

Abstract
West Nile virus (WNV) is a RNA virus of the family Flaviviridae and the leading cause of mosquito-borne encephalitis in the United States. Humoral immunity is essential for protection against WNV infection; however, the requirements for initiating effective antibody responses against WNV infection are still unclear. CD22 (Siglec-2) is expressed on B cells and regulates B cell receptor signaling, cell survival, proliferation, and antibody production. In this study, we investigated how CD22 contributes to protection against WNV infection and found that CD22 knockout (Cd22−/−) mice were highly susceptible to WNV infection and had increased viral loads in the serum and central nervous system (CNS) compared to wild-type (WT) mice. This was not due to a defect in humoral immunity, as Cd22−/− mice had normal WNV-specific antibody responses. However, Cd22−/− mice had decreased WNV-specific CD8+ T cell responses compared to those of WT mice. These defects were not simply due to reduced cytotoxic activity or increased cell death but, rather, were associated with decreased lymphocyte migration into the draining lymph nodes (dLNs) of infected Cd22−/− mice. Cd22−/− mice had reduced production of the chemokine CCL3 in the dLNs after infection, suggesting that CD22 affects chemotaxis via controlling chemokine production. CD22 was not restricted to B cells but was also expressed on a subset of splenic DCIR2+ dendritic cells that rapidly expand early after WNV infection. Thus, CD22 plays an essential role in controlling WNV infection by governing cell migration and CD8+ T cell responses.
INTRODUCTION
West Nile virus (WNV) is a member of the Flaviviridae family, and protective immunity against WNV requires both innate and adaptive immune responses. Type I and type II interferon (IFN) production and signaling, as well as humoral and CD4+/CD8+ T effector responses (1), are required to protect against lethal WNV infection. B cell-deficient (μMT) mice are highly susceptible to WNV infection (2), and notably, mice that are unable to secrete IgM are also highly susceptible (3). Passive transfer of immune sera protects μMT mice from rapidly succumbing to WNV infection (2, 4), underscoring the importance of humoral immunity. In addition, adoptive transfer of purified B cells from immune mice partially rescues immunodeficient Rag1−/− mice, which lack functional B and T cells, from succumbing to WNV infection (2). Together, these findings highlight the importance of antibody and B cell effector responses for protection against encephalitic disease and prompted us to further investigate the requirements for generating protective B cell responses to WNV.
CD22 (Siglec-2) is a cell surface receptor with expression in humans and mice predominantly restricted to B cells (5–7). It is approximately 135 kDa in size and contains an intracellular immunoreceptor tyrosine-based inhibitory motif within its cytoplasmic tail (8, 9). CD22 can function in trans to mediate adhesion between B cells and other cell types (6, 10, 11) and also in cis with the B cell receptor (BCR) to modulate BCR signaling pathways (12–14). The proximal extracellular portion of CD22 binds to glycoproteins that contain α2,6-sialic acid linkages (15), and a number of cell types express ligands that bind to CD22, including T cells, B cells, and dendritic cells (DCs) (16, 17). However, relatively little is known about how CD22 signaling affects these non-B cell populations.
CD22 is important for regulation of B cell antibody production and other functions in activated B cells. Cd22−/− mice produce normal antibody responses to T cell-dependent protein antigens but have diminished responses to T cell-independent antigens and reduced numbers of marginal zone B cells, demonstrating a role for CD22 in generating antibody responses (16, 18). In addition, Cd22−/− B cells exhibit increased apoptosis and dysregulated proliferation upon BCR ligation (19–22). Although CD22 regulates multiple B cell functions, the role of CD22 in protection against viral pathogens is unclear. For example, Cd22−/− mice infected with lymphocytic choriomeningitis virus (23), vesicular stomatitis virus (23), or Staphylococcus aureus (24) have no differences in survival compared to wild-type (WT) mice. As B cells and antibody responses are essential for protective immunity against WNV infection, we hypothesized that the loss of CD22 would impair anti-WNV humoral responses and protection from infection.
In this study, we investigated the role of CD22 in protection against WNV infection. Cd22−/− mice were highly susceptible and succumbed to WNV infection, but surprisingly, this was not due to defects in antibody responses. Rather, we found a defect in accumulation of antigen-specific CD8+ T cells in the spleens and the brains of Cd22−/− mice, which was due to impaired cell proliferation and cell migration to infected tissues. We conclude that expression of CD22 regulates CD8+ T cell-mediated immune responses and cell migration after WNV infection.
MATERIALS AND METHODS
Mice and WNV infection.
Cd22−/− mice (Cd22tm1Eac) (21) were generated and backcrossed to C57BL/6J mice for more than 10 generations. B cell-deficient μMT breeders (B6.129S2-Ighmtm1Cgn/J) were kindly provided by David Rawlings (University of Washington). C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and subsequently bred in-house. All mice were housed and maintained in a specific-pathogen-free (SPF) facility at the University of Washington. Female and male 6- to 10-week-old, age- and sex-matched mice were injected under anesthesia in one or both hind footpads with 10 μl of virus diluted in a vehicle of Hank's buffered salt solution (HBSS) containing 1% fetal calf serum (FCS). All procedures were approved and conducted according to regulations of the Institutional Animal Care and Use Committee (IACUC) of the University of Washington (Seattle, WA).
Virus.
All virus stocks were maintained and characterized by the Virology Core within the University of Washington's Center for Flavivirus Immunity. The WNV isolate TX-2002 HC (WNV-TX) strain (25) was used at 103 PFU/dose, as determined by a plaque assay using baby hamster kidney (BHK) cells. In some experiments, a viral molecular clone generated from plaque-purified WNV-TX was used at the dose described above (26). No differences in pathogenesis have been observed between the biological isolate and the infectious cloned viruses used in this study.
Viral tissue burden quantification.
Mice were sacrificed and perfused with 30 ml of phosphate-buffered solution (PBS), and tissues were harvested into centrifuge tubes containing 1.4-mm PreCellys ceramic beads (Cayman Chemical, Ann Arbor, MI) and 500 μl of PBS. All tissues were homogenized in a PreCellys-24 homogenizer (Bertin Technologies, Paris, France) at 5,000 rpm for 20 s, except brain tissues which went through two cycles of homogenization. Samples were spun down, and the supernatants were analyzed by a standard plaque assay using BHK cells. Viral titers in sera and popliteal draining LNs (dLNs) were determined by real-time quantitative PCR (RT-qPCR) as previously described (27). Briefly, peripheral blood samples were taken from infected mice at various time points, and viral RNA was extracted using a QiaAMP viral RNA extraction kit (Qiagen, Valencia, CA). Total RNA was extracted from dLNs using RNeasy kits (Qiagen). Sequences for primers and TaqMan probes used were as previously published (28).
Quantification of WNV-specific antibody responses.
To determine relative quantities of WNV-specific antibodies, a sandwich enzyme-linked immunosorbent assay (ELISA) was performed as previously described using WNV envelope (E) protein purified from BL21 bacterial colonies (29). A 5-μg/ml concentration of WNV E protein was diluted in carbonate binding buffer (sodium carbonate in PBS, pH 7.2) and used to coat ELISA plates (Nalge Nunc International, Rochester, NY) at 4°C overnight. Serum samples were inactivated for virus by exposure to UV light for 30 min, serially diluted 3-fold starting at 1/20 in 0.1% milk casein in PBS, and then incubated on the plate for at least 1 h. Goat anti-mouse IgM, total IgG, IgG2b, and IgG2c antibodies conjugated to horseradish peroxidase (HRP) (Southern Biotech, Birmingham, AL) were added at a dilution of 1:2,000 or 1:4,000 as recommended by the manufacturer's protocol. Absorbance values were read at 450-nm and 570-nm wavelengths to correct for machine background on a Bio-Rad Micromanager plate reader (version 1.2). To determine relative quantities, a positive reading was determined to be a value above the cutoff value determined from the mean of negative-control wells (naive and/or mock-infected mice) + the standard deviation of the mean × the standard deviation multiplier, f (30). Negative-control wells contained serial dilutions from at least three individual mice per experiment.
