

Abstract
Earlier studies have shown that active MEK blocks the activation of protein kinase R (PKR), a component of antiviral innate immune responses. In this report we show that the herpes simplex virus 1 virion host shutoff (VHS) RNase protein and MEK (mitogen-activated protein kinase kinase) act cooperatively in blocking the activation of PKR. This conclusion is based on the following. (i) In contrast to viral gene expression in the parental cell line or a cell line expressing a constitutively active MEK, the replication of a VHS mutant is particularly impaired in cells expressing dominant negative MEK. In this cell line PKR is activated by phosphorylation, and the accumulation of several viral proteins is delayed. (ii) In transfected cells, wild-type VHS blocked the activation of PKR, whereas PKR was activated in cells transfected with a mutant VHS or with plasmids encoding the VHS RNase and VP16 and VP22, the two viral proteins that neutralize the RNase activity of VHS. The results suggest that early in infection the VHS RNase degrades RNAs that activate PKR. Coupled with published data, the results suggest that inhibition of activation of PKR or its effect on viral replication is staged early in infection by VHS, postsynthesis of VP16 and VP22 by the γ134.5 protein, and very late in infection by the US11 protein.
INTRODUCTION
Protein kinase R (PKR) is one of the major innate immune mechanisms resident in the cytoplasm of eukaryotic cells (reviewed in reference 1). In the presence of double-stranded RNA or interferon, the PKR is activated by phosphorylation, dimerizes, and activates two defense mechanisms. Specifically it phosphorylates the translation elongation factor eIF-2α, and it activates NF-κB (2–4). The consequence of phosphorylation of eIF-2α (eIF-2α∼P) is the total shutoff of protein synthesis. Activation of NF-κB results in activation of numerous stress response antiviral genes that ultimately can shut off viral replication (5–8). Virtually all viruses block activation or the consequences of activation of PKR. For most of these viruses, it is unclear if the multiple PKR inhibitory mechanisms are redundant roles or are necessary functions to regulate PKR activity at different stages in the virus life cycle (reviewed in reference 9). In the case of herpes simplex virus 1 (HSV-1), a viral gene, designated γ134.5, whose expression is partially dependent on the onset of viral DNA synthesis encodes a protein, ICP34.5, that recruits protein phosphatase 1a to dephosphorylate eIF-2α∼P (10–12). HSV-1 also encodes a second protein, US11 which, if expressed prior to the activation of PKR, binds to it and prevents its activation (13). In this report we show that PKR activation is also blocked by the virion host shutoff (VHS) protein, an RNase encoded by the UL41 gene of HSV-1.
The studies reported here were based on two observations. First, VHS-null (ΔVHS) mutant viruses exhibit nearly normal growth in continuous cell lines such as HEp-2 or Vero. However, studies carried out in primary cultures showed significant growth differences between ΔVHS mutant viruses and wild-type parent viruses (14). Additionally, ΔVHS mutants are severely compromised in experimental animal systems (15, 16). Interestingly, this defect is partially overcome when interferon (IFN) signaling is absent, suggesting that VHS plays a significant role in blocking activation of interferon-dependent defense mechanisms for virus replication and pathogenesis (17–19). Earlier studies using a cell line stably expressing a constitutively activated form of MEK (caMEK) have shown that MEK plays a key role in the suppression of PKR activation during viral replication (20). Since PKR is activated by RNA, the question arose whether activated MEK recruits an RNase and, more specifically, the VHS RNase to block reactivation.
MATERIALS AND METHODS
Cell culture.
Vero cells (American Type Culture Collection) were propagated in minimal essential medium (MEM) supplemented with 6% fetal bovine serum (FBS). HT1080 cells having lost the activated mutant N-ras allele (obtained from E. J. Stanbridge, Irvine, CA) and clonal transfectants derived from the HT1080 parent cell line, designated HT-caMEK and HT-dnMEK (where dn is dominant negative) and referred to as clones HT84-4 and HT92-6, were described elsewhere (20). The HT1080 cells and derived cells lines were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco/Invitrogen Corporation, Grand Island, NY) supplemented with 10% fetal bovine serum (Lonza, Belgium).
Cell infection and viruses.
HSV-1(F) is the prototype HSV-1 strain used in Roizman laboratory (21). The mutant ΔVHS virus R2621 has been described elsewhere (22). Cells were seeded onto 12-well plates at 3 × 105 cells per dish or in 25-cm2 flasks at 1.5 × 106 per flask. The next day, cells were exposed to 10 or 1 PFU per cell of wild-type or R2621 mutant virus for 1 h at 37°C and then removed and replaced with medium. The infection continued at 37°C for the length of time indicated for each experiment. Cells were either harvested for immunoblotting or collected for assaying viral recovery on Vero cell monolayers.
