

Abstract
Highly pathogenic Nipah virus (NiV) infections are transmitted via airway secretions and urine, commonly via the respiratory route. Epithelial surfaces represent important replication sites in both primary and systemic infection phases. NiV entry and spread from polarized epithelial cells therefore determine virus entry and dissemination within a new host and influence virus shedding via mucosal surfaces in the respiratory and urinary tract. To date, there is no knowledge regarding the entry and exit sites of NiV in polarized epithelial cells. In this report, we show for the first time that NiV can infect polarized kidney epithelial cells (MDCK) from both cell surfaces, while virus release is primarily restricted to the apical plasma membrane. Substantial amounts of basolateral infectivity were detected only after infection with high virus doses, at time points when the integrity of the cell monolayer was largely disrupted as a result of cell-to-cell fusion. Confocal immunofluorescence analyses of envelope protein distribution at early and late infection stages suggested that apical virus budding is determined by the polarized sorting of the NiV matrix protein, M. Studies with stably M-expressing and with monensin-treated cells furthermore demonstrated that M protein transport is independent from the glycoproteins, implying that the M protein possesses an intrinsic apical targeting signal.
INTRODUCTION
Nipah virus (NiV) is a highly pathogenic member of the genus Henipavirus within the family Paramyxoviridae, originating from fruit bats (1). NiV was first discovered in Malaysia and Singapore in 1999 during an outbreak of severe respiratory disease in pigs and fatal encephalitis in humans (2). Since 2001, smaller outbreaks of NiV infections in India and Bangladesh have been regularly reported (3). Although human infections have been described only in Southeast Asia so far, there is recent evidence for the existence of Henipavirus-related viruses in African fruit bats (4, 5).
Like all paramyxoviruses, NiV is an enveloped virus with a negative-stranded RNA genome (6). Cell infections start with binding of the viral surface glycoprotein G to cellular ephrin-B2 or ephrin-B3 receptors (7–10). After receptor binding, the fusion protein, F, mediates pH-independent fusion of the viral envelope with the host cell membrane to allow virus entry (for a review, see reference 11). While the NiV surface glycoproteins G and F are essential for virus entry processes and later on for cell-to-cell fusion, the third NiV envelope-associated protein, the matrix protein, M, plays an essential role in virus assembly and budding. Similar to many viral matrix proteins, NiV M is a cytoplasmic protein which rapidly associates with cellular membranes. M organizes the assembly of cytoplasmic nucleocapsids and surface glycoproteins at the plasma membrane and is thus needed for efficient release of progeny virus. Conclusive evidence has been provided that late-domain-like motifs and ubiquitin-regulated nuclear-cytoplasmic trafficking are required for NiV M-mediated budding processes (12–14).
The most common route of transmission of NiV is through the oronasopharyngeal route. After initial replication in the airways, NiV disseminates systemically. In the viremic phase of infection, NiV targets endothelial cells in many organs. The pronounced infection of microvascular endothelial cells in the central nervous system (CNS), causing vasculitis, thrombosis, and necrosis, is finally the cause of the multifocal encephalitis generally observed in humans and some animal species (for a review, see reference 15). While CNS pathology was primarily responsible for clinical disease in humans, pigs naturally infected during the first NiV outbreak in Malaysia showed mainly respiratory symptoms, with widespread lesions in the lung epithelium (16). Studies of experimentally infected pigs presented similar results, with the highest virus contents in the upper and lower respiratory tract (15). Studies of NiV infection in a hamster model furthermore showed that the respiratory epithelium is the initial area for NiV replication before it comes to a more systemic spread of infection (17, 18). In agreement with the reported NiV shedding in airway secretions and urine, epithelial cells in the respiratory tract, the kidneys, and the bladder were found to be infected in late stages of natural or experimental NiV infections (reviewed in reference 19). These immunohistological data of in vivo infections clearly demonstrate that NiV efficiently infects epithelial cells in mucosal surfaces.
Epithelial cells differ from most other cell types in their polarized phenotype and their barrier function. The most important feature is their apical and basolateral plasma membrane domains that are strictly separated by tight junctions. Due to specialized protein-sorting machineries in these cells, the two membrane domains differ substantially in their compositions (20, 21). Protein sorting, maintaining the polarity and the specialized functions of epithelial cells, can also influence virus infections. While the polarized distribution of the viral receptor can restrict virus entry to one surface domain, sorting of viral proteins can lead to a vectorial virus release (22–26). Since the handling of NiV is restricted to biosafety level 4 (BSL-4) laboratories, knowledge about the molecular mechanisms underlying the interactions of NiV with epithelial cells based on studies with live virus is extremely limited. We have shown in a previous study that both NiV surface glycoproteins possess tyrosine-dependent sorting signals responsible for the basolateral targeting of the proteins upon single expression in polarized MDCK cells. However, the localization of G and F proteins in infected polarized MDCK cells was found to be bipolar, with most of the glycoproteins concentrating at the apical membrane (27). As it is known for several viruses that the glycoprotein distribution does not necessarily determine the site of virus budding (28–31), the impact of the NiV glycoprotein distribution is not yet known. The aim of this study was thus to elucidate the virus entry and exit pathways in polarized epithelial cells and to clarify the role of vectorial sorting of the NiV envelope proteins in virus spread and release from epithelial cells.
MATERIALS AND METHODS
Cell culture.
Vero76 and MDCK cells were cultivated in Dulbecco's modified Eagle's medium (DMEM; Gibco) and Eagle's minimal essential medium (MEM; Gibco), respectively, with 10% fetal calf serum (FCS; Life Technologies), 100 U of penicillin/ml, 0.1 mg of streptomycin/ml, and 4 mM l-glutamine (Gibco). For studies of polarized cells, MDCK cells were seeded onto permeable Transwell filter membranes (ThinCerts tissue culture inserts; Greiner Bio-One) with a 1.0-μm or 0.4-μm pore size and cultured until full polarization was reached. To measure polarity, transepithelial resistance (TER) was controlled daily by using an EVOM2 instrument (World Precision Instruments). Only cells with a TER above 180 Ω cm2 were used for our analyses.
Virus infections.
All experiments with live NiV were performed under BSL-4 conditions at the Institute of Virology, Philipps University of Marburg. The NiV strain used in this study was a human isolate and was propagated as described previously (32). For infection of polarized cells, MDCK cells were cultivated on filter supports for 5 days, and cell polarity was controlled daily by measuring the TER. Fully polarized cell cultures were then incubated with NiV at either a low multiplicity of infection (MOI) (0.01) or a high MOI (10) from either the apical or the basal side for 1 h at 37°C. After virus adsorption, cells were washed five times and incubated in cell culture medium at 37°C. To analyze virus growth and polarity of virus release, samples from the apical and basal media were taken at different time points, and titers were determined by the 50% tissue culture infection dose (TCID50) method on Vero76 cells, using an automated pipetting device (Freedom EVO; Tecan). To determine the polarity of virus release in nonpolarized cells, confluent Vero76 cells grown on filter supports were infected at an MOI of 0.01, and apical and basal media were titrated by the TCID50 method. For immunofluorescence analysis, NiV-infected cells were inactivated for 48 h with 4% paraformaldehyde (PFA; Sigma-Aldrich) in DMEM and further processed under BSL-2 conditions.
Ephrin-B2/-B3 surface staining.