To determine neutralizing antibody titers, serum samples were analyzed in a plaque reduction neutralization titer (PRNT) assay as previously described (27). Briefly, serum samples were diluted in Dulbecco's modified essential medium (DMEM) and complement inactivated by incubation at 56°C for 30 min. Samples and 102 PFU of virus suspended in DMEM were incubated for 1 h at 37°C prior to being plated onto BHK cells in 6-well plates and incubated for another hour before being overlaid with 2 ml of 0.5% agarose.
WNV epitope-specific peptides and major histocompatibility complex (MHC) class I tetramer.
For in vitro restimulation, 1 μM CD8+ T cell-specific NS4B 9-mer SSVWNATTA (31) or CD4+ T cell-specific NS32066–2080 15-mer RRWCFDGPRTNTILE (32) peptide (Genemed Synthesis Inc., San Antonio, TX) was added to 4 × 106 splenocytes cultured with GolgiPlug containing brefeldin A (BD Biosciences, San Diego, CA) at 37°C for 5 or 16 h, respectively. Cells were then spun down and used for intracellular cytokine staining (ICS) as described below.
To generate an MHC class I tetramer, monomeric subunits were generated from NS4B 9-mer peptide at the Fred Hutchinson Immune Monitoring facility (Seattle, WA). Monomers were subsequently tetramerized using streptavidin-phycoerythrin (streptavidin-PE) (BD Biosciences). All tetramer batches were titrated and tested prior to use.
Cell isolation.
Spleens and popliteal dLNs were harvested and resuspended in serum-free RPMI 1640 medium (Thermo Scientific, Waltham, MA) in the presence of Liberase collagenase mix (Roche, Pleasanton, CA) and DNase I (Roche). Tissues were digested at 37°C for 45 min with mechanical disruption using a magnetic stir bar. Cells were then washed with FCS-containing RPMI 1640 medium and spleens lysed with 1× RBC lysis buffer (BioLegend, San Diego, CA) prior to staining for flow cytometry.
For isolation of lymphocytes from the brain, tissues were harvested and finely chopped with scissors over a wire screen mesh in cold 5% FCS-containing PBS. Cells were washed twice with serum-free PBS before being resuspended in 30% Percoll (Sigma-Aldrich, St. Louis, MO). A 70% Percoll layer was underlaid, and cells were spun down for 20 min at room temperature. Lymphocytes were obtained from the 30 to 70% interface and washed with serum-containing RPMI 1640 medium prior to staining for flow cytometry.
Flow cytometry.
At various time points postinfection (p.i.), popliteal dLNs or spleens were harvested from mice and made into a single cell suspension. The following rat anti-mouse antibodies obtained from eBioscience (San Diego, CA), Miltenyi Biotec (Auburn, CA), or BD Biosciences were used: DCIR2-biotin (eBioscience; clone 33D1) with streptavidin-PE or streptavidin PE Cy7 (eBioscience), CD3ε-PerCP Cy5.5 (eBioscience; clone 145-2C11) or CD3ε-PE Cy7 (eBioscience; clone 145-2C11), CD45R/B220-eFluor450 (eBioscience; clone RA3-6B2) or CD45R/B220-peridinin chlorophyll protein (CD45R/B220-PerCP) (BD; clone RA3-6B2), CD11c-allophycocyanin (CD11c-APC) (eBioscience; clone N418), NK1.1-PerCP Cy5.5 (eBioscience; clone PK136), mPDCA1-PE (Miltenyi; clone JFO5-1C2.4.1), CD44-PE Cy7 (eBioscience; clone IM7), CD8α-APC Cy7 (BD; clone 53-6.7), CD4-APC (BD; RM4-5), CD22-fluorescein isothiocyanate (CD22-FITC) or CD22-PE (BD; clone Cy34.1), and Ly5.1-APC (eBioscience; clone A20). Anti-DCAL2 antibody clones were generated in our laboratory and conjugated to APC fluorochromes (33). All viable-cell events were acquired on an LSR II flow cytometer (Becton, Dickinson, Franklin Lakes, NJ) and analyzed with Flowjo software (Treestar Incorporated, Ashland, OR). To determine the absolute cell numbers of a specific cell population, total cell numbers of harvested tissues were counted and multiplied by the frequency of a specific population (expressed as a percentage of total acquired events). To detect WNV-specific CD8+ T cells, cells were first stained with surface markers and washed before being stained with a 1:100 to 1:250 dilution of MHC class I tetramer-PE at 4°C for 25 min. All samples were stained in PBS containing 2% FCS and 0.05% azide (FACS buffer) at 4°C and fixed with PBS containing 4% paraformaldehyde before acquisition.
For ICS, cells were spun out of medium containing WNV-specific peptide and brefeldin A, stained for surface markers, fixed with PBS containing 4% paraformaldehyde, and then permeabilized and stained in FACS buffer containing 0.1% saponin with gamma interferon (IFN-γ)-Pacific blue (eBioscience; clone XMG1.2), tumor necrosis factor alpha (TNF-α)-PE (eBioscience; clone MP6-XT22), perforin-PE (eBioscience; clone eBioOMAK-D), or respective isotype control antibodies.
In vivo cytotoxicity assays.
Procedures for in vivo cytotoxicity assays are described elsewhere (34). Briefly, Ly5.1+ (CD45.1+) splenocytes from C57BL/6 mice were incubated in the presence or absence of 1 μM NS4B peptide for 1 h at 37°C. Cells were labeled with 0.3 μM (unpulsed) or 3 μM (NS4B pulsed) carboxyfluorescein succinimidyl ester (CFSE; Sigma-Aldrich) and then adoptively transferred at a 1:1 ratio (10 × 106 cells total) by intravenous (i.v.) injection either into naive WT or Cd22−/− mice or into WT or Cd22−/− mice (Ly5.2+/CD45.2+) infected 5 days earlier with WNV. Spleens were harvested 2 h after adoptive transfer and splenocytes analyzed by flow cytometry.
Annexin V staining.
Splenocytes were harvested and processed as described above, and cells were stained for surface markers, annexin V-Pacific blue (eBioscience), and Live/Dead-APC (Invitrogen) according to the manufacturer's protocol (eBioscience). Cells were fixed in 1% paraformaldehyde and events immediately acquired on a flow cytometer.
Cell sorting of DC subsets.
DCIR2+ DCs were sorted on a BD Aria machine as previously described (33). Briefly, single-cell suspensions of splenocytes from WT and Cd22−/− mice were positively selected using mouse anti-CD11c magnetic beads and LS magnetic columns (Miltenyi Biotech, Auburn, CA). Selected cells were then incubated with monoclonal antibodies for 30 min on ice: B220-FITC, NK1.1-PE Cy7, CD11c-PE, and CD8α-APC Cy7/Alexa eFluor 780 (eBioscience) and anti-DCAL2-APC, clone P2E7 (33). DCIR2+ DC subsets were B220− NK1.1− CD11chi DCAL2− CD8α−, DCAL2+ DC subsets were B220− NK1.1− CD11chi DCAL2+ CD8α−, and CD8α+ DC subsets were B220− NK1.1− CD11chi DCAL2+ CD8α+. DCIR2+ DC subsets were verified for DCIR2 expression after sorting. Cells were washed twice and sorted. The cell purity was >96% after sorting.
Adoptive-transfer experiments.
For transfer of DCIR2+ DCs, 1.4 × 106 B220− NK1.1− CD11chi DCIR2+ DCAL2− CD8α− DCs were sorted as described above and washed twice with cold PBS before being injected i.v. into age- and sex-matched WT and Cd22−/− recipients. Recipient mice were infected subcutaneously (s.c.) 3 h after transfer with 103 PFU of WNV-TX, and spleens were harvested 7 days postinfection for flow cytometry analysis. For transfer of Ly5.1+ splenocytes, female Ly5.1+ spleens from C57BL/6 mice were harvested and digested with collagenase as described above. After red cell lysis, cells were washed and resuspended at 7.5 × 107/ml in sterile cold PBS. A total of 200 μl of single-cell suspension was injected i.v. into tail veins of recipient WT or Cd22−/− mice (Ly5.2+). One day after transfer, the left hind footpad was injected s.c. with 103 PFU of WNV-TX, while the right hind footpad was left uninjected (naive) or injected s.c. with 10 μl of 1% FCS-HBSS vehicle (mock). Twenty-four hours after infection, left and right popliteal dLNs were harvested and processed for flow cytometry.