Extraction of viral DNA.
Confluent cultures of HT1080 and HT92-6 (dnMEK) cells grown in 25-cm2 flasks were infected with 1 PFU of HSV-1(F) or R2621 per cell. Twenty-four hours after infection, the infected cells were resuspended in 1 ml of TRIzol RNA/DNA/protein isolation reagent and used for DNA extraction, according to the manufacturer's instructions. Briefly, from all samples the remaining aqueous phase overlying the interphase was removed. The DNA was precipitated from the interphase and organic phase with ethanol by the addition of 0.3 ml of 100% ethanol per 1 ml of TRIzol reagent used for the initial homogenization. Next, the samples were stored at room temperature (RT) for 2 to 3 min, and DNA was sedimented by centrifugation at 2,000 × g for 5 min at 4°C. The DNA pellet was rinsed twice in a solution containing 0.1 M sodium citrate in 10% ethanol. At each rinse, the DNA pellet was stored in the washing solution for 30 min at room temperature with periodic mixing and centrifuged at 2,000 × g for 5 min at 4°C. Next, the DNA pellet was suspended in 75% ethanol, stored for 10 to 20 min at room temperature with periodic mixing, and centrifuged at 2,000 × g for 5 min at 4°C. To remove the ethanol, the DNA pellet was air-dried by keeping tubes open for 3 to 5 min at RT. The DNA, derived from 106 cells, was dissolved in 0.1 ml of 8 mM NaOH. The insoluble material was removed by centrifugation at 12,000 × g for 10 min. Following solubilization in 8 mM NaOH, the pH of the DNA sample was adjusted to 8.4 with HEPES.
Real-time PCR.
Real-time PCR was carried out in a 25-μl reaction mixture containing 1 μl of DNA preparation, 0.5 μM each forward and reverse primer (For-59-CATCACCGACCCGGAGAGGGAC; Rev-59-GGGCCAGGCGCTTGTTGGTGTA), 300 nM TaqMan probe (59-6FAM-CCGCCGAACTGAGCAGACACCCGCGC-TAMRA, where 6FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine), and 12.5 μl of Maxima Probe qPCR Master Mix (2×) (Maxima Probe qPCR; Fermentas Life Sciences). The amplification was carried out with the aid of a Cepheid SmartCycler II System (Cepheid Europe, France) under the following conditions: incubation for 10 min at 95°C, followed by 40 cycles of 30s at 95°C, 30s at 55°C, and 30s at 72°C, with a final cycle of 5 min at 72°C. Each amplification run contained two negative controls and 10-fold serially diluted reference DNA obtained from bacterial artificial chromosome (BAC)-HSV in order to generate the standard curve. Viral load was derived from the threshold cycle (CT) using the standard curve generated in parallel, and the result is expressed as concentration in μg of DNA/μl (23).
Cytoplasmic protein extraction and immunoblotting.
Infected cells were subjected to cytoplasmic protein extraction for Western blot analysis (24). Cell pellets containing approximately 3 × 106 cells were collected at the time indicated for each experiment, washed in 1× phosphate-buffered saline (PBS), and resuspended in 150 μl of hypotonic buffer A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT], 0.2 mM phenylmethylsulfonyl fluoride [PMSF]). The cell suspensions were then incubated on ice for 15 min and lysed by 15 passages through a 25-gauge needle. Cytoplasm fractions were collected by centrifugation at 12,000 rpm for 5 min at 4°C. An equal amount of the cytoplasmic fraction was subjected to electrophoresis in 10% denaturing polyacrylamide gels, transferred to nitrocellulose membranes (Bio-Rad Life Science Research, Hercules, CA), blocked, and reacted with primary antibody followed by the appropriate secondary antibody.
Antibodies.
Polyclonal antibodies against the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the total and phosphorylated forms of PKR (at Thr446) were purchased from Ambion Life Technologies (Grand Island, NY) and Cell Signaling Technology (Beverly, MA), respectively. Monoclonal antibodies to glycoprotein D and ICP0 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The rabbit anti-glutathione S-transferase (GST)-UL49 polyclonal antibody was described elsewhere (25). Secondary antibodies anti-rabbit and anti-mouse IgG conjugated to peroxidase were also from Santa Cruz Biotechnology. Protein bands were visualized using SuperSignal West Pico as a chemiluminescent substrate (Thermo Scientific, Rockford, IL).
Transient transfection.