Staining of ephrin-B2 on the cell surface of polarized MDCK cells was performed as previously described (33). MDCK cells grown for 5 days on filter supports were fixed with 4% PFA for 10 min and then incubated with recombinant mouse EphB4/Fc, a soluble ephrin-B2 (EB2) receptor fused to the Fc region of human IgG (R&D Systems), at a concentration of 2 μg/ml at 4°C for 2 h. Bound EphB4/Fc was stained with goat-derived rhodamine-conjugated anti-human IgG antibodies at 4°C for 2 h (dilution, 1:100; Jackson ImmunoResearch). To enhance the signal, donkey-derived Alexa Fluor 568 (AF 568)-conjugated anti-goat IgG antibodies (Life Technologies) were used for 2 h at 4°C (dilution, 1:250). E-cadherin was stained after permeabilization with 0.1% Triton X-100 (Sigma-Aldrich) for 15 min. Cells were then treated with monoclonal anti-E-cadherin antibodies (BD Biosciences) at a dilution of 1:100 for 2 h at 4°C and with Alexa Fluor 488 (AF 488)-conjugated anti-mouse IgG antibodies from chicken for 2 h at 4°C (dilution, 1:100; Life Technologies). For costaining of the apical MDCK cell marker protein gp114 (canine CEACAM1; CD66) and E-cadherin, cells were fixed with PFA, permeabilized with 0.2% Triton X-100, and incubated with anti-E-cadherin mouse antibodies and a rabbit antiserum against gp114/CD66 (anti-CEACAM1 H-136; Santa Cruz Biotech) at a dilution of 1:20. Primary antibodies were detected with AF 488-conjugated anti-mouse and AF 568-labeled anti-rabbit IgG antibodies. Samples were mounted with Mowiol 4-88 (Calbiochem) and analyzed with a confocal laser scanning microscope (SP 5 II; Leica).
Electron microscopy.
For transmission electron microscopic analysis of NiV-infected cells, filter-grown cells were infected at an MOI of 10. At 24 h postinfection (p.i.), cells were fixed with 4% PFA and 0.1% glutaraldehyde in 0.1 M PHEM buffer [60 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 25 mM HEPES, 2 mM MgCl2, 10 mM EGTA (pH 6.9)] for 30 min at room temperature. Cells were then incubated overnight with 4% PFA in 0.1 M PHEM buffer at 4°C. Samples were removed from the BSL-4 laboratory in fresh 4% PFA in DMEM, postfixed for 60 min with 1% osmium tetroxide in 50 mM HEPES buffer (pH 7.5), washed, and stained overnight at 4°C in a 2% aqueous uranyl acetate solution. Cells were dehydrated in a graded series of ethanol, flat embedded in a mixture of Epon and Araldite, and polymerized at 60°C for 24 h (34). Ultrathin sections (60 to 90 nm) of the cell monolayers were cut perpendicular to the plane of the monolayer with a Leica Ultracut UCT microtome (Leica Microsystems, Germany). Sections were stained with Reynolds' lead citrate and analyzed by using a Zeiss 109 transmission electron microscope at 80 kV.
Confocal immunofluorescence analysis.
For immunostaining of the NiV envelope-associated proteins on Transwell filters, infected cells were inactivated for 48 h with 4% PFA. To detect the viral glycoproteins, cells were incubated from the apical and basal sides with monoclonal rabbit antibodies against NiV F (mab92) or against NiV G (mab26, kindly provided by Benhur Lee) at a dilution of 1:250 for 2 h at 4°C. M protein was stained after permeabilization with methanol (Sigma-Aldrich) for 2 h at 4°C by M-specific monoclonal antibody F45G5 (dilution, 1:500) (35). Primary antibodies were detected with AF 568-conjugated secondary antibodies (dilution, 1:250). For costaining of the G and M proteins, fixed cells were incubated with rabbit anti-G antibodies and AF 568-labeled secondary antibodies, as described above. The cells were then permeabilized, and the M protein was stained with the F45G5 mouse antibodies and AF 488-conjugated secondary antibodies. Samples were mounted in Mowiol 4-88 and were analyzed by using a Zeiss 510Meta confocal laser scanning microscope. For quantification of the overall apical/basolateral protein distribution, fluorescence intensities in apical (xy sections 1 to 45) and basolateral (xy sections 46 to 100) horizontal sections were quantified by using ImageJ (http://rsbweb.nih.gov/ij) and corrected for background. Since M staining was done in permeabilized cells, basolateral sections also contained cytoplasmic fluorescence signals. To analyze the colocalization of G and M in apical and basolateral sections, the ImageJ colocalization threshold plugin was used.
Immunostaining of NiV-infected cells.
To detect virus-positive cells after infection from the apical or basolateral side, filter-grown infected cells were fixed with PFA and incubated with the NiV-specific antiserum gp4 (kindly provided by Heinz Feldmann) and AF 488-labeled secondary antibodies. To monitor the increasing size of syncytia, the cells were inactivated with PFA at different time points p.i., permeabilized with methanol, and incubated with M-specific F45G5 antibodies and AF 568-conjugated secondary antibodies, as described above.
Permeability assay.
The permeability of polarized MDCK cell monolayers was analyzed at 24, 48, and 72 h after infection by a horseradish peroxidase (HRP) flowthrough assay. As described previously (36), the culture medium in the apical filter chamber was exchanged with serum-free MEM with HRP (Sigma) at a concentration of 5 μg/ml. After 1 h at 37°C, samples from the basal medium were taken, and 20 μl was mixed with 150 μl substrate buffer (0.1 M KH2PO4 and 0.05 M citric acid [pH 5] with freshly added 0.012% H2O2 and 400 μg/ml o-phenylenediamine). HRP activity was determined colorimetrically by determining the absorbance at 450 nm using a microplate reader (Phomo). HRP activity was normalized to the HRP activity in the basal medium of an empty filter without cells.
Plasmid constructs and stable and transient protein expression in MDCK cells.
cDNA fragments spanning the F, G, and M genes of the NiV genome (GenBank accession number AF212302) were cloned into expression plasmids. The NiV F gene was subcloned from the pczCFG5-NiV F vector (37) into the pCAGGS vector (38), using the restriction enzymes EcoRI and BamHI for the insert and BglII and EcoRI for the vector (pCAGGS NiV F). The BD In-Fusion PCR cloning system (Becton, Dickinson and Company) was used to clone the M gene into a pCG vector (39) (pCG NiV M). The C-terminal flag epitope tag was introduced into the M gene also by using the BD In-Fusion PCR cloning system (pCG NiV Mflag). The open reading frame of the NiV G protein was codon optimized and synthesized by Geneart AG (Germany). The codon-optimized NiV G gene was cloned into the pCAGGS vector, using the KpnI and SacI restriction sites (pCAGGS NiV G).
Stable expression of flag epitope-tagged M protein in MDCK cells was achieved by cotransfecting pCG NiV Mflag together with a plasmid containing the neomycin resistance gene, using Lipofectamine 2000 transfection reagent (Life Technologies), as described previously (27). Transfected cells were selected by adding 0.75 mg G418 (Gibco) per ml to the medium. Stable cell lines containing 25 to 60% M-positive cells, as determined by indirect immunofluorescence analysis, were established.
Monensin treatment of transfected and infected MDCK cells.