In vivo BrdU incorporation.
In vivo bromodeoxyuridine (BrdU) uptake by cell populations was performed using a commercially available BrdU kit according to the manufacturer's protocol (BD Biosciences). Briefly, 24 h prior to harvest of tissues, infected mice were injected with 100 μg/ml of BrdU resuspended in sterile PBS intraperitoneally (i.p.). Tissues were harvested and digested with Liberase as described above, and single cell suspensions were stained for various surface markers. Intracellular BrdU staining was performed the following day after fixation and permeabilization, and events were immediately acquired on a flow cytometer.
RNA isolation and RT-qPCR.
Total mRNA was isolated from whole LNs using Qiagen RNeasy minispin columns and the manufacturer's protocol. Viral RNA was extracted from serum samples using a QiaAMP viral RNA extraction kit (Qiagen). In brief, LNs were homogenized and treated with RLT lysis buffer to obtain lysates for RNA isolation. Whole-blood samples were spun down in Vacutainer serum separator tubes (BD Biosciences), and serum samples were collected and stored at −80°C until use. Isolated RNA was reverse transcribed using a cloned avian myeloblastosis virus (AMV) reverse transcriptase kit (Invitrogen, Grand Island, NY) to make cDNA for RT-qPCR analysis by TaqMan (Applied Biosystems Inc., Foster City, CA) or Sybr green (Roche). Thermocycler conditions and primers for IFN-β and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene analysis using Sybr green were previously described (35); primers were purchased from Invitrogen. Threshold cycle (CT) values from samples analyzed by TaqMan were made relative to 18S rRNA and normalized to WT uninfected samples. CT values from samples analyzed by Sybr green were made relative to the GAPDH housekeeping gene. TaqMan primers for determination of chemokine and 18S expression from whole lymph nodes were designed and purchased from Applied Biosystems Inc. The primers and thermocycler conditions for determination of viral RNA in the serum were as previously described (27, 28).
Type I IFN bioassay.
Serum samples were collected at various time points p.i. and complement inactivated by incubation at 56°C for 30 min. Samples were analyzed using a type I IFN bioassay with encephalomyocarditis virus on BHK cells as previously described (27). All samples were run in duplicate in 96-well plates.
Statistical analysis.
All statistical analyses were performed using GraphPad Prism 4 software, and tests were chosen with consideration for the normality of the data. Unless otherwise indicated, statistical analyses were conducted using Student's t test with Welch's correction.
Ethics statement.
All animal experiments were performed in accordance to NIH guidelines listed in the Guide for the Care and Use of Laboratory Animals (36), the Animal Welfare Act, and U.S. federal law. All experiments were approved by the University of Washington Institutional Animal Care and Use (IACUC) committee, animal welfare assurance number A3464-01. The University of Washington Animal Care and Use Program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC), accreditation number 000523, and registered with the USDA, certificate number 91-R-0001.
RESULTS
Cd22−/− mice have increased susceptibility to WNV-TX infection.
To determine whether CD22 plays a role in protection against WNV infection, we assessed susceptibility of WT and Cd22−/− mice to infection after subcutaneous (s.c.) inoculation with 103 PFU of WNV-TX. Eighty-eight percent of Cd22−/− mice succumbed to infection (mean survival time, 9.5 days), compared to 15% of infected WT mice (P < 0.0001) (Fig. 1A). Moreover, while symptoms began to resolve in WT mice by 7 days postinfection (p.i.), encephalitic disease and paralysis-like conditions were significantly enhanced in Cd22−/− mice (Fig. 1B). Similar to the case for Cd22−/− mice, WNV-TX was highly virulent and caused comparable clinical progression of disease in μMT mice (Fig. 1A and C), further demonstrating the importance of B cells in protection against WNV infection (2) and suggesting B cell defects in WNV-infected Cd22−/− mice.
Fig 1.
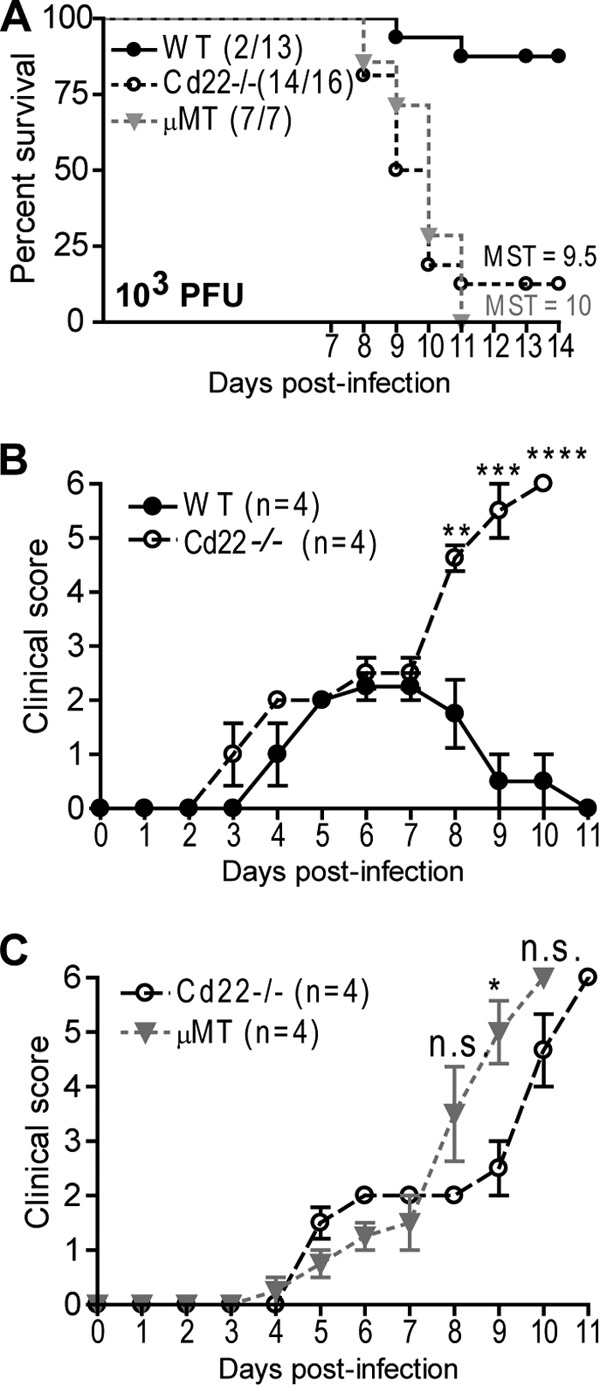
Cd22−/− mice are more susceptible to WNV infection than WT mice. WT, Cd22−/−, and μMT mice were inoculated s.c. with 103 PFU of WNV-TX and monitored daily for survival (A) and morbidity (B and C). (A) Cd22−/− mice had a mean survival time (MST) of 9.5 days, and μMT mice had an MST of 10 days. Statistics were performed using a log rank test for significance, comparing the percentage of surviving WT mice to that of Cd22−/− mice (P < 0.0001). Numbers in parentheses indicate numbers of mice that succumbed out of total mice infected. (B and C) Clinical scores for mice inoculated s.c. with 103 PFU of WNV-TX (infected) are represented. Scoring is as follows: 1, ruffled fur, lethargy, hunched posture, no paresis; 2, very mild to mild paresis; 3, frank paresis involving at least one hind limb and/or conjunctivitis or mild paresis in two hind limbs; 4, severe paresis while still retaining feeling and possibly limbic; 5, true paralysis; and 6, moribund. Error bars represent variance of clinical scores. Statistics were performed using Student's t test to compare means of clinical scores at individual time points. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. n.s., not significant. Data show one representative of three independent experiments with four mice per group.
WNV-infected Cd22−/− mice have enhanced virus replication in peripheral and CNS compartments.