HT1080 cells were transfected with the plasmids pVHS, pVHSm, pVP16, and pVP22 expressing the wild-type UL41, nuclease-defective mutant UL41, UL48, and UL49 open reading frames (ORFs), respectively, as described elsewhere (26). The construct MTS1 (27), derived from the pAcSG2 transfer vector (PharMingen), was used as a control. A total of 2 × 105 cells were seeded in 12-well plates in the presence of RPMI medium (Lonza) supplemented with 10% FBS (Lonza). After overnight incubation, 1.5 μg of total DNA representing the plasmids above was incubated with Lipofectamine Reagent Plus (Invitrogen) and in the presence of OptiMEM (Gibco). The DNA-Lipofectamine mixture was then added to cultured cells and incubated for 4 h at 37°C. The medium was then replaced with OptiMEM supplemented with 10% FBS. Forty-eight hours after the medium was changed, the cells were collected and tested for total and phosphorylated forms of PKR by Western blot analysis.
RESULTS
Characterization of virus growth in a cell line expressing a dominant negative form of MEK.
In this series of experiments, we compared the replication of a ΔVHS mutant virus (R2621) with wild-type HSV-1(F) virus in cell lines derived from human fibrosarcoma HT1080 cells stably expressing a dominant negative form of MEK (dnMEK). Parental HT1080 and dnMEK (HT92-6) cell lines were either mock infected or exposed to 10 PFU of HSV-1(F) or R2621 virus per cell. Viral replication was assessed at several levels, such as appearance of cytopathic effect (CPE), accumulation of viral DNA, and protein and virus yields. The results were as follows.
HT1080 and HT92-6 cells exposed to HSV-1(F) both exhibited pronounced CPE (Fig. 1b and e) 12 h after virus exposure, suggesting that the wild-type virus replicated similarly in the two cell lines. Conversely, while the HT1080 cells exposed to R2621 showed the characteristic signs of infection (Fig. 1c), CPE was not observed in HT92-6 cells exposed to the ΔVHS mutant virus (Fig. 1f).
Fig 1.
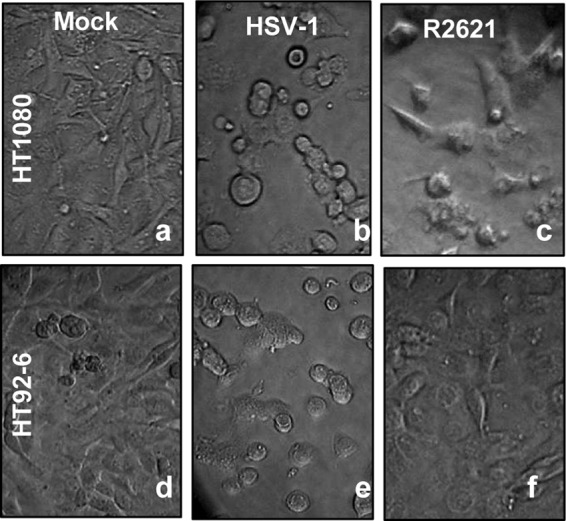
Cytopathic effects in infected parental HT1080 and dnMEK (HT92-6) cell lines. Replicate cultures of HT-1080 and HT92-6 cells were either mock infected (a and d) or infected with 10 PFU of HSV-1(F) (b and e) or R2621 mutant (c and f) virus per cell. Images were taken at 12 h after infection.
To determine whether the absence of CPE was due to lack of viral DNA synthesis, replicate cultures of HT1080 or HT92-6 cells were exposed to 10 PFU of HSV-1(F) or R2621 virus per cell. The cells were harvested 24 h after infection and analyzed by real-time PCR for the presence of viral DNA, as described in Materials and Methods. As shown in Fig. 2, even though fewer viral DNA copies were detected in HT92-6 cells infected by wild-type HSV-1(F) than in the HSV-1(F)-infected HT1080 parental cells, a striking reduction in viral DNA accumulation was observed in HT92-6 cells infected by R2621 mutant virus. We conclude that the HT1080 wild-type human fibrosarcoma cells are permissive to R2621 viral replication, whereas the HT92-6 cells derived from HT1080 cells and ectopically expressing a dominant negative form of MEK restrict the replication of the ΔVHS mutant R2621.
Fig 2.