MDCK cells grown for 24 h on coverslips were cotransfected with plasmids coding for NiV M and F or NiV M and G. Monensin (Sigma-Aldrich) was added 2 h after transfection at a concentration of 20 μM. At 24 h posttransfection (p.t.), the cells were fixed with 4% PFA in DMEM, permeabilized with methanol, and incubated for 1 h with monoclonal antibodies from rabbits directed against the individual envelope proteins (anti-M antibody F45G5 [35], anti-NiV F antibody mab92, and anti-NiV G antibody mab26 [kindly provided by Benhur Lee]). Primary rabbit antibodies (mab26 and mab92; dilution, 1:250) were detected with AF 488-conjugated secondary antibodies. Monoclonal mouse anti-M antibodies (F45G5; dilution, 1:500) were detected with AF 568-coupled secondary antibodies. The samples were mounted in Mowiol 4-88.
For monensin treatment of infected cells, MDCK cells were seeded onto coverslips and were infected the next day with NiV (MOI of 0.01). Monensin was added to the cells at 1 h p.i., at a concentration of 20 μM. At 24 h p.i., the cells were inactivated for 48 h with 4% PFA in DMEM and permeabilized with methanol. Coimmunostaining was performed as described above.
RESULTS
Ephrin receptor expression and NiV entry are bipolar.
Although there is conclusive evidence from immunohistological studies that NiV infects epithelial cells in the respiratory and urinary tract in vivo (18, 40–42), the site of virus entry into polarized epithelial cells has not been directly determined. Since entry receptor expression is an indispensable prerequisite for virus infections from the apical and/or basolateral surface domain, we first analyzed the distribution of the NiV entry receptors using polarized MDCK cells, an epithelial kidney cell line readily supporting NiV infection (27, 43). To determine the localization of ephrin-B2/-B3 on the surface of polarized MDCK cells, the cells were cultured on permeable filter supports until full polarization was reached. After fixation with paraformaldehyde (PFA), cells were incubated with the soluble ephrin-B2/-B3 ligand EphB4 fused to human Fc (EphB4/Fc) and fluorescently labeled secondary antibodies. Adherens junctions in the polarized MDCK cell monolayer were visualized by using a monoclonal antibody directed against E-cadherin. As shown in Fig. 1A, ephrin staining was detected in apical, central, and basal sections through the monolayer, while E-cadherin was visible only in the central section. In the vertical section, an almost equal ephrin distribution around the polarized cells can be seen (Fig. 1B), clearly demonstrating a bipolar localization of NiV receptors in polarized MDCK cells. Although nonpolarized expression of the NiV receptor is necessary, it is not automatically sufficient for bipolar infection. Virus entry might nonetheless be polarized because an essential second receptor is lacking or because the actin cortex cannot be overcome at both plasma membrane domains (23, 44–46). To determine the polarity of NiV entry, polarized MDCK cells were infected with NiV by adding virus to either the apical or basal filter chamber. At 48 h p.i., the cells were inactivated with 4% PFA, and NiV-positive cells were detected by immunostaining. As shown in Fig. 2, NiV-induced foci were detected after infection from both the apical and basal sides, demonstrating that NiV entry into polarized epithelial cells is not restricted. As with the kidney cell line MDCK, infection of primary human bronchial epithelial cells was also found to be bipolar (data not shown).
Fig 1.
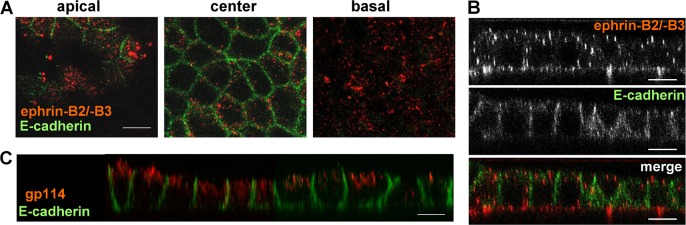
Surface distribution of ephrin receptors. Polarized MDCK cells grown on filter supports were fixed with 4% PFA and incubated with recombinant mouse EphB4/Fc, a soluble ephrin-B2/-B3 ligand fused to the Fc region of human IgG. Bound EphB4/Fc was stained with rhodamine- and AF 568-conjugated secondary antibodies (red). Adherens junctions were visualized after permeabilization by antibodies directed against E-cadherin and AF 488-conjugated secondary antibodies (green). (A and B) Confocal images representing apical, center, and basal horizontal sections through the cell monolayer (A) and a vertical section (B) are shown. (C) To control MDCK cell polarity, E-cadherin and the apical marker protein gp114 (canine CEACAM1) were detected after cell permeabilization by specific primary and according secondary antibodies. Bars, 10 μm.
Fig 2.
Polarity of NiV entry. Polarized MDCK cells were infected with NiV (MOI of 10) from either the apical (A) or basal (B) filter chamber. At 48 h p.i., the cells were fixed with 4% PFA, and virus-positive cells were stained with a NiV-specific antiserum and AF 488-labeled secondary antibodies. Bars, 100 μm.
Virus release from polarized epithelial cells is restricted mostly to the apical plasma membrane.
While virus entry is determined mainly by the distribution of viral receptors, virus release is directed by the localization of the viral surface glycoproteins or the matrix protein (29, 31, 34, 47–50). To analyze budding polarity in epithelial cells, polarized MDCK cells were infected with high and low doses of NiV, and virus titers in the cell culture media from the apical and basal filter chambers were determined. As shown in Fig. 3A, infection at a low MOI (0.01) resulted in a predominantly apical virus release, with virus titers in the apical medium being 2 to 3 log steps higher than basal virus titers. Infection with a 1,000-fold-higher MOI resulted in a more rapid increase in overall virus titers, as expected (Fig. 3B). Nonetheless, virus release was predominantly apical within the first 48 h p.i. Only late in infection (72 h p.i.) did the difference between virus titers in apical and basal media decrease to 1.5 log steps (Fig. 3B). Moreover, the side of virus entry also did not influence the budding polarity. When we infected polarized MDCK cells from either the apical or the basal side, over 98% of the released infectious virus particles were found in the apical filter chamber (Fig. 3C and D). We also analyzed virus release from nonpolarized Vero cells and subconfluent MDCK cells. Here, virus release was bipolar, with a <1-log difference between the apical and basal virus titers (data not shown). Selective apical NiV release was also confirmed by electron microscopy. For this purpose, NiV-infected filter-grown MDCK monolayers were embedded and sectioned for electron microscopic analysis at 24 h p.i. Alignments of nucleocapsids and virus buds were seen only at the apical plasmalemma (Fig. 3E).
Fig 3.

NiV release from infected polarized MDCK cells. (A and B) MDCK cells were cultivated on permeable filter supports for 5 days and infected from the apical side at an MOI of 0.01 (A) or an MOI of 10 (B). Cell-free virus in the apical and basal media was titrated by the TCID50 method at 24, 48, and 72 h p.i. (n = 3). (C and D) Polarized MDCK cells were infected from either the apical (C) or basal (D) side at an MOI of 10. Virus yields in the apical and basal media were determined at 24 and 48 h p.i. The percentages of total infectivity in the apical and basal media (n = 3) are shown. Error bars indicate standard deviations. (E) NiV-infected polarized MDCK cells were fixed and processed for transmission electron microscopy at 24 h p.i. The top left panel shows an overview of a NiV-positive syncytium. Top right and bottom panels show higher magnifications of boxed areas showing an apical virus bud (1), nucleocapsids (RNPs) aligned at the apical plasma membrane (2), and a viral inclusion (3). Arrows indicate cross-sectioned viral RNPs.