WNV infection in mammalian hosts is acquired through peripheral inoculation (1). Infected cells migrate from the skin to dLNs, where the virus infects and activates both myeloid and lymphoid cells; this is followed by viremia and viral entry into peripheral tissues such as the spleen, kidneys, and liver. Six to 7 days after initial infection, virus is detected in the central nervous system (CNS); its presence there can lead to encephalitis symptoms in the host. To define the role of CD22 in controlling WNV in vivo, we inoculated WT and Cd22−/− mice s.c. with 103 PFU of WNV-TX and then monitored viral burden within peripheral and CNS tissues using a plaque assay on BHK cells. We observed similar kinetics and peak virus replication in the spleens of infected WT and Cd22−/− mice. Infected Cd22−/− mice exhibit normal tissue tropism (Fig. 2A and B). Analysis of virus loads in sera and dLNs is difficult using standard plaque assays, but real-time PCR is a suitable alternative (3, 37). Thus, we determined virus burdens in sera and dLNs using real-time PCR. There were no significant differences in viral burdens in dLNs between WT and Cd22−/− mice at days 1, 2, and 4 p.i. (Fig. 2C). Infected Cd22−/− mice, unlike WT mice, exhibited increased virus in the serum on day 4 p.i. However, serum viral loads in the Cd22−/− mice returned to normal levels by day 7 p.i. (Fig. 2D). This increase in the Cd22−/− mice serum viral loads at day 4 preceded significantly higher viral loads on day 7 p.i. in the brain and spinal cord than in infected WT mice (Fig. 2E and F). The detection of increased virus in the CNS coincided with the enhanced clinical scores on day 7 p.i. observed in infected Cd22−/− mice (Fig. 1B). These data demonstrate that CD22 is dispensable for controlling virus replication in the periphery (spleen and kidneys) but is essential in controlling viremia at early times during infection and virus replication in the CNS.
Fig 2.

Increased viral titers in the CNS and serum of WNV-infected Cd22−/− mice. Viral titers were determined using a standard plaque assay (A, B, E, and F) or quantification using real-time qPCR (C and D). Mice were inoculated as described in the text and tissues harvested at the indicated days p.i. Graphs show PFU of virus plaqued per gram of tissue or copies of viral RNA. Dotted lines show limits of detection. Statistics were performed using a Mann-Whitney U test of the median. *, P < 0.05; **, P < 0.01. n.s., not significant; n.d., not detected. Data are shown for 6 to 10 mice per time point per group.
WNV-specific antibody responses are not defective in Cd22−/− mice.
The enhanced viral burden in the serum and CNS compartments of infected Cd22−/− mice is consistent with previous observations in mice unable to secrete IgM (3), suggesting that CD22 may function to regulate antibody production during WNV infection. As CD22-deficient mice have lower antibody responses to T-independent antigens, we hypothesized that Cd22−/− mice would also have decreased antibody responses to WNV. However, by and large, the levels of WNV-specific IgM and IgG to WNV envelope (E) protein in serum were similar in infected WT and Cd22−/− mice as quantified by ELISA (Fig. 3A). At day 4 p.i. in Cd22−/− mice, there was a significant increase in serum WNV-specific IgM antibodies, but at day 7 p.i. IgM antibody levels were similar to levels in WT mice. WT and Cd22−/− mice had similar WNV-specific serum IgG2b and IgG2c levels (Fig. 3A) and, furthermore, had similar neutralizing antibody titers at day 7 p.i. (Fig. 3B). Thus, humoral immunity is not defective in WNV-infected Cd22−/− mice.
Fig 3.

Virus-specific antibody responses in WNV-infected Cd22−/− mice. (A) WNV E-protein-specific sandwich ELISAs were performed on serum samples from infected WT or Cd22−/− mice. Each symbol shows relative titers of antigen-specific antibody from an individual mouse. Data show samples of mice from three independent infections, where n > 9 individual mice for IgM and total IgG and n = 5 to 9 for IgG subclasses. (B) PRNT assays were used to quantify antibody titers with 50% virus neutralizing capacity (expressed as 1/PRNT50). Data show one out of three independent infections with at least four mice per group per time point. Statistics were performed using Student's t test. *, P < 0.05. n.s., not significant.
Cd22−/− mice have decreased CD8+ T cell responses to WNV.
Since there were no apparent defects in antibody responses in Cd22−/− mice, we next considered if other defects in adaptive immunity might contribute to the increased susceptibility of Cd22−/− mice to WNV. During WNV infection, CD4+ or CD8+ T cells are essential in controlling viral titers in the CNS at late times after infection (38, 39). Therefore, we examined WNV-specific T cell responses in the absence of CD22. We first enumerated WNV-specific effector CD8+ T cells using a MHC class I tetramer specific for the immunodominant peptide NS4B (31), specific to CD8+ T cells (see Fig. S1A in the supplemental material), and an antibody specific for the T cell activation/differentiation marker CD44. On day 5 p.i., WNV-specific effector CD8+ T cells had expanded to similar levels in the spleens of infected WT and Cd22−/− mice (Fig. 4A). However, by day 7 p.i., there were 2.5-fold-fewer WNV-specific CD8+ T cells in infected Cd22−/− mice.
Fig 4.

Impaired WNV-specific CD8+ T cell responses in the spleens of infected Cd22−/− mice. Splenocytes were harvested from naive or infected WT or Cd22−/− mice at various time points after infection. (A) Tetramer staining. Total splenocytes were stained with surface markers for CD8+ T cells, NS4B-specific MHC I tetramer, and CD44. Tetramer+ CD44+ CD8+ T cells are quantified in representative dot plots as frequency of CD8+ cells (left dot plots) or in cell numbers (right bar graph). (B) CD8+ T cell ICS. Total splenocytes were restimulated ex vivo with 1 μM NS4B peptide as described in Materials and Methods. Data are shown as IFN-γ+ TNF-α+ cells as a frequency of total CD8+ T cells (left dot plots) and total cell numbers of CD8+ IFN-γ+ TNF-α+ populations (right bar graphs). (C) In vivo cytotoxicity assays were performed at described in Materials and Methods. CFSE-labeled unpulsed (CFSE-low) and NS4B-pulsed (CFSE-high) Ly5.1+ splenocytes were transferred into naive WT or Cd22−/− mice or into WT or Cd22−/− mice infected 5 days earlier with WNV; 2 h later, splenocytes were isolated and stained for surface Ly5.1 expression. (Left histograms) Representative histograms show CFSE-low and -high populations as a frequency of total Ly5.1+ cells. (Right graph) Each symbol represents percent killing of NS4B-pulsed Ly5.1+ target cells in an infected WT or Cd22−/− mouse. (D) In vivo BrdU incorporation assay. Naive and day 5 and 7 infected WT and Cd22−/− mice were injected with BrdU i.p. as described in Materials and Methods. Data are shown as BrdU+ tetramer+ cells as a frequency of total CD8+ T cells (left dot plots) and frequency of CD8+ tetramer+ cells that are BrdU+ (right bar graphs). (E) CD4+ T cell ICS. Total splenocytes were restimulated ex vivo with overnight with 1 μM NS3 peptide prior to ICS to detect IFN-γ in CD4+ T cells. Data are shown as IFN-γ+ cells as a frequency of total CD4+ T cells (left dot plots) and cell numbers of CD4+ IFN-γ+ populations (right bar graphs). Data show one representative of three independent experiments with at least three mice per group per time point. Statistics were performed using Student's t test. *, P < 0.05. n.s., not significant.