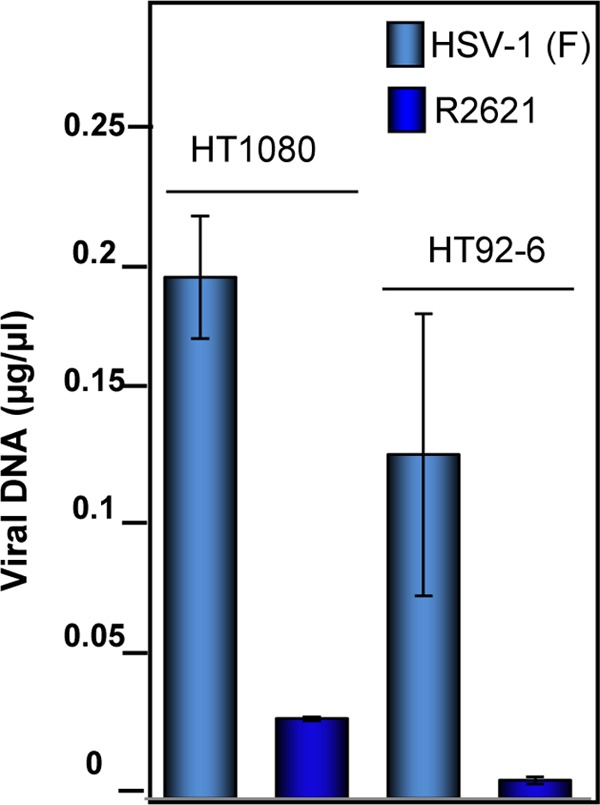
Viral replication is variable in parental HT1080, dnMEK (HT92-6), and caMEK (HT84-4) cell lines. Cells were exposed to 1 PFU of HSV-1(F) or R2621 mutant virus per cell in serum-free medium. At 24 h after infection, the amounts of viral DNA were determined by real-time PCR using a TaqMan probe specific for UL30.
To investigate the role of MEK kinases in enabling the replication of R2621 mutant virus, we employed a cell line expressing a constitutively active form of MEK (caMEK), designated HT84-4. Two series of experiments were carried out to determine the effect of caMEK on viral growth. In the first, we measured the viral yields obtained in infected parental HT1080 cells and in the derivative HT84-4 (caMEK) and HT92-6 (dnMEK) cells. The results shown in Table 1 highlight two major points. (i) Both wild-type and ΔVHS mutant viruses showed impaired replication in HT92-6 cells compared to replication in the parental HT1080 cells, suggesting that the inhibition of MEK activity by expression of a dnMEK affects negatively the ability of HSV-1 to replicate. This conclusion is corroborated by the observation that the replication of both viruses is enhanced in cells expressing caMEK. (ii) The growth of ΔVHS mutant virus is substantially impaired in all three cell lines compared to that of wild-type HSV-1, indicating that VHS is required to overcome antiviral responses mounted in those cells.
Table 1.
Virus yields in different cell lines infected with wild-type HSV-1 or R2621
| Cell line | Virus yield (no. of PFU/ml)a |
|
|---|---|---|
| HSV-1(F) | R2621 | |
| HT1080 | 2.2 × 108 ± 3.5 × 107 | 4.0 × 106 ± 1.4 × 106 |
| HT92-6 | 7.3 × 107 ± 7.0 × 105 | 9.0 × 105 ± 4.2 × 105 |
| HT84-4 | 3.9 × 108 ± 4.9 × 107 | 1.7 × 107 ± 1.9 × 107 |
PFU counts were determined by a plaque assay on Vero cells. The data are shown as means of duplicates ± standard deviations.
To determine whether virus yield correlated with the expression of viral proteins, we analyzed viral protein accumulation either in parental cells or in those expressing dnMEK or caMEK upon infection with HSV-1(F) or R2621. Replicate cultures of HT1080, HT92-6, and HT84-4 cells were mock infected or exposed to 10 PFU of HSV-1(F) or R2621 virus per cell. Equal amounts of the lysates of infected cells harvested 9 or 24 h after infection were electrophoretically separated in denaturing polyacrylamide gels, transferred to nitrocellulose membranes, and probed with antibodies to representative α (ICP0) and γ (gD and VP22) viral proteins. The results shown in Fig. 3 were the following. (i) The levels of accumulation of the viral proteins in cells infected with HSV-1(F) appeared to be similar in the three cell lines (Fig. 3A, lanes 1 to 6). (ii) As could be predicted from the studies on the accumulation of viral DNA, the levels of accumulation of viral proteins in the three cell lines infected with the R2621 mutant virus varied considerably. At 9 h after infection with R2621, the accumulation of ICP0 in the caMEK (HT84-4) line was approximately four times higher than that in parental cell line (HT1080). ICP0 was not detected in HT92-6 cells (Fig. 3B, lanes 1 to 3). Similarly gD was not detected in HT92-6 cells (Fig. 3B, lane 2), and VP22 was not detected in either HT1080 or HT92-6 cells. At 24 h after infection, we detected smaller amounts of ICP0, gD, and VP22 in all cell lines. The decreases in the amounts of VP22 and ICP0 in HT92-6 cells infected with R2621 mutant virus are greater than the apparent decrease in total protein loaded in that lane.
Fig 3.