NiV infection results in growing focus formation and an increase in transepithelial permeability.
Despite the clear apical NiV release, virus titers in the basal medium of cells infected at a high MOI increased up to 105 TCID50/ml at 48 h p.i. and even to 107 TCID50/ml at 72 h p.i. We hypothesized that this is due to an increasing disruption of the cell monolayer as a result of ongoing virus replication and cell-to-cell fusion. This idea is supported by the finding that NiV-induced focus formation increased drastically over time (Fig. 4) (27) and raised the question of to what extent the transepithelial permeability of polarized cell monolayers is affected by infection. To address this, we performed a horseradish peroxidase (HRP) flowthrough assay by adding HRP to the apical medium of infected polarized MDCK cells at different times after infection. In cells infected at a low MOI of 0.01, giving maximal basal virus titers below 104 TCID50/ml (Fig. 3A), we did not see an increase in the transepithelial permeability (Fig. 5). In contrast, infection at a 1,000-fold-higher MOI, which produced relatively high basal virus titers, resulted in a clear increase in transepithelial permeability (Fig. 5). Although most of the infectivity was still found in the apical medium (Fig. 3B), it must be assumed that NiV-induced syncytium formation together with other virus-induced cytopathic effects interfere with the correct distribution of cellular membrane and junction proteins or affect signaling pathways, finally disrupting the cell layer to such an extent that a substantial amount of virus can overcome the epithelial barrier and reach the basolateral side. In agreement with the view that junctions are disrupted only at late time points p.i., electron microscopy images and immunostainings with the marker zonula occludens protein 3 (ZO-3) showed that tight junctions between NiV-positive syncytia and neighboring uninfected cells were intact at 24 h p.i. (data not shown).
Fig 4.
Kinetics of NiV spread in polarized MDCK cells. MDCK cells were grown on permeable filter supports until they had reached full polarity. The cells were then infected with NiV at an MOI of 10. At the indicated time points after infection, the cells were fixed with 4% PFA, permeabilized with methanol, and incubated with a monoclonal antibody directed against the NiV M protein and AF 568-conjugated secondary antibodies. Images were recorded with a Leica SP 5 II confocal laser scanning microscope. Bar, 100 μm.
Fig 5.

Permeability of NiV-infected polarized MDCK cell monolayers. MDCK cells were cultivated for 5 days on permeable filter supports and were infected from the apical side at an MOI of 0.01 (low-dose NiV) or an MOI of 10 (high-dose NiV). Horseradish peroxidase (HRP) was added to the apical filter chamber of uninfected (mock) or NiV-infected cells at 24, 48, and 72 h p.i. After 1 h, HRP flowthrough from the apical to the basal medium was quantified by measuring the HRP activity in the basal medium. Values were normalized to HRP activity in the basal medium of an empty filter without cells (equal to 1). Error bars indicate standard deviations (n ≥ 3). Mock is shown as the mean of the values obtained at 24, 48, and 72 h in five independent experiments. OD, optical density.
Distribution of envelope-associated proteins changes during infection.
The finding that larger amounts of basolateral virus were found only late during high-dose infections, when the permeability of the cell monolayer was drastically increased, suggests that NiV is not substantially released from basolateral membranes. As we have shown that both NiV glycoproteins must be expressed basolaterally to mediate spread of infection via cell-to-cell fusion (27), vectorial apical virus release is unlikely determined by the expression pattern of the NiV surface glycoproteins G and F. To evaluate the idea that apical budding is defined by the third viral envelope protein, the M protein, we determined the localization of the NiV envelope proteins in infected polarized MDCK cells at early and late infection stages. Since syncytium formation was already seen at the earliest time point that viral proteins could be detected (14 h p.i.) (Fig. 4), single, nonfused cells in early infection phases were not found. Early-infected cells were detected only at the edges of syncytia at any time point of infection. Cells in the center of a syncytium had already undergone fusion with neighboring cells and therefore represent cells late in infection. To analyze protein distributions, infected cells were inactivated with 4% PFA at 24 h p.i. and either stained from both sides of the filter insert with NiV-specific antibodies detecting the G and F glycoproteins (Fig. 6A and B) or permeabilized and subsequently incubated with M-specific monoclonal antibodies (Fig. 6C). To determine the colocalization of the glycoproteins and the M protein, costaining of the G and M proteins was performed (Fig. 6D). Confocal vertical sections from the apical-to-basal direction through the NiV-positive syncytia (Fig. 6, Gallery) were taken and superimposed to give a composite stack of all images (Fig. 6, Hyperstack). The blue pseudocolor in the hyperstack, representing the fluorescence in apical sections, is concentrated mostly in the middle of the syncytia. The red/white pseudocolor illustrating the signals in the lateral and basal sections is weaker and seen predominantly at the edges of the syncytia, suggesting that protein amounts and distributions differ in distinct parts of a syncytium. To evaluate this idea, vertical sections along lines through cells at the edge (lines 1) and the center (lines 2) of the syncytia were recorded. Figure 6 (right, panels 1) shows the protein distribution in early stages of the infection. Here, cells were not yet evidently fused, and the NiV glycoproteins F and G were found almost exclusively at the basolateral side, while the M protein was still localized predominantly in the cytoplasm (Fig. 6A to C, panels 1). A colocalization of M and G was not observed (Fig. 6D, section 1). In the sections through the center of the syncytium, virus-mediated cell-to-cell fusion had already disrupted the lateral cell membranes. Here, much larger protein amounts were detected, and G, F, and M accumulated almost exclusively at apical membranes and showed a clear colocalization (late infection) (Fig. 6A to D, panels 2). The changes in the protein distributions suggest that the early infection phase is short and transient because G/F-mediated fusion with neighboring cells is initiated rapidly. Due to increasing fusion and virus-induced cytopathic effects, lateral membranes are disrupted, and basolateral sorting is abolished. As a consequence, G and F proteins synthesized in late infection phases accumulate predominantly at apical cell surfaces, where they meet the M protein. Consequently, the number of early-infected cells is always limited, and most proteins are apically expressed in cells in late infection stages. This view was supported when we quantified the total protein amounts by measuring the signal intensities above background fluorescence levels in apical sections (sections 1 to 45 of the gallery) and basolateral sections (sections 46 to 100). Eighty-eight percent of the total G, 86% of the total F, and 95% of the total M signals were detected in apical sections. The colocalization of G and M in basolateral sections (Fig. 6D) was below 1%. The predominant apical expression of all three NiV envelope-associated proteins is in full agreement with the observed apical NiV release.
Fig 6.

Distribution of the NiV envelope proteins in early-infected cells and in late stages of infection. MDCK cells grown on filter supports were infected with NiV (MOI of 10) after they reached full polarity. At 24 h p.i., the cells were fixed with 4% PFA. (A and B) NiV glycoproteins were stained with specific antibodies against the F protein (A) or the G protein (B). (C) M protein was detected after permeabilization with methanol by incubation with an anti-M monoclonal antibody. All primary antibodies were visualized with AF 568-conjugated secondary antibodies. For costaining of the G and M proteins, fixed cells were incubated with G-specific antibodies and AF 568-labeled anti-rabbit antibodies (red). The cells were then permeabilized, and the M protein was stained with M-specific and AF 488-conjugated antibodies (green). (D) Merged images are shown. Left panels (Gallery) show confocal horizontal xy sections from top (top left) to bottom (bottom right). Sections were superimposed by using a customized NIH ImageJ hyperstack function to give a pseudocolored composite stack of all sections (Hyperstack). The color bar represents the pseudocolor code used for apical (blue) to basal (red/white) sections. White lines in the hyperstack indicate the regions along which vertical sections of early-infected, nonfused cells at the edges of syncytia (lines 1) or fused cells late in infection in the center of foci (lines 2) were recorded. Right panels (Side view) show vertical xz sections through early-infected cells (panels 1) and cells late in infection (panels 2). Images were recorded with a Zeiss 510Meta confocal laser scanning microscope. Bars, 10 μm.