We also examined whether WNV-specific CD8+ T cells in infected Cd22−/− mice had defects in cytokine production or cytotoxic activity. Upon restimulation with the NS4B peptide, infected Cd22−/− mice had ∼3-fold-fewer IFN-γ+ TNF-α+-secreting CD8+ T cells at day 7 p.i. (Fig. 4B; see also Fig. S1B in the supplemental material). However, WT and Cd22−/− mice had no differences in their frequencies of CD8+ TNF-α+ single cytokine-producing cells. CD8+ T cell lysis of WNV-infected cells depends on perforin (40). However, at day 7 p.i., WT and Cd22−/− mice had similar levels of perforin-producing WNV-specific CD8+ T cells (see Fig. S1C in the supplemental material), suggesting that cytotoxic CD8+ T cells function normally in Cd22−/− mice. To test if WT and Cd22−/− mice differ in cytotoxic T cell activity in vivo, we pulsed CFSE-labeled Ly5.1+ splenocytes with NS4B peptide (CFSEhi) so they could serve as WNV target cells and then adoptively transferred them along with unpulsed splenocytes (CFSElow) into Ly5.2+ WT and Cd22−/− mice that had or had not been infected with WNV 5 days earlier. We selected day 5 p.i. as the time to conduct an in vivo cytotoxicity assay since day 5 p.i. was when frequencies and numbers of WNV-specific CD8+ T cells were similar between infected WT and Cd22−/− mice (Fig. 4A and B). Two hours after injection of the labeled target cells, spleens were analyzed for levels of Ly5.1+ CFSEhi target cells compared to Ly5.1+ CFSElow cells that were not targets (Fig. 4C). There were no differences in percent killing of Ly5.1+ NS4B-pulsed target cells in WT versus Cd22−/− mice (Fig. 4C), which is consistent with the similar levels of perforin production in splenic CD8+ T cells from WT and Cd22−/− mice (see Fig. S1C). Taken together, our data show that Cd22−/− mice have decreased numbers of WNV-specific CD8+ IFN-γ/TNF-α-secreting but not perforin-producing T cells in the spleen at day 7 p.i. and have no cell-intrinsic defects in CD8+ T cell killing of WNV target cells.
CD22 regulates B cell proliferation and turnover (21), so we wondered if the absence of CD22 might affect proliferation or turnover of cell types such as CD8+ T cells that express ligands for CD22. We examined whether decreased numbers of WNV-specific CD8+ T cells in infected Cd22−/− mice were due to either increased cell death or dysregulated cell proliferation. Using annexin V staining to detect dying cells, we found no differences in frequency of early apoptotic WNV-specific CD8+ T cells in the spleens of infected WT and Cd22−/− on either day 5 or 7 p.i. (see Fig. S2B in the supplemental material). In contrast, using BrdU incorporation as a measure of T cell proliferation in vivo, we found that at the earliest time point when WNV-specific CD8+ T cells could be clearly detected by MHC I tetramer staining (day 5 p.i.), the frequency of BrdU+ tetramer+ CD8+ T cells at the peak of cell division (day 5 p.i.) was reduced in infected Cd22−/− mice compared to WT mice (Fig. 4D).
In contrast to the clear differences in CD8+ T cell responses, there were no significant differences between WT and Cd22−/− mice in the numbers of splenic WNV-specific IFN-γ-secreting CD4+ T cells (Fig. 4E) or in the frequency of splenic BrdU+ B cells, CD4+ cells, or total CD8+ T cells (see Fig. S3A to C in the supplemental material). These results demonstrate that decreased numbers of WNV-specific CD8+ T cells in the spleens of infected Cd22−/− mice are due in part to reduced CD8+ T cell proliferation rather than to an increased frequency of cell death and that CD22 regulates CD8+ T cell proliferation during WNV infection.
WNV-infected Cd22−/− mice have decreased expansion of CD8+ T cells, myeloid DCs, and NK cells.
CD22 interacts with ligands on antigen-presenting cell populations such as DCs (16) that, in turn, are responsible for priming and regulating antiviral T cell responses. Splenic myeloid DCs can be divided broadly into CD8α+ and CD8α− populations (41), and CD8α− DCs can be further subdivided into two subsets based on their expression of either DCIR2 (dendritic cell immunoreceptor 2/33D1) or DCAL2 (dendritic cell-associated C-type lectin 2, myeloid inhibitory C-type lectin, Clec12a)-expressing subsets (33) (see Fig. S4 in the supplemental material). CD8α+ DC subsets are superior to CD8α− DCs in cross-presenting antigen to CD8+ T cells (42). In addition, our laboratory has shown that the DCAL2+ (DCIR2−) and CD8α+ DC subsets preferentially induce T helper 1 responses, while DCIR2+ DCs predominantly induce T helper 2 responses (33). In other words, these populations may have different roles in the generation of immune responses to WNV. Thus, we enumerated both DCs and lymphocytes in the footpad dLNs and spleens of infected WT and Cd22−/− mice to determine how CD22 affects cell expansion and recruitment after WNV infection. One day p.i., total cell numbers in the dLNs were expanded to similar levels in infected WT and Cd22−/− mice (Fig. 5A); there were no significant differences in either myeloid DC or lymphocyte populations (Fig. 5B and C). However, there were nearly 2-fold-fewer NK cells in the dLNs of infected Cd22−/− mice than in those of WT mice (Fig. 5B). Interestingly, little or no CD8α− DCIR2+ DCs were detectable in the dLNs of uninfected WT or Cd22−/− mice, but their numbers increased significantly in dLNs in both genotypes as early as 1 day p.i. (Fig. 5B).
Fig 5.

CD8+ T cells, NK cells, and myeloid DC populations are decreased in the dLNs and spleens of WNV-infected Cd22−/− mice. Pooled popliteal dLNs (A, B, and C) and spleens (D and E) were harvested from naive mice or mice infected with 103 PFU of WNV-TX at the indicated days p.i. Total cells were stained for the indicated leukocyte populations. Total cell numbers for various populations are shown per dLN 1 day postinfection (A, B, and C) or per spleen (D and E) at the indicated time points. Statistics was performed using Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. n.s., not significant; n.d., not detected. Data show one representative of 3 independent experiments with three mice per group per time point (dLNs) or mice from three independent experiments where n > 9 per group per time point (spleen).
The numbers of total splenocytes in infected WT mice expanded 1.3-fold compared to those in uninfected mice by 5 and 7 days p.i., yet total cell numbers remained relatively unchanged in the spleens of infected Cd22−/− mice (Fig. 5D). Compared to WT mice, WNV-infected Cd22−/− mice had no significant differences in the numbers of splenic B cells but did have fewer total CD3+ T cells at day 7 p.i. (Fig. 5D). This decrease in splenic T cell numbers could be attributed mainly to CD8+ T cells, which were 1.5-fold lower in Cd22−/− mice than in WT mice at day 7 p.i. While splenic NK cells and myeloid DC subsets all expanded after infection of WT mice, they remained relatively unchanged in Cd22−/− mice (Fig. 5D and E). Specifically, there were significant increases in DCIR2+ and CD8α+ DCs in WT mice after infection but not in Cd22−/− mice (Fig. 5E). Nonlymphoid, non-DC populations, which include cells such as monocytes and macrophages, also did not expand in Cd22−/− mice (Fig. 5E). Taken together, these data show that CD22 regulates changes in NK cell, CD8+ T cell, and DC numbers in the spleen in WNV-infected mice, resulting in an inability of these populations to expand, be recruited, or be retained in the spleens of Cd22−/− mice after infection.
Based on these findings, we examined BrdU uptake by NK cell and myeloid DC populations to assess whether there was a defect in expansion of these cell types after infection in Cd22−/− mice. Surprisingly, the frequencies of BrdU+ NK cells and myeloid DC subsets in the spleen were similar between infected WT and Cd22−/− mice (see Fig. S3D in the supplemental material; other data not shown). Annexin V staining revealed no significant differences in early apoptotic death in NK cell or myeloid DC populations in the spleen (see Fig. S2E to G in the supplemental material). Thus, lower numbers of splenic NK cells and myeloid DC subsets in WNV-infected Cd22−/− mice were not simply due to reduced proliferation or increased cell death, raising the possibility that the reduced numbers were due to a defect in cell migration.