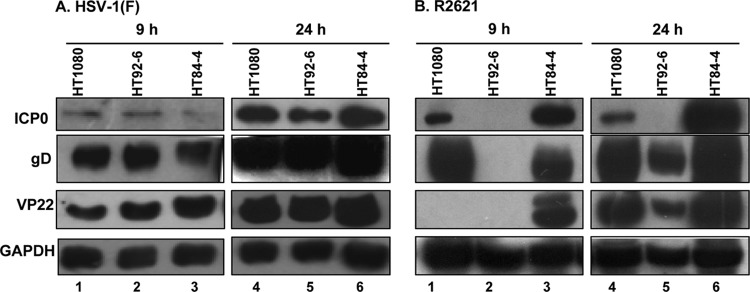
Viral gene accumulation in parental HT1080, dnMEK (HT92-6), and caMEK (HT84-4) infected cells. To determine the expression of viral genes in presence or absence of MEK protein, replicate cultures of HT1080, HT92-6, and HT84-4 cells were exposed to 10 PFU per cell of HSV-1(F) wild-type virus (A) or R2621 mutant virus (B). Cells were harvested at 9 and 24 h after infection and processed as described in Materials and Methods. Electrophoretically separated proteins were immunoblotted with antibodies for the α protein ICP0, β/γ protein gD, and γ protein VP22. GAPDH was used as a loading control.
PKR is phosphorylated in cells infected with ΔVHS mutant virus.
As noted above, MEK plays a fundamental role in blocking activation of PKR activity during HSV-1 replication. Furthermore, increased permissiveness to herpes simplex virus infections is correlated to the overactivation of Ras-dependent pathways that alter the activity of PKR through MEK (28). Based on this evidence, we next investigated the status of PKR activation in the three cell lines infected with wild-type or ΔVHS-null mutant virus. Replicate cultures of HT1080, HT92-6, or HT84-4 cells were exposed to 10 PFU of HSV-1(F) or R2621 virus per cell. The cells were harvested at 3, 9, or 24 h after infection and processed as described in Materials and Methods. The cytoplasmic proteins were electrophoretically separated in denaturing polyacrylamide gels, transferred to nitrocellulose membranes, and probed with antibodies directed to total and phosphorylated forms of PKR. GAPDH served as a loading control. The results (Fig. 4) were as follows. As expected, infection of the HT1080 parental cell line or of HT84-4 cells by wild-type virus did not result in accumulation of phosphorylated PKR (Fig. 4A, p-PKR, lanes 1 to 6 and 13 to 18). However, cells expressing dnMEK showed a basal level of PKR phosphorylation (Fig. 4A, lanes 7, 9, and 11) that was slightly increased starting 9 h after HSV-1(F) infection (Fig. 4A, lanes 10 and 12). Interestingly, phosphorylated PKR was detected in the three cell lines infected by R2621 (Fig. 4B, lanes 4, 6, 10, 12, 16, and 18). Quantitative analysis of the level of phosphorylation revealed a higher level of PKR activation in cells expressing dnMEK than in the parental or caMEK-expressing cells. These findings suggest that VHS acts cooperatively with MEK to modify the level of activation of PKR and consequently plays an important role in controlling the innate defense mechanism of the host cell. Indeed, PKR can achieve its function in innate antiviral defense when MEK and VHS are not functional.
Fig 4.
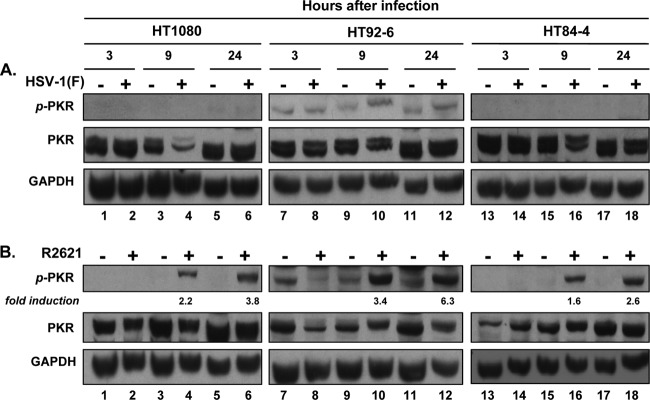
PKR activation in infected parental HT1080, dnMEK (HT92-6), and caMEK (HT84-4) cells. To determine the influence of mutant MEK expression on PKR activation, replicate cultures of HT1080, HT92-6, and HT84-4 cells were exposed to 10 PFU per cell of HSV-1(F) wild-type virus (A) or R2621 mutant virus (B). Cells were harvested at 3, 9, and 24 h after infection, and cytoplasmic extracts were prepared as described in Materials and Methods. Electrophoretically separated proteins were immunoblotted with antibodies for total PKR and the phosphorylated form of PKR (on threonine 446) (p-PKR). GAPDH was used as a loading control. The fold induction of the phosphorylated form of PKR was determined using the TINA program to measure the intensity of the band normalized to GAPDH.