M is not cotransported with the NiV glycoproteins.
The lack of M accumulation at basolateral surfaces at which the NiV glycoproteins were readily detected (Fig. 6A to C), together with the only partial colocalization of M and NiV glycoproteins at apical surface membranes later in infection (Fig. 6D), suggested independent apical M trafficking. To evaluate the hypothesis of a glycoprotein-independent surface transport of M, we analyzed the distribution of the M protein in monensin-treated cells. Monensin is an ionophore that blocks transport of transmembrane proteins in the medial-Golgi compartment (51, 52). To determine if a transport block of one or both NiV glycoproteins affects M trafficking, MDCK cells were cotransfected with plasmids encoding NiV M and either the F or G protein or were infected with NiV. One to two hours later, monensin was added for 24 h at a concentration of 20 μM. The transfected or infected cells were then fixed, permeabilized, and incubated with specific monoclonal antibodies to costain the M protein and either the F protein (Fig. 7A) or the G protein (Fig. 7B). In untreated cells, both NiV glycoproteins and the M protein showed membrane accumulation and marked colocalization (Fig. 7A and B). Upon treatment with monensin, the colocalization was lost. Glycoprotein F was retained intracellularly, while M protein localization was not influenced in both M- and F-cotransfected cells and NiV-infected cells (Fig. 7A). Similarly, the G protein was found to be accumulating in perinuclear intracellular compartments in monensin-treated cells, while the M protein was still localized at the plasma membrane (Fig. 7B). These results clearly indicate that intracellular retention of the NiV glycoproteins had no influence on the M surface localization, supporting the model that M transport occurs independently of the viral glycoproteins.
Fig 7.

NiV envelope protein transport in the presence of monensin. Nonpolarized MDCK cells grown on coverslips were cotransfected with M and either the NiV F or G gene or were infected with NiV (MOI of 0.01). At 2 h after transfection or at 1 h p.i., 20 μM monensin was added to the medium. Twenty-four hours later, the cells were fixed and permeabilized with methanol. The envelope proteins were stained by using M-, F-, or G-specific antibodies. The glycoproteins were then visualized with AF 488-labeled anti-rabbit IgG antibodies (green), and the M protein was detected with AF 568-conjugated anti-mouse IgG antibodies (red). (A) Colocalization of M and F proteins in the absence of monensin (control w/o monensin) and in the presence of monensin (M/F cotransfection + monensin and NiV infection + monensin). (B) Colocalization of M and G proteins in the absence of monensin (control w/o monensin) and in the presence of monensin (M/G cotransfection + monensin and NiV infection + monensin). The insets represent expansions of the merged images. Bars, 10 μm.
Apical M targeting in stably expressing MDCK cells.
To finally determine if not only M transport to the cell surface per se but also selective targeting to apical surfaces in polarized epithelial cells is an intrinsic property, we analyzed the M protein distribution in polarized cells in the absence of NiV infection. For this purpose, we generated MDCK cell clones stably expressing the NiV M gene. Constitutively M-expressing cells were seeded onto Transwell filters, and the M distribution at different time points after seeding was analyzed by coimmunostaining of the M protein and the junction marker E-cadherin. Until 4 days after seeding, the cells were not yet polarized, as indicated by a low transepithelial resistance (TER) and the lack of distinct E-cadherin staining at the cell junctions (data not shown). At 4 days postseeding, cells had established a polarized monolayer giving a TER of >180 Ω cm2 and displaying correct junctional E-cadherin staining. In such polarized cell cultures, the M protein was expressed predominantly in patches at the apical cell membrane (Fig. 8). This indicates targeted apical M accumulation that does not depend on other NiV proteins or infection-induced processes.
Fig 8.
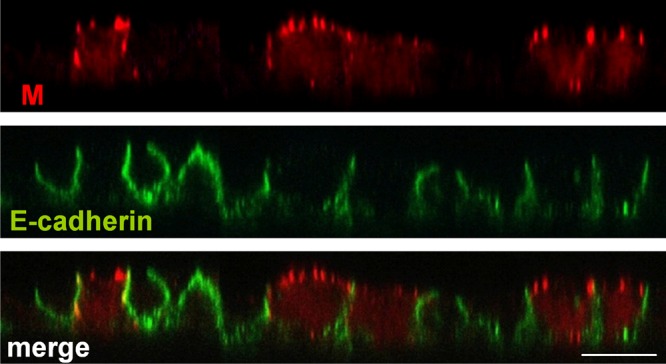
Distribution of NiV M protein upon stable expression. MDCK cells stably expressing flag-tagged NiV M were seeded onto permeable filter supports. The cells were fixed, permeabilized, and stained at 4 days after seeding by using a monoclonal antibody directed against the flag epitope and AF 568-labeled secondary antibodies (red). The cellular adherence junction protein E-cadherin was detected with specific antibodies and AF 488-conjugated secondary antibodies (green). Bar, 10 μm.
DISCUSSION
In this study, we report for the first time that the entry of NiV into epithelial cells is bipolar, while virus egress is mainly restricted to the apical cell surface. The latter is the result of an almost exclusive apical accumulation of the cytoplasmic M protein which does not depend on other viral proteins. While the M protein was located in the cytoplasm or associated with apical plasma membranes, the two NiV surface glycoproteins were found to be expressed basolaterally early in infection mediating cell-to-cell fusion, thereby allowing a rapid spread of infection. As a consequence of the pronounced cell-to-cell fusion and ongoing virus replication, the cell polarity was lost at later time points of infection, and the NiV glycoproteins were no longer sorted basolaterally. The resulting G and F retargeting to the apical surface allows them to meet the M protein, which finally leads to efficient apical virus budding. Since cell-to-cell fusion not only interferes with basolateral glycoprotein sorting but also disrupts epithelial cell integrity and increases transepithelial permeability, apically released virus particles can later overcome the epithelial barrier.
Entry of NiV is bipolar.
Many mammalian viruses initially gain access to their hosts by crossing epithelial barriers in the respiratory, digestive, or reproductive tract either with or without infection of the epithelial cells themselves. Since epithelial tissues act as barriers between body cavities and underlying tissues, viruses have to adapt to find ways across. To penetrate the epithelial barrier, viruses such as HIV, Epstein-Barr virus, hepatitis C virus, or measles virus use cells of the immune system with innate capacities to cross epithelia (macrophages and dendritic cells) as “Trojan horses” (53–56). Adenoviruses exploit cytokine secretion activities of macrophages associated with the apical surface of the epithelium to induce a redistribution of their basolateral receptors to the apical surface, where they mediate virus entry (57). As a further example, coxsackie B virus binds and clusters apical CD55 receptors, which do not mediate virus entry but activate signaling and actin reorganization that leads to translocation of virus particles to tight junctions, where the viruses then engage their basolateral coxsackie-adenovirus receptor (CAR) entry receptors (58). We have shown here that NiV can use a less sophisticated way by directly and rapidly infecting epithelial cells from either the apical or basolateral side. Supporting our model of bidirectional NiV entry, primary human bronchial epithelial cells could also be infected from both sides (L. Sauerhering and S. Erbar, unpublished data). Along with observations in vivo describing epithelial cell infections in different infection phases (18, 59, 60), our finding of bipolar entry of NiV into epithelial cells suggests the model that infection from the apical (luminal) side of epithelia in upper airways supports the establishment of an infection in a new host. During the viremic phase, entry via basolateral plasma membranes into respiratory (or urinary) epithelial cells most likely allows efficient infection and virus shedding from mucosal surfaces.