The splenic defect in WNV-specific CD8+ T cell responses in Cd22−/− mice also correlated with decreased cellular infiltrates into the brains of infected Cd22−/− mice 7 days p.i. (Fig. 6). A decrease was evident in CD4+ T cells, CD8+ T cells, and WNV-specific CD8+ T cells (Fig. 6B and D) but not in NK cells (Fig. 6E). The numbers of infiltrating nonlymphoid populations, which likely include infiltrating macrophages and resident microglial cells, were also not significantly different between infected WT and Cd22−/− mice (Fig. 6F). Thus, the CD22-dependent defect in migration of cells into the CNS predominantly affects T cells. It has been established that infiltrating T cells are important for controlling virus replication in the CNS and, ultimately, survival of the host from succumbing to encephalitic disease (43, 44). Thus, the diminished number of WNV-specific CD8+ T cells in the spleens of Cd22−/− mice along with the decreased CD8+ T cell infiltrates and inability to control viral replication in the brain likely accounts for Cd22−/− mice succumbing to disease after WNV infection.
Fig 6.

Decreased T cell infiltrates into the brains of WNV-infected Cd22−/− mice. Mice were inoculated s.c. with WNV-TX and perfused with PBS on day 7 p.i. Brains were harvested, and lymphocytes were isolated as described in the text. Cells were enumerated after flow cytometry analysis for total leukocytes (A), total CD4+ T cells (B), total CD8+ T cells (C), CD8+ tetramer+ T cells (D), total NK cells (E), and nonlymphoid cells (CD19− CD3− NK1.1−) (F). Symbols indicate individual mice from at least three independent experiments. Statistics were performed using Student's t test. *, P < 0.05. n.s., not significant.
Cd22−/− mice have a defect in cell recruitment.
The overall normal rates of cell proliferation and cell death in infected Cd22−/− mice suggested that decreased CD8+ T cells and certain subsets of myeloid DCs in infected Cd22−/− mice may be due to defective cell migration into infected tissues. Subcutaneous infection of the footpad with WNV predictably induces entry of leukocytes from circulation into dLNs (45), which thus has the potential to provide a reliable assay to examine leukocyte migration into a site of infection. To directly test whether cell migration into infected tissues is altered in Cd22−/− mice in vivo, we adoptively transferred congenically marked WT Ly5.1+ splenocytes into Ly5.2+ WT and Cd22−/− recipients by intravenous (i.v.) injection and subsequently infected them with WNV. We then harvested individual dLNs of hind footpads that had been inoculated with WNV or with a vehicle alone or left injected, and we quantified, as an indicator of cell migration (46, 47), the numbers of Ly5.1+ subsets 24 h p.i. (Fig. 7). Both lymphocytes and DCs migrated into the dLNs of WNV-infected WT and Cd22−/− mice. The infected dLNs from Cd22−/− mice had significantly fewer Ly5.1+ NK cells, CD3+ T cells, CD4+ T cells, and CD8+ T cells than did those of WT mice (Fig. 7A to D). There was no significant difference in the number of migrating Ly5.1+ CD11c+ DCs in the dLNs of Cd22−/− mice compared to WT mice (Fig. 7E) and no differences in the number of B cells that had entered into dLNs (Fig. 7F). Thus, loss of CD22 expression alters cell migration of NK cells and T cells into WNV-infected tissues.
Fig 7.

Impaired recruitment of T cells and NK cells into infected dLNs in Cd22−/− mice. Ly5.1+ total splenocytes from WT C57BL/6 mice were adoptively transferred by i.v. injection into Ly5.2+ WT or Cd22−/− mice. The next day, individual footpads of recipient mice were left uninjected (naive), injected with 10 μl of a 1% FCS-containing HBSS vehicle (mock), or injected with 103 PFU of WNV-TX (infected). Twenty-four hours p.i., individual dLNs were harvested and processed separately before staining for the following populations: CD3+ T cells (A), NK cells (B), total CD4+ T cells (C), total CD8+ T cells (D), CD11chi DCs (E), and B cells (F). Symbols represent individual dLNs from mice from three independent experiments. Statistics were performed using a Mann-Whitney U test of the median. *, P < 0.05; **, P < 0.01. n.s., not significant.
We hypothesized that dysregulation of chemokine production within the dLNs of infected Cd22−/− mice may lead to impaired migration of cells from the circulation. CCR5 and its ligands CCL3, CCL4, and CCL5 have been implicated in the pathogenesis of WNV infection and are important for recruitment of T cells into the CNS for protection from encephalitis (48). Thus, we isolated dLNs early after infection to determine whether expression of chemokine genes was altered in Cd22−/− mice (Fig. 8). Compared to expression in dLNs from WT mice, the dLNs from Cd22−/− mice had decreased mRNA expression of Ccl3 and Ccl5 6 h after WNV infection (Fig. 8A to C). However, this difference was no longer present at 12 h p.i. Expression of Ccl3 and Ccl4 was induced 5- to 35-fold in WT dLNs at 6 and 12 h p.i. (Fig. 8A and B), while Ccl5 levels did not increase significantly (Fig. 8C). This defect of chemokine gene expression in the Cd22−/− dLNs was restricted to Ccl3 and Ccl5; there was no difference between WT and Cd22−/− mice in induction of Ccl4 expression or in expression of other cytokine genes such as Ifnα, Ifnß, Ifnγ, Tnfα, Cxcl10, Cxcl11, and Ccl2 (Fig. 8D and data not shown). Levels of type I IFN in serum also were not different between infected WT and Cd22−/− mice (Fig. 8E). These data show that the expression of CCR5 ligands was selectively altered in the dLNs of WNV-infected Cd22−/− mice even though the numbers of T cells and DCs, populations that can produce CCR5 agonists (49), were similar between infected WT and Cd22−/− mice (Fig. 5B and C).
Fig 8.

CD22 expression regulates production of specific chemokines early in the WNV-infected dLNs. (A to D) Mice were inoculated and dLNs were harvested at indicated time points p.i. Relative expression levels of the indicated genes were quantified and graphed as fold change using TaqMan (A, B, and C) or Sybr green (D) as described in Materials and Methods. Data show one representative of three independent experiments with at least two mice per group per time point. (E) Mice were inoculated and serum samples taken at the indicated time points. Type I IFN production was quantified from serum samples using a standard bioassay. Data are represented as international units (IU) per ml of serum. Data show one representative of two independent experiments with three mice per group per time point. Statistics were performed using Student's t test. *, P < 0.05; **, P < 0.01. n.s., not significant.
Myeloid DCIR2+ CD8α− DCs express CD22.
Although CD22 is predominantly expressed on B cells, B cell numbers, B cell migration into infected tissues, and antibody production during WNV infection were all unaffected in Cd22−/− mice. Antiviral effector CD8+ T cell responses are primed and maintained by accessory cells, including myeloid DCs (50), which, in contrast to B cells, after WNV infection were significantly decreased in number within the spleens of Cd22−/− mice compared to WT mice (Fig. 5E). Given the CD8+ T cell defect in infected Cd22−/− mice, it was possible that DC functions may be defective in Cd22−/− mice and thereby contribute to the decreased WNV-specific CD8+ T cell activation and expansion. Previously, Edwards et al. reported that CD22 is expressed at low levels on CD8α− DCs in the spleen but not on CD8α+ DCs (51). We reexamined the expression of CD22 on splenic DC subsets and found that CD22 was expressed as expected on B cells (Fig. 9A) but was also present on the subset of CD8α− DCs expressing DCIR2 (Fig. 9B). CD22 was not detected on CD8α− DCAL2+ DCs, CD8α+ (DEC205+) DCs, or plasmacytoid DCs (Fig. 9A and B). As in the spleen, the DCIR2+ DCs are the only myeloid DC subset that expresses CD22 in infected dLNs (Fig. 9C).
Fig 9.

DCIR2+ DCs express CD22. Shown is flow cytometry analysis of CD22 expression on splenic populations in WT C57BL/6 and Cd22−/− mice. Histograms show staining with anti-mouse CD22 antibody (bold) or isotype control antibody (shaded) and are representative of one of four independent experiments with at least 3 mice per group. (A) Spleen cells were isolated from naive WT C57BL/6 mice, and the indicated cell types were stained for CD22 expression. (B) Dot and contour flow plots show the gating scheme for myeloid DC subsets: B220− NK1.1− CD11chi populations were subdivided into DCIR2+, DCIR2− (DCAL2+), and CD8α+ DCs and analyzed for expression of CD22. (C) Cells from popliteal dLNs of infected WT and Cd22−/− mice were stained for CD22 expression on days 1 and 2 p.i. (D and E) Sorted DCIR2+ DCs from WT and Cd22−/− mice were adoptively transferred into naive Cd22−/− recipient groups i.v. Cd22−/− mice that received no DCs (−), WT DCs, or Cd22−/− DCs were inoculated with 103 PFU of WNV-TX and spleens harvested 7 days p.i. Numbers of total splenocytes (D) and total CD8+ T cells (E) are graphed. Each symbol shows an individual mouse from three independent experiments. Statistics were performed using Student's t test, with some P values reported. *, P < 0.05; ***, P < 0.001. n.s., not significant.