HSV-1 VHS selectively suppresses expression of PKR in HT1080 cell lines.
In order to verify the involvement of the VHS in the control of PKR, HT1080 cells were transfected with plasmids expressing wild-type or catalytically inactive VHS protein under the control of the cytomegalovirus (CMV) promoter either alone or along with plasmids expressing two other tegument proteins, UL48 (VP16) and UL49 (VP22), previously reported to regulate the function of VHS (26, 29). The cells were harvested 48 h after transfection, and lysates were electrophoretically separated in a denaturing polyacrylamide gel, transferred to nitrocellulose membranes, and probed with antibodies against total and phosphorylated forms of PKR and against GAPDH as a loading control. The results reported in Fig. 5 show that phosphorylated PKR was not detected in cells transfected with wild-type VHS (lane 2). This phenomenon is most likely dependent on the VHS RNase activity inasmuch as it was not observed in cells transfected with the RNase-defective form of VHS (lane 3) or those coexpressing the tegument proteins VP22 and VP16 (Fig. 5, lane 4). These results suggest that VHS controls the activation of PKR during viral infection by degrading the RNA that ultimately activates PKR.
Fig 5.
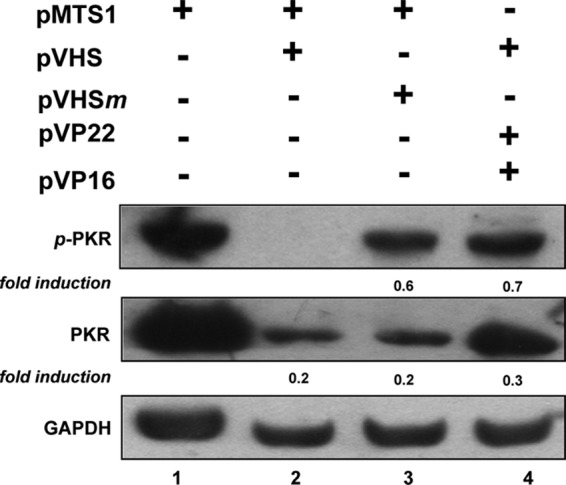
Transient transfection of VHS suppresses the expression of phospho-PKR (p-PKR) in HT1080 cell lines. Cells were transfected with 1.5 μg of plasmid DNA expressing wild-type (pVHS) or catalytically inactive (pVHSm) VHS protein under the control of the CMV promoter either alone or in the presence of plasmids expressing two other tegument proteins, UL48 (pVP16) and UL49 (pVP22). Plasmid pMTS1 was used as a control. The transfected cells were incubated for 48 h before being harvested for detection of total PKR and phosphorylated forms of PKR on threonine 446. The fold induction of both the phosphorylated form of PKR and PKR were determined separately using the TINA program to measure the intensity of the band normalized to GAPDH.
DISCUSSION
In earlier studies, Pasieka et al. (32) reported that ΔVHS mutants accumulated increased amounts of interferon-stimulated transcripts and larger amounts of phosphorylated eIF-2α and postulated that cell infected with ΔVHS mutants accumulated to increased levels of double-stranded RNA. The key findings reported here complement and extend the studies reported by Pasieka et al. (32) and may be summarized as follows: wild-type virus and ΔVHS mutants replicate to nearly equal levels in most continuous cell lines. In these cell lines, as exemplified by the HT1080 line, the replication of ΔVHS mutants is affected by the status of MEK. The viruses grow well in cell lines expressing constitutively active MEK and very poorly in cell lines expressing a dominant negative MEK.
Earlier studies have shown that constitutively active MEK blocks activation of PKR (20). This report demonstrated that the VHS RNase plays a key role in activation of PKR. Specifically, whereas wild-type virus enhanced slightly the activation of PKR in cells expressing dnMEK, the ΔVHS mutant activated PKR in both the parental cell lines and those expressing dnMEK and even in the cell lines expressing constitutively active MEK. This finding leads to the conclusion that VHS plays a significant role in blocking activation of PKR at early times after infection.
PKR is known to be activated by double-stranded DNA or by interferon (1–4). In the case of the VHS RNase, the possible target for blocking activation of PKR is RNA. In the case of HSV-1-infected cells, likely sources of double-stranded RNA are complementary RNAs transcribed during infection or RNAs that fold and self-anneal to long stretches of double-stranded RNA. In studies published many years ago, viral RNAs that were deliberately self annealed and then digested with single-strand-specific RNase were complementary to as much as 50% of the viral DNA (30, 31). In principle, the VHS RNase could clear both cellular and viral RNAs capable of self-annealing to produce double-stranded stretches capable of activating PKR.