NiV release occurs via apical surfaces.
The site of virus release is assumed to influence the spread of an infection. Apically released viruses cause mainly local infections restricted to mucosal surfaces at the viral entry site, while basolateral or bipolar virus shedding from epithelia facilitates systemic spread. Although NiV causes systemic infection, virus particles are released predominantly from apical surfaces, not only in MDCK cells (Fig. 3) but also in primary human bronchial epithelial cells (Sauerhering and Erbar, unpublished). Overcoming of the epithelial barrier is thus not mediated by basolateral or bipolar virus release from epithelial cells. Our data rather suggest that local NiV infection of the first epithelial target cells, likely in the oronasal cavity and the upper respiratory tract, disrupts the integrity of the polarized epithelial cell layer, allowing the virus to pass the epithelial barrier and to infect other cells present in the mucosa. Immune cells then likely play a crucial role in further virus dissemination (59, 61). Similar to what has been shown for measles virus, canine distemper virus, or HIV (54, 62–65), hijacking of immune cells might be primarily responsible for the initiation of systemic NiV infection. In the viremic phase, NiV reaches mucosal surfaces of the lower respiratory and urinary tracts by the hematogenous route and infects epithelial cells via basal membranes. Apical virus release subsequently supports efficient virus shedding by airway secretions and urine.
M selectively accumulates at apical plasma membranes.
Viral matrix proteins are initially soluble cytoplasmic proteins. Their intracellular transport mechanisms must therefore differ from the well-characterized secretory trafficking pathways of transmembrane glycoproteins. A previous study revealed that NiV M crosses the nucleus, where it becomes monoubiquitinated before it is exported to the cytoplasm and reaches the plasma membrane (14). Surface transport was proposed to depend on ubiquitination and two potential late (L) domain motifs in the M protein, although the exact transport route is not yet known (12–14). Our finding that M accumulated at apical membranes suggests that the NiV M protein is targeted apically via a robust transport machinery that is not affected by virus-mediated cytopathic effects. One might hypothesize that M does not use a directed vesicle-based polarized transport pathway, which is assumed to be largely disturbed in syncytial cells, but rather traffics randomly through the cell. The observed apical accumulation may then be due to binding to a highly stable apical binding partner, which is not affected by NiV-induced syncytium formation. This could be a component of the apical cytocortex, such as a member of the ezrin-radixin-moesin (ERM) family (66). It remains to be elucidated if NiV M-driven apical budding needs actin dynamics or a direct or indirect interaction with the actin cytoskeleton, as was demonstrated previously for a number of enveloped viruses (46).
Polarized basolateral localization of the NiV glycoproteins is lost in late infection stages.
In some virus infections, surface distributions of glycoproteins and matrix proteins overlap only partly in polarized epithelial cells (47, 67). Envelope proteins can even be expressed at opposing membranes. Here, partial retargeting of the glycoproteins by the matrix protein to the site of budding is believed to be required for virus release (25, 29, 30, 34, 68, 69). Resembling measles virus (29), NiV M, F, and G proteins possess opposing targeting signals. While the M protein accumulates at apical surfaces, the two glycoproteins possess tyrosine-dependent transport signals that mediate basolateral targeting upon single expression. The basolateral sorting is lost upon coexpression of G and F, suggesting that cell-to-cell fusion causes a loss of cell polarization to such an extent that G and F are no longer correctly targeted (27). In agreement with this model, basolateral sorting of the glycoproteins was seen only in early stages of NiV infection, when cells had not yet undergone fusion (Fig. 6A and B, section 1). In syncytia, the basolateral distribution was lost. While minor amounts of the G/F proteins were still found at basolateral membranes of small syncytia (27), both glycoproteins almost exclusively accumulated apically in large syncytia (Fig. 6A and B, section 2). This indicates that the distribution of G and F changes depending on the infection stage, from mainly basolateral (early phase) to predominantly apical (late infection phases). Late in infection, ongoing cell-to-cell fusion and virus replication cause extensive cytopathic effects and likely disrupt all basolateral trafficking pathways. The consecutive accumulation of the glycoproteins at apical surfaces might be triggered by an apical G/F retargeting promoted by the M protein, as proposed previously for other viruses (25, 29, 30, 34, 68, 69). However, there is no evidence so far that G, F, and M meet and interact intracellularly before reaching the plasma membrane. Cotransport to apical membranes is therefore highly unlikely. Apical accumulation of the glycoproteins might rather be due to apical targeting signals in G and F, which are functional only if the dominant basolateral signals are not recognized (70). The existence of such apical signals is assumed from the finding that G and F mutants with mutated basolateral targeting signals are not expressed bipolarly but are rather sorted to apical membranes (27). Further studies are planned to elucidate the molecular requirements for the predominantly apical G/F accumulation in the latest stages of NiV infection.
Model for NiV replication in epithelial cells.
Based on the presented data, we propose the following model for NiV entry, spread, and exit from polarized cells. When NiV enters a new host by the respiratory route, airway epithelia are targeted from the luminal (apical) side. Later, during the viremic phase of infection, when the virus reaches the mucosa of the respiratory or urinary tract from the blood, NiV must enter epithelial cells from the submucosal (basolateral) side. In agreement with this in vivo situation, NiV entry into polarized epithelial cells was found to be bipolar (Fig. 9A). After successful infection of epithelial cells, NiV F and G are initially expressed basolaterally, thereby allowing rapid cell-to-cell fusion and spread of infection within the monolayer. In these early stages of infection, M is still localized in the cytoplasm (Fig. 9B). Later, when syncytia are formed, the M protein and both glycoproteins accumulate at apical surfaces, resulting in an efficient apical release of infectious virus particles (Fig. 9C). The productive virus replication together with the pronounced G/F-mediated fusion processes finally cause a disruption of the epithelial monolayer that later allows a transmigration of apically released viral particles to the basal side (Fig. 9D).
Fig 9.

Schematic model for NiV entry, spread, and release and envelope protein distribution in polarized epithelial cells. (A) NiV can enter the epithelium from either the luminal (apical) or basolateral side. (B) Early in infection, viral G and F proteins are localized basolaterally, while the M protein is still localized in the cytoplasm. Basolaterally expressed viral glycoproteins lead to cell-to-cell fusion, facilitating lateral spread of infection. (C) Later in infection, all three envelope proteins (M, G, and F) concentrate at apical plasma membranes, leading to apical virus release. (D) Virus replication and cell-to-cell fusion finally disrupt epithelial cell integrity and allow subsequent spread of apical virus through the partly disrupted epithelial barrier.
ACKNOWLEDGMENTS
We are grateful to Heinz Feldmann (Hamilton, MT), and Benhur Lee (UCLA) for providing anti-NiV antibodies. We thank Anja Heiner and Sonja Heck for excellent technical assistance. We also thank Carolin Weise for help with the confocal immunofluorescence microscopy. All work with NiV was performed in the BSL-4 facility of the Philipps University of Marburg.