We then directly evaluated the role of CD22+ DCIR2+ DCs on splenic CD8+ T cell levels in WNV-infected mice. We adoptively transferred purified DCIR2+ DCs from WT or Cd22−/− mice into naive Cd22−/− recipients and then examined total splenocyte and CD8+ T cell numbers 7 days after WNV infection. Infected Cd22−/− mice had fewer total splenocytes (Fig. 9D) and CD8+ T cells (Fig. 9E) than WT mice. Infected Cd22−/− mice that received WT DCIR2+ DCs had splenocyte levels restored to normal and that were significantly higher than in Cd22−/− mice that received no DCs (Fig. 9D). CD8+ T cell numbers were also increased in Cd22−/− mice after transfer of WT DCIR2+ DCs, but this increase was not significant (Fig. 9E). In contrast, adoptive transfer of CD22− DCIR2+ DCs into WNV-infected Cd22−/− mice had little or no effect on splenocyte or CD8+ T cell expansion. Thus, DCIR2+ DCs may contribute to splenocyte and CD8+ T cell expansion after WNV infection through a CD22-dependent process.
DISCUSSION
We have shown a direct role for CD22 in the generation of protective immune responses against WNV and have determined that CD22 is expressed on and may regulate a subset of splenic DCIR2+ CD8α− DCs. CD22 contributes to adaptive immunity by more than simply regulating B cell-mediated antibody responses: it regulates the proliferation of WNV-specific CD8+ T cells, as well as cell migration into infected sites such as the brain and dLNs.
Although CD22 has been reported to be required for normal T cell-independent antibody responses (19, 21), we detected no defects in WNV-specific antibody responses in infected Cd22−/− mice (Fig. 3). After WNV infection, both μMT mice and mice unable to secrete IgM have significantly elevated viral loads in the spleen and kidneys (2, 3). This was not the case in WNV-infected Cd22−/− mice (Fig. 2). Furthermore, Cd22−/− mice had normal numbers of B cells in both the dLNs and spleen both before and after WNV infection (Fig. 5), and Cd22−/− B cells also migrated into dLNs at the same levels as WT B cells (Fig. 7F). Thus, unlike the B cell-deficient and IgM-deficient mice, the increased susceptibility in Cd22−/− mice does not appear to be due to a defect in humoral immunity.
Susceptibility of Cd22−/− mice to WNV coincides with increased viral entry into the CNS (Fig. 2E and F) and inability to resolve neurological complications (Fig. 1B and C). CD8+ T cells are the predominant cell type responsible for lysis of WNV-infected neurons (52, 53), and unimpaired CD8+ T cell entry into the CNS is critical for controlling WNV viral replication and preventing encephalitis (40, 54). Infected Cd22−/− mice had decreased numbers of WNV-specific CD8+ T cells (tetramer binding or IFN-γ/TNF-α secreting) in the spleens and brains day 7 p.i. (Fig. 4 and 6) but no cellular defects in perforin production (see Fig. S1C in the supplemental material). They also had normal cytotoxic T cell killing of WNV target cells in vivo at a time (day 5 p.i.) when the numbers and frequencies of WNV-specific CD8+ T cells in the spleen were similar between infected WT and Cd22−/− mice (Fig. 4C). CD8+ T cells showed an inability to expand (Fig. 4D) and to migrate to sites of infection within Cd22−/− mice in vivo (Fig. 5D and 6C and D). Together, these data suggest that the CD8+ T cell numbers within infected tissues are deficient in Cd22−/− mice but that the CD8+ T cells present have normal cytolytic activity.
While the requirement for CD8+ T cells in controlling virus in the CNS has been well established, the role of CD4+ T cells is less clear. One study suggested that CD4+ T cells also mediate in vivo killing of WNV-infected cells (32), while another study argued that CD4+ T cells mainly function to support antibody responses and that CD8+ T cell migration to the CNS and alone is insufficient to protect from WNV encephalitis (39). Although CD4+ T cell numbers were reduced in the CNS of Cd22−/− mice (Fig. 6), a defect in the CD4+ T cell response is unlikely to be responsible for the increased susceptibility of Cd22−/− mice. Our data show that CD4+ T numbers and WNV-specific responses were normal in the spleens and dLNs of Cd22−/− mice (Fig. 4E and 5). Moreover, unlike CD4+ T cell-deficient mice, Cd22−/− mice had no defects in WNV-specific humoral responses (39). Overall, pathological characteristics of Cd22−/− mice more closely resemble those of CD8+ T cell-deficient mice (38). Collectively, these data support the model that impaired WNV-specific CD8+ T cell entry into the CNS is a major contributing factor for uncontrolled virus replication and encephalitis in Cd22−/− mice.
The exact mechanism (s) by which CD22 functions to mediate protection against WNV infection requires further investigation, but it may involve regulation of cell migration. Infected Cd22−/− mice have significantly decreased T cell migration into the brain (Fig. 6) and decreased T cell, NK cell, but not B cell trafficking into dLNs (Fig. 7), suggesting that CD22 regulates migration of specific cell types during WNV infection. CD22 has been shown to control the migration of certain B cells (19), but unlike in this study, the effects of CD22 on migration were B cell intrinsic. For example, Cd22−/− mice and mice with mutated CD22 ligand binding domains have reduced numbers of long-lived B cells and recirculating B cells in the bone marrow in vivo (19, 55). However, T cells express surface glycans that contain α2,6-sialic linkages that can bind to CD22 (6, 56, 57). Furthermore, NK cells can bind CD22 (D. Y. Ma, unpublished data). Thus, T cells and NK cells could be regulated via direct binding to CD22 in trans. In addition, mice lacking the ST6Gal I glycosyltransferase, which adds CD22 ligand α2,6-sialic acid linkages to N-glycans, have a reduction in virus-specific CD8+ but not CD4+ T cells in the spleen after influenza virus infection (58). Thus, it is possible that a lack of binding to CD22 by T cells and NK cells in Cd22−/− mice leads to defective entry into or reduced retention of these cells within sites of WNV infection. The fact that both CD8+ T cell proliferation (Fig. 4C) and migration (Fig. 7D) are reduced in infected Cd22−/− mice suggests that the decreased numbers of WNV-specific CD8+ T cells in the spleens and brains of Cd22−/− mice are due to defects in both proliferation and cell recruitment into infected tissues.
CD22 may also affect cell recruitment into infected tissues through regulation of chemokine production in vivo, as we observed an early reduction in expression of Ccl3 and Ccl5 but not Ccl4 in the dLNs of infected Cd22−/− mice (Fig. 8A to C). CCR5 and its ligands play an important role in the recruitment of T cells to sites of inflammation (59, 60), and CCR5 is especially significant in the context of WNV infection (48). Murine CCR5 is required for T and NK cell entry into the WNV-infected CNS (61), and loss-of-function mutations in human CCR5 expression have been correlated with increased presentation of neurological symptoms after WNV infection (62). Thus, defective production of CCR5 agonists in the infected dLNs of Cd22−/− mice may account for fewer T and NK cell entering the dLNs and other sites of infection.
