsdorff CH, Nassal M, Fuller SD. 1995. Evolutionary conservation in the hepatitis B virus core structure: comparison of human and duck cores. Structure 3:1009–1019 [DOI] [PubMed] [Google Scholar]
- 9. Stannard LM, Hodgkiss M. 1979. Morphological irregularities in Dane particle cores. J. Gen. Virol. 45:509–514 [DOI] [PubMed] [Google Scholar]
- 10. Zlotnick A, Cheng N, Conway JF, Booy FP, Steven AC, Stahl SJ, Wingfield PT. 1996. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry 35:7412–7421 [DOI] [PubMed] [Google Scholar]
- 11. Dhason MS, Wang JC-Y, Hagan MF, Zlotnick A. 2012. Differential assembly of hepatitis B virus core protein on RNA and DNA correlates with the virus lifecycle. Virology 430:20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ceres P, Zlotnick A. 2002. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 41:11525–11531 [DOI] [PubMed] [Google Scholar]
- 13. Endres D, Zlotnick A. 2002. Model-based analysis of assembly kinetics for virus capsids or other spherical polymers. Biophys. J. 83:1217–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Katen SP, Zlotnick A. 2009. Thermodynamics of virus capsid assembly. Methods Enzymol. 455:395–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zlotnick A, Johnson JM, Wingfield PW, Stahl SJ, Endres D. 1999. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry 38:14644–14652 [DOI] [PubMed] [Google Scholar]
- 16. Packianathan C, Katen SP, Dann CE, 3rd, Zlotnick A. 2010. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J. Virol. 84:1607–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen C, Wang JC-Y, Zlotnick A. 2011. A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics. PLoS Pathog. 7:e1002388 doi:10.1371/journal.ppat.1002388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bourne C, Lee S, Venkataiah B, Lee A, Korba B, Finn MG, Zlotnick A. 2008. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J. Virol. 82:10262–10270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Katen SP, Chirapu SR, Finn MG, Zlotnick A. 2010. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem. Biol. 5:1125–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. U. S. A. 102:8138–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ceres P, Stray SJ, Zlotnick A. 2004. Hepatitis B virus capsid assembly is enhanced by naturally occurring mutation F97L. J. Virol. 78:9538–9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan TT, Sahu GK, Whitehead WE, Greenberg R, Shih C. 1999. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J. Virol. 73:5731–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan TT, Tai PC, Shih C. 1999. Subtype-independent immature secretion and subtype-dependent replication deficiency of a highly frequent, naturally occurring mutation of human hepatitis B virus core antigen. J. Virol. 73:10122–10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bourne C, Finn MG, Zlotnick A. 2006. Global structural changes in hepatitis B capsids induced by the assembly effector HAP1. J. Virol. 80:11055–11061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stray SJ, Zlotnick A. 2006. BAY 41-4109 has multiple effects on hepatitis B virus capsid assembly. J. Mol. Recognit. 19:542–548 [DOI] [PubMed] [Google Scholar]
- 27. Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. 2003. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 299:893–896 [DOI] [PubMed] [Google Scholar]
- 28. King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. 1998. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta-l-2′,3′-dideoxy-3′-thiacytidine. Antimicrob. Agents Chemother. 42:3179–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perni RB, Conway SC, Ladner SK, Zaifert K, Otto MJ, King RW. 2000. Phenylpropenamide derivatives as inhibitors of hepatitis B virus replication. Bioorg. Med. Chem. Lett. 10:2687–2690 [DOI] [PubMed] [Google Scholar]
- 30. Stoltefuss J, Goldmann S, Krämer T, Schlemmer K-H, Niewöhner U, Paessens A, Lottmann S, Deres K, Weber O. 1 October 1999. New dihydropyrimidine derivatives and their corresponding mesomers useful as antiviral agents. WO patent 9954326
- 31. Stolting J, Stoltefuss J, Goldmann S, Kramer T, Schlemmer K-H, Niewohner U, Paessens A, Graef E, Lottmann S, Deres K, Weber O. 2000. Preparation of 4-aryl-2-pyridinyl-1,4-dihydropyrimidine-5-carboxylates for treatment of hepatitis B infection. WO patent 2000-EP2327; 2000058302
- 32. Hacker HJ, Deres K, Mildenberger M, Schroder CH. 2003. Antivirals interacting with hepatitis B virus core protein and core mutations may misdirect capsid assembly in a similar fashion. Biochem. Pharmacol. 66:2273–2279 [DOI] [PubMed] [Google Scholar]
- 33. Zlotnick A, Ceres P, Singh S, Johnson JM. 2002. A small molecule inhibits and misdirects assembly of hepatitis B virus capsids. J. Virol. 76:4848–4854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Porterfield JZ, Zlotnick A. 2010. A simple and general method for determining the protein and nucleic acid content of viruses by UV absorbance. Virology 407:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P. 1979. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646–650 [DOI] [PubMed] [Google Scholar]
- 36. Abraham TM, Lewellyn EB, Haines KM, Loeb DD. 2008. Characterization of the contribution of spliced RNAs of hepatitis B virus to DNA synthesis in transfected cultures of Huh7 and HepG2 cells. Virology 379:30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lewellyn EB, Loeb DD. 2011. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 85:1298–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maguire ML, Loeb DD. 2010. cis-Acting sequences that contribute to synthesis of minus-strand DNA are not conserved between hepadnaviruses. J. Virol. 84:12824–12831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bourne CR, Katen SP, Fulz MR, Packianathan C, Zlotnick A. 2009. A mutant hepatitis B virus core protein mimics inhibitors of icosahedral capsid self-assembly. Biochemistry 48:1736–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson JM, Tang J, Nyame Y, Willits D, Young MJ, Zlotnick A. 2005. Regulating self-assembly of spherical oligomers. Nano Lett. 5:765–770 [DOI] [PubMed] [Google Scholar]
- 41. Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. 2007. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res. 76:168–177 [DOI] [PubMed] [Google Scholar]
- 42. Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202 doi:10.1371/journal.pone.0017202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hilmer JK, Zlotnick A, Bothner B. 2008. Conformational equilibria and rates of localized motion within hepatitis B virus capsids. J. Mol. Biol. 375:581–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delaney W, Edwards R, Colledge D, Shaw T, Furman P, Painter G, Locarnini S. 2002. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 46:3057–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]