B.L. and L.S. were supported by fellowships of the Studienstiftung des Deutschen Volkes and the Jürgen-Manchot-Stiftung, respectively. This work was supported by grants of the German Research Foundation (DFG) to A.M. (GK 1216/2 and MA 1886/6-1).
Footnotes
Published ahead of print 2 January 2013
REFERENCES
- 1. Yob JM, Field H, Rashdi AM, Morrissy C, van der Heide B, Rota P, bin Adzhar A, White J, Daniels P, Jamaluddin A, Ksiazek T. 2001. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg. Infect. Dis. 7:439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–1435 [DOI] [PubMed] [Google Scholar]
- 3. Lo MK, Lowe L, Hummel KB, Sazzad HM, Gurley ES, Hossain MJ, Luby SP, Miller DM, Comer JA, Rollin PE, Bellini WJ, Rota PA. 2012. Characterization of Nipah virus from outbreaks in Bangladesh, 2008-2010. Emerg. Infect. Dis. 18:248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Rasche A, Yordanov S, Seebens A, Oppong S, Adu Sarkodie Y, Pongombo C, Lukashev AN, Schmidt-Chanasit J, Stöcker A, Carneiro AJ, Erbar S, Maisner A, Fronhoffs F, Buettner R, Kalko EK, Kruppa T, Franke CR, Kallies R, Yandoko ER, Herrler G, Reusken C, Hassanin A, Krüger DH, Matthee S, Ulrich RG, Leroy EM, Drosten C. 2012. Bats host major mammalian paramyxoviruses. Nat. Commun. 3:796 doi:10.1038/ncomms1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayman DT, Suu-Ire R, Breed AC, McEachern JA, Wang L, Wood JL, Cunningham AA. 2008. Evidence of henipavirus infection in West African fruit bats. PLoS One 3:e2739 doi:10.1371/journal.pone.0002739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harcourt BH, Tamin A, Ksiazek TG, Rollin PE, Anderson LJ, Bellini WJ, Rota PA. 2000. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 271:334–349 [DOI] [PubMed] [Google Scholar]
- 7. Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, Bishop KA, Choudhry V, Dimitrov DS, Wang LF, Eaton BT, Broder CC. 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. U. S. A. 102:10652–10657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Mol. Biol. 15:567–572 [DOI] [PubMed] [Google Scholar]
- 9. Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405 [DOI] [PubMed] [Google Scholar]
- 10. Negrete OA, Wolf MC, Aguilar HC, Enterlein S, Wang W, Muhlberger E, Su SV, Bertolotti-Ciarlet A, Flick R, Lee B. 2006. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog. 2:e7 doi:10.1371/journal.ppat.0020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ciancanelli MJ, Basler CF. 2006. Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J. Virol. 80:12070–12078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patch JR, Han Z, McCarthy SE, Yan L, Wang LF, Harty RN, Broder CC. 2008. The YPLGVG sequence of the Nipah virus matrix protein is required for budding. Virol. J. 5:137 doi:10.1186/1743-422X-5-137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Wolf MC, Holbrook MR, Freiberg AN, Lee B. 2010. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 6:e1001186 doi:10.1371/journal.ppat.1001186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weingartl HM, Berhane Y, Czub M. 2009. Animal models of henipavirus infection: a review. Vet. J. 181:211–220 [DOI] [PubMed] [Google Scholar]
- 16. Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, Ksiazek TG, Zaki SR, Paul G, Lam SK, Tan CT. 1999. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 354:1257–1259 [DOI] [PubMed] [Google Scholar]
- 17. de Wit E, Bushmaker T, Scott D, Feldmann H, Munster VJ. 2011. Nipah virus transmission in a hamster model. PLoS Negl. Trop. Dis. 5:e1432 doi:10.1371/journal.pntd.0001432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rockx B, Brining D, Kramer J, Callison J, Ebihara H, Mansfield K, Feldmann H. 2011. Clinical outcome of henipavirus infection in hamsters is determined by the route and dose of infection. J. Virol. 85:7658–7671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maisner A, Neufeld J, Weingartl H. 2009. Organ- and endotheliotropism of Nipah virus infections in vivo and in vitro. Thromb. Haemost. 102:1014–1023 [DOI] [PubMed] [Google Scholar]
- 20. Rodriguez-Boulan E, Nelson WJ. 1989. Morphogenesis of the polarized epithelial cell phenotype. Science 245:718–725 [DOI] [PubMed] [Google Scholar]
- 21. Simons K, Wandinger-Ness A. 1990. Polarized sorting in epithelia. Cell 62:207–210 [DOI] [PubMed] [Google Scholar]
- 22. Connolly-Andersen AM, Magnusson KE, Mirazimi A. 2007. Basolateral entry and release of Crimean-Congo hemorrhagic fever virus in polarized MDCK-1 cells. J. Virol. 81:2158–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dylla DE, Michele DE, Campbell KP, McCray PB., Jr 2008. Basolateral entry and release of New and Old World arenaviruses from human airway epithelia. J. Virol. 82:6034–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuller S, von Bonsdorff CH, Simons K. 1984. Vesicular stomatitis virus infects and matures only through the basolateral surface of the polarized epithelial cell line, MDCK. Cell 38:65–77 [DOI] [PubMed] [Google Scholar]
- 25. Maisner A, Klenk H, Herrler G. 1998. Polarized budding of measles virus is not determined by viral surface glycoproteins. J. Virol. 72:5276–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 76:5654–5666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weise C, Erbar S, Lamp B, Vogt C, Diederich S, Maisner A. 2010. Tyrosine residues in the cytoplasmic domains affect sorting and fusion activity of the Nipah virus glycoproteins in polarized epithelial cells. J. Virol. 84:7634–7641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Batonick M, Oomens AG, Wertz GW. 2008. Human respiratory syncytial virus glycoproteins are not required for apical targeting and release from polarized epithelial cells. J. Virol. 82:8664–8672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Naim HY, Ehler E, Billeter MA. 2000. Measles virus matrix protein specifies apical virus release and glycoprotein sorting in epithelial cells. EMBO J. 19:3576–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanger C, Muhlberger E, Ryabchikova E, Kolesnikova L, Klenk HD, Becker S. 2001. Sorting of Marburg virus surface protein and virus release take place at opposite surfaces of infected polarized epithelial cells. J. Virol. 75:1274–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zimmer G, Zimmer KP, Trotz I, Herrler G. 2002. Vesicular stomatitis virus glycoprotein does not determine the site of virus release in polarized epithelial cells. J. Virol. 76:4103–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moll M, Diederich S, Klenk HD, Czub M, Maisner A. 2004. Ubiquitous activation of the Nipah virus fusion protein does not require a basic amino acid at the cleavage site. J. Virol. 78:9705–9712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thiel L, Diederich S, Erbar S, Pfaff D, Augustin HG, Maisner A. 2008. Ephrin-B2 expression critically influences Nipah virus infection independent of its cytoplasmic tail. Virol. J. 5:163 doi:10.1186/1743-422X-5-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kolesnikova L, Ryabchikova E, Shestopalov A, Becker S. 2007. Basolateral budding of Marburg virus: VP40 retargets viral glycoprotein GP to the basolateral surface. J. Infect. Dis. 196(Suppl 2):S232–S236 doi:10.1086/520584 [DOI] [PubMed] [Google Scholar]
- 35. Berhane Y, Berry JD, Ranadheera C, Marszal P, Nicolas B, Yuan X, Czub M, Weingartl H. 2006. Production and characterization of monoclonal antibodies against binary ethylenimine inactivated Nipah virus. J. Virol. Methods 132:59–68 [DOI] [PubMed] [Google Scholar]