DCIR2+ DCs may play essential roles in governing immune responses during WNV infection. This distinct subset of splenic DCs expresses CD22 (Fig. 9), and while not present in dLNs of uninfected mice, these DCs increase in number in the dLNs after WNV infection (Fig. 5B). DCIR2+ DCs are detectable in the dLNs within 24 h after infection (Fig. 5B) and thus are in a position to encounter and rapidly respond to WNV and help initiate adaptive immune responses. Adoptive transfer of CD22+ DC1R2+ DCs but not CD22− DCIR2+ DCs restored splenocyte and CD8+ T cell expansion to normal levels in infected Cd22−/− mice (Fig. 9D and E). Our data indicate that expression of CD22 on DCIR2+ DCs affects splenocyte expansion during WNV infection; however, the exact role of DCIR2+ DCs during WNV infection is still unclear. The loss of CD22 may also affect other responses, such as cell migration and cytokine production, that may also influence the susceptibility of these mice to infection. Our preliminary data suggest that DCIR2+ DCs may help regulate cell migration or retention of CCR5+ cells at sites of infection, since unlike CD8α+ DCs and DCAL2+ CD8α− DCs, they can produce all three of the major ligands for CCR5: CCL3, CCL4, and CCL5 (Ma, unpublished). Further studies are required to elucidate whether DC-dependent production of chemokines affects recruitment of lymphocytes to infected tissues after WNV challenge and how CD22 may play a role in these processes.
In summary, our data support a working model that during acute WNV infection, CD22 functions to promote CD8+ T cell expansion and leukocyte migration to sites of infection. This model is supported by the fact that absence of CD22 disrupts early chemokine production, which, in turn, affects the migration of specific CCR5+ cells to sites of viral replication. A reduced capacity to proliferate combined with dysregulated chemokine production may, in turn, impair CD8+ T cell entry into the CNS to control viral replication, accounting for the increased susceptibility of infected Cd22−/− mice. CD22 clearly is important for protection against a viral pathogen, WNV, and this protection does not operate through regulation of antigen-specific antibody responses. Further studies are necessary to evaluate the role of other possible factors, such as cell migration, that may influence susceptibility to WNV in Cd22−/− mice. Furthermore, since CD22 is expressed on DCIR2+ DCs, it will be important to define how CD22 regulates both CD8+ T cell migration and DCIR2+ DC functions and whether CD22 and CD22+ DCIR2+ DCs play similar roles in protective immune responses to other pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marianne Bryan, Jay Chaplin, Kevin Draves, Erin Mehlhop, and Hilario Ramos for technical help. We also thank Keith Elkon for helpful discussions. We thank members of the University of Washington U19 Consortium for Study of West Nile Virus for helpful discussions and technical help. Finally, we thank Michael Diamond at Washington University at St. Louis for providing WNV envelope protein.
This study was supported by NIH grants AI83019 and AI52203 and HHMI grant 56006710.
D.Y.M., M.G., and E.A.C. conceived and designed the experiments. D.Y.M., M.S.S., and S.K. performed the experiments. D.Y.M., M.S.S., S.K., M.G., and E.A.C. analyzed the data. D.Y.M., M.S.S., S.K., and M.G. contributed reagents, other materials, and analysis tools. D.Y.M. and E.A.C. wrote the manuscript.
We report no competing financial interests.
Footnotes
Published ahead of print 9 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02368-12.
REFERENCES
- 1. Samuel MA, Diamond MS. 2006. Pathogenesis of West Nile virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 80:9349–9360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. 2003. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 77:2578–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 198:1853–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Engle MJ, Diamond MS. 2003. Antibody prophylaxis and therapy against West Nile virus infection in wild-type and immunodeficient mice. J. Virol. 77:12941–12949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dörken B, Moldenhauer G, Pezzutto A, Schwartz R, Feller A, Kiesel S, Nadler LM. 1986. HD39 (B3), a B lineage-restricted antigen whose cell surface expression is limited to resting and activated human B lymphocytes. J. Immunol. 136:4470–4479 [PubMed] [Google Scholar]
- 6. Stamenkovic I, Sgroi D, Aruffo A, Sy MS, Anderson T. 1991. The B lymphocyte adhesion molecule CD22 interacts with leukocyte common antigen CD45RO on T cells and alpha 2–6 sialyltransferase, CD75, on B cells. Cell 66:1133–1144 [DOI] [PubMed] [Google Scholar]
- 7. Torres RM, Law CL, Santos-Argumedo L, Kirkham PA, Grabstein K, Parkhouse RM, Clark EA. 1992. Identification and characterization of the murine homologue of CD22, a B lymphocyte-restricted adhesion molecule. J. Immunol. 149:2641–2649 [PubMed] [Google Scholar]
- 8. Law CL, Sidorenko SP, Chandran KA, Zhao Z, Shen SH, Fischer EH, Clark EA. 1996. CD22 associates with protein tyrosine phosphatase 1C, Syk, and phospholipase C-gamma(1) upon B cell activation. J. Exp. Med. 183:547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schulte RJ, Campbell MA, Fischer WH, Sefton BM. 1992. Tyrosine phosphorylation of CD22 during B cell activation. Science 258:1001–1004 [DOI] [PubMed] [Google Scholar]
- 10. Clark EA. 1993. CD22, a B cell-specific receptor, mediates adhesion and signal transduction. J. Immunol. 150:4715–4718 [PubMed] [Google Scholar]
- 11. Stamenkovic I, Seed B. 1990. The B-cell antigen CD22 mediates monocyte and erythrocyte adhesion. Nature 345:74–77 [DOI] [PubMed] [Google Scholar]
- 12. Pezzutto A, Dorken B, Moldenhauer G, Clark EA. 1987. Amplification of human B cell activation by a monoclonal antibody to the B cell-specific antigen CD22, Bp 130/140. J. Immunol. 138:98–103 [PubMed] [Google Scholar]
- 13. Pezzutto A, Rabinovitch PS, Dorken B, Moldenhauer G, Clark EA. 1988. Role of the CD22 human B cell antigen in B cell triggering by anti-immunoglobulin. J. Immunol. 140:1791–1795 [PubMed] [Google Scholar]
- 14. Tuscano J, Engel P, Tedder TF, Kehrl JH. 1996. Engagement of the adhesion receptor CD22 triggers a potent stimulatory signal for B cells and blocking CD22/CD22L interactions impairs T-cell proliferation. Blood 87:4723–4730 [PubMed] [Google Scholar]
- 15. Law CL, Aruffo A, Chandran KA, Doty RT, Clark EA. 1995. Ig domains 1 and 2 of murine CD22 constitute the ligand-binding domain and bind multiple sialylated ligands expressed on B and T cells. J. Immunol. 155:3368–3376 [PubMed] [Google Scholar]
- 16. Santos L, Draves KE, Boton M, Grewal PK, Marth JD, Clark EA. 2008. Dendritic cell-dependent inhibition of B cell proliferation requires CD22. J. Immunol. 180:4561–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tedder TF, Tuscano J, Sato S, Kehrl JH. 1997. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol. 15:481–504 [DOI] [PubMed] [Google Scholar]
- 18. Samardzic T, Marinkovic D, Danzer CP, Gerlach J, Nitschke L, Wirth T. 2002. Reduction of marginal zone B cells in CD22-deficient mice. Eur. J. Immunol. 32:561–567 [DOI] [PubMed] [Google Scholar]
- 19. Nitschke L, Carsetti R, Ocker B, Kohler G, Lamers MC. 1997. CD22 is a negative regulator of B-cell receptor signalling. Curr. Biol. 7:133–143 [DOI] [PubMed] [Google Scholar]
- 20. O'Keefe TL, Williams GT, Davies SL, Neuberger MS. 1996. Hyperresponsive B cells in CD22-deficient mice. Science 274:798–801 [DOI] [PubMed] [Google Scholar]
- 21. Otipoby KL, Andersson KB, Draves KE, Klaus SJ, Farr AG, Kerner JD, Perlmutter RM, Law CL, Clark EA. 1996. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature 384:634–637 [DOI] [PubMed] [Google Scholar]
- 22. Poe JC, Haas KM, Uchida J, Lee Y, Fujimoto M, Tedder TF. 2004. Severely impaired B lymphocyte proliferation, survival, and induction of the c-Myc:Cullin 1 ubiquitin ligase pathway resulting from CD22 deficiency on the C57BL/6 genetic background. J. Immunol. 172:2100–2110 [DOI] [PubMed] [Google Scholar