Activation of PKR is a very important innate immune response targeted by most of the viruses that infect and multiply in eukaryotic cells (9). A fundamental strategy of HSV to combat innate immune responses is to target multiple functions to block them. Indeed, it may be argued that the number of functions targeted by the virus to block a specific response reflects the importance of the innate response as a threat to virus replication and dissemination (10–13). In the case of PKR, three fundamental viral strategies have become apparent. First, as illustrated in Fig. 6, early in infection the VHS RNase degrades RNA that could potentially activate PKR. In due course, with the onset of synthesis of VP16 and VP22, VHS is neutralized and no longer degrades RNA. At this point ICP34.5, the product of the γ134.5 gene, appears, becomes active, and reverses the effect of activated PKR by dephosphorylating eIF-2α∼P to block disruption of protein synthesis in the infected cells. Finally, late in infection, US11, a very late viral protein, binds to inactive PKR and blocks its activation. The results reported here and in preceding studies show that activated MEK plays a major role in blocking activation of PKR. It is not clear whether HSV can activate MEK to add to the functions that block PKR.
Fig 6.
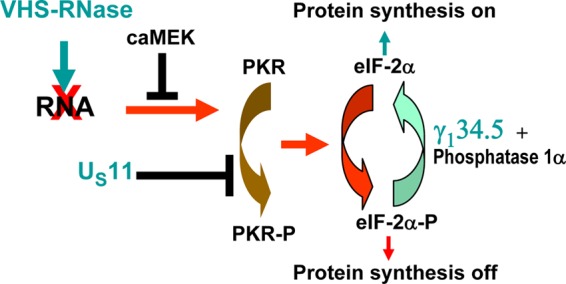
Schematic representations of the viral and cellular pathways that affect activation of protein kinase R (PKR). Early in infection the VHS RNase introduced into cells during the course of infection degrades the RNA that activates PKR. The VHS RNase is neutralized by the viral DNA synthesis-dependent onset of synthesis of VP16 (UL48) and VP22 (UL49). With the onset of synthesis of viral DNA, there is augmented synthesis of ICP34.5, the product of the γ134.5 gene; ICP34.5 recruits protein phosphatase 1α to dephosphorylate eIF-2α, there by enabling continuous protein synthesis in the infected cells. Late in infection, US11 may play a role in blocking PKR activation by binding to it. In addition, activated MEK blocks activation of PKR. PKR-P, phosphorylated PKR.
It should be noted that degradation of RNA would block the activation of PKR. In contrast, by the time ICP34.5 enters into the fray, PKR is phosphorylated and active. By dephosphorylating eIF-2α, ICP34.5 and PP1α deal with the consequences of activation of PKR but not with the active PKR per se. Earlier studies have shown that one function of activated PKR is to activate NF-κB and that HSV-1 requires some products of genes induced by NF-κB for optimal replication (18, 19).
ACKNOWLEDGMENTS
These studies were aided by the Italian Ministry of University and Research, Research Projects of National Interest 2008, and by a grant from the National Cancer Institute (R01CA088860).
Footnotes
Published ahead of print 9 January 2013
REFERENCES
- 1. García MA, Meurs EF, Esteban M. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811 [DOI] [PubMed] [Google Scholar]
- 2. Levin D, London IM. 1978. Regulation of protein synthesis: Activation by double-stranded RNA of a protein kinase that phosphorylates eukaryotic initiation factor 2. Proc. Natl. Acad. Sci. U. S. A. 75:1121–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vattem KM, Staschke KA, Wek RC. 2001. Mechanism of activation of the double-stranded RNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2). Eur. J. Biochem. 268:3674–3684 [DOI] [PubMed] [Google Scholar]
- 4. Williams BR. 1999. PKR: a sentinel kinase for cellular stress. Oncogene 18:6112–6120 [DOI] [PubMed] [Google Scholar]
- 5. Gil J, Rullas J, Garcia MA, Alcami J, Esteban M. 2001. The catalytic activity of dsRNA-dependent protein kinase, PKR, is required for NF-κB activation. Oncogene 20:385–394 [DOI] [PubMed] [Google Scholar]
- 6. Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. 2000. NF-κB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-κB-inducing kinase and IκB kinase. Mol. Cell. Biol. 20:1278–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dev A, Iyer S, Razani B, Cheng G. 2011. NF-κB and innate immunity. Curr. Top. Microbiol. Immunol. 349:115–143 [DOI] [PubMed] [Google Scholar]
- 8. Bonnet MC, Daurat C, Ottone C, Meurs EF. 2006. The N-terminus of PKR is responsible for the activation of the NF-κB signaling pathway by interacting with the IKK complex. Cell Signal. 18:1865–1875 [DOI] [PubMed] [Google Scholar]
- 9. Langland JO, Cameron JM, Heck MC, Jancovich JK, Jacobs BL. 2006. Inhibition of PKR by RNA and DNA viruses. Virus Res. 119:100–110 [DOI] [PubMed] [Google Scholar]