- 36. Erbar S, Maisner A. 2010. Nipah virus infection and glycoprotein targeting in endothelial cells. Virol. J. 7:305 doi:10.1186/1743-422X-7-305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moll M, Kaufmann A, Maisner A. 2004. Influence of N-glycans on processing and biological activity of the Nipah virus fusion protein. J. Virol. 78:7274–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199 [DOI] [PubMed] [Google Scholar]
- 39. Cathomen T, Buchholz CJ, Spielhofer P, Cattaneo R. 1995. Preferential initiation at the second AUG of the measles virus F mRNA: a role for the long untranslated region. Virology 214:628–632 [DOI] [PubMed] [Google Scholar]
- 40. Middleton DJ, Westbury HA, Morrissy CJ, van der Heide BM, Russell GM, Braun MA, Hyatt AD. 2002. Experimental Nipah virus infection in pigs and cats. J. Comp. Pathol. 126:124–136 [DOI] [PubMed] [Google Scholar]
- 41. Mungall BA, Middleton D, Crameri G, Bingham J, Halpin K, Russell G, Green D, McEachern J, Pritchard LI, Eaton BT, Wang LF, Bossart KN, Broder CC. 2006. Feline model of acute Nipah virus infection and protection with a soluble glycoprotein-based subunit vaccine. J. Virol. 80:12293–12302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wong KT, Grosjean I, Brisson C, Blanquier B, Fevre-Montange M, Bernard A, Loth P, Georges-Courbot MC, Chevallier M, Akaoka H, Marianneau P, Lam SK, Wild TF, Deubel V. 2003. A golden hamster model for human acute Nipah virus infection. Am. J. Pathol. 163:2127–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Diederich S, Sauerhering L, Weis M, Altmeppen H, Schaschke N, Reinheckel T, Erbar S, Maisner A. 2012. Activation of the Nipah virus fusion protein in MDCK cells is mediated by cathepsin B within the endosome-recycling compartment. J. Virol. 86:3736–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delorme-Axford E, Coyne CB. 2011. The actin cytoskeleton as a barrier to virus infection of polarized epithelial cells. Viruses 3:2462–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Realpe M, Espinosa R, López S, Arias CF. 2010. Rotaviruses require basolateral molecules for efficient infection of polarized MDCKII cells. Virus Res. 147:231–241 [DOI] [PubMed] [Google Scholar]
- 46. Taylor MP, Koyuncu OO, Enquist LW. 2011. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 9:427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Owens RJ, Dubay JW, Hunter E, Compans RW. 1991. Human immunodeficiency virus envelope protein determines the site of virus release in polarized epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 88:3987–3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodriguez Boulan E, Pendergast M. 1980. Polarized distribution of viral envelope proteins in the plasma membrane of infected epithelial cells. Cell 20:45–54 [DOI] [PubMed] [Google Scholar]
- 49. Schlie K, Maisa A, Freiberg F, Groseth A, Strecker T, Garten W. 2010. Viral protein determinants of Lassa virus entry and release from polarized epithelial cells. J. Virol. 84:3178–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tashiro M, McQueen NL, Seto JT, Klenk HD, Rott R. 1996. Involvement of the mutated M protein in altered budding polarity of a pantropic mutant, F1-R, of Sendai virus. J. Virol. 70:5990–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Griffiths G, Quinn P, Warren G. 1983. Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus. J. Cell Biol. 96:835–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mollenhauer HH, Morré DJ, Rowe LD. 1990. Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim. Biophys. Acta 1031:225–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Izquierdo-Useros N, Naranjo-Gómez M, Erkizia I, Puertas MC, Borràs FE, Blanco J, Martinez-Picado J. 2010. HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? PLoS Pathog. 6:e1000740 doi:10.1371/journal.ppat.1000740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yüksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog. 7:e1001263 doi:10.1371/journal.ppat.1001263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shannon-Lowe CD, Neuhierl B, Baldwin G, Rickinson AB, Delecluse HJ. 2006. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 103:7065–7070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stamataki Z, Shannon-Lowe C, Shaw J, Mutimer D, Rickinson AB, Gordon J, Adams DH, Balfe P, McKeating JA. 2009. Hepatitis C virus association with peripheral blood B lymphocytes potentiates viral infection of liver-derived hepatoma cells. Blood 113:585–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lütschg V, Boucke K, Hemmi S, Greber UF. 2011. Chemotactic antiviral cytokines promote infectious apical entry of human adenovirus into polarized epithelial cells. Nat. Commun. 2:391 doi:10.1038/ncomms1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131 [DOI] [PubMed] [Google Scholar]
- 59. Middleton DJ, Weingartl HM. 2012. Henipaviruses in their natural animal hosts. Curr. Top. Microbiol. Immunol. 359:105–121 [DOI] [PubMed] [Google Scholar]
- 60. Rockx B, Winegar R, Freiberg AN. 2012. Recent progress in henipavirus research: molecular biology, genetic diversity, animal models. Antiviral Res. 95:135–149 [DOI] [PubMed] [Google Scholar]
- 61. Mathieu C, Pohl C, Szecsi J, Trajkovic-Bodennec S, Devergnas S, Raoul H, Cosset FL, Gerlier D, Wild TF, Horvat B. 2011. Nipah virus uses leukocytes for efficient dissemination within a host. J. Virol. 85:7863–7871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Swart RL, Ludlow M, de Witte L, Yanagi Y, van Amerongen G, McQuaid S, Yuksel S, Geijtenbeek TB, Duprex WP, Osterhaus AD. 2007. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. 3:e178 doi:10.1371/journal.ppat.0030178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Piguet V, Steinman RM. 2007. The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. 28:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rudd PA, Cattaneo R, von Messling V. 2006. Canine distemper virus uses both the anterograde and the hematogenous pathway for neuroinvasion. J. Virol. 80:9361–9370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. von Messling V, Milosevic D, Cattaneo R. 2004. Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc. Natl. Acad. Sci. U. S. A. 101:14216–14221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morales FC, Takahashi Y, Kreimann EL, Georgescu MM. 2004. Ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc. Natl. Acad. Sci. U. S. A. 101:17705–17710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mora R, Rodriguez-Boulan E, Palese P, Garcia-Sastre A. 2002. Apical budding of a recombinant influenza A virus expressing a hemagglutinin protein with a basolateral localization signal. J. Virol. 76:3544–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dietzel E, Anderson DE, Castan A, von Messling V, Maisner A. 2011. Canine distemper virus matrix protein influences particle infectivity, particle composition, and envelope distribution in polarized epithelial cells and modulates virulence. J. Virol. 85:7162–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Riedl P, Moll M, Klenk HD, Maisner A. 2002. Measles virus matrix protein is not cotransported with the viral glycoproteins but requires virus infection for efficient surface targeting. Virus Res. 83:1–12 [DOI] [PubMed] [Google Scholar]
- 70. Matter K, Mellman I. 1994. Mechanisms of cell polarity: sorting and transport in epithelial cells. Curr. Opin. Cell Biol. 6:545–554 [DOI] [PubMed] [Google Scholar]