- 10. Chou J, Chen JJ, Gross M, Roizman B. 1995. Association of a Mr 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2α and premature shutoff of protein synthesis after infection with γ134.5− mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 92:10516–10520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cassady KA, Gross M, Roizman B. 1998. The second-site mutation in the herpes simplex virus recombinants lacking the γ134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2α. J. Virol. 72:7005–7011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He B, Gross M, Roizman B. 1998. The γ134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 273:20737–20743 [DOI] [PubMed] [Google Scholar]
- 13. Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 72:8620–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzutani T, Nagamine M, Shibaki T, Ogasawara M, Yoshida I, Daikoku T, Nishiyama Y, Azuma M. 2000. The role of the UL41 gene of herpes simplex virus type 1 in evasion of non-specific host defence mechanisms during primary infection. J. Gen. Virol. 81:1763–1771 [DOI] [PubMed] [Google Scholar]
- 15. Strelow L, Smith T, Leib D. 1997. The virion host shutoff function of herpes simplex virus type 1 plays a role in corneal invasion and functions independently of the cell cycle. Virology 231:28–34 [DOI] [PubMed] [Google Scholar]
- 16. Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 189:663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 77:9337–9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taddeo B, Luo TR, Zhang W, Roizman B. 2003. Activation of NF-κB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100:12408–12413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taddeo B, Zhang W, Lrkeman F, Roizman B. 2004. Cells lacking NF-κB or in which NF-κB is not activated vary with respect to ability to sustain herpes simplex virus 1 replication and are not susceptible to apoptosis induced by a replication-incompetent mutant virus. J. Virol. 78:11615–11621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith KD, Mezhir JJ, Bickenbach K, Veerapong J, Charron J, Posner MC, Roizman B, Weichselbaum RR. 2006. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Δγ134.5 mutants of herpes simplex virus 1. J. Virol. 80:1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364 [DOI] [PubMed] [Google Scholar]
- 22. Poon AP, Roizman B. 1997. Differentiation of the shutoff of protein synthesis by virion host shutoff and mutant γ134.5 genes of herpes simplex virus 1. Virology 229:98–105 [DOI] [PubMed] [Google Scholar]
- 23. Kessler HH, Mühlbauer G, Rinner B, Stelzl E, Berger A, Dörr HW, Santner B, Marth E, Rabenau H. 2000. Detection of herpes simplex virus DNA by real-time PCR. J. Clin. Microbiol. 38:2638–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sciortino MT, Medici MA, Marino-Merlo F, Zaccaria D, Giuffrè-Cuculletto M, Venuti A, Grelli S, Mastino A. 2008. Involvement of HVEM receptor in activation of nuclear factor κB by herpes simplex virus 1 glycoprotein D. Cell. Microbiol. 10:2297–2311 [DOI] [PubMed] [Google Scholar]
- 25. Blaho JA, Mitchell C, Roizman B. 1994. An amino acid sequence shared by the herpes simplex virus 1 alpha regulatory proteins 0, 4, 22, and 27 predicts the nucleotidylylation of the UL21, UL31, UL47, and UL49 gene products. J. Biol. Chem. 269:17401–17410 [PubMed] [Google Scholar]
- 26. Taddeo B, Sciortino MT, Zhang W, Roizman B. 2007. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. U. S. A. 104:12163–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sciortino MT, Taddeo B, Poon AP, Mastino A, Roizman B. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. U. S. A. 99:8318–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Farassati F, Yang AD, Lee PW. 2001. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat. Cell Biol. 3:745–750 [DOI] [PubMed] [Google Scholar]
- 29. Sciortino MT, Taddeo B, Giuffrè-Cuculletto M, Medici MA, Mastino A, Roizman B. 2007. Replication-competent herpes simplex virus 1 isolates selected from cells transfected with a bacterial artificial chromosome DNA lacking only the UL49 gene vary with respect to the defect in the UL41 gene encoding host shutoff RNase. J. Virol. 81:10924–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kozak M, Roizman B. 1975. RNA synthesis in cells infected with herpes simplex virus. IX. Evidence for accumulation of abundant symmetric transcripts in nuclei. J. Virol. 15:36–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jacquemont B, Roizman B. 1975. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and double-stranded RNA prepared from them. J. Virol. 15:707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pasieka RJ, Lu B, Crosby SD, Wylie KM, Morrison LA, Alexander DE, Menachery VD, Leib DA. 2008. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J. Virol. 82:5527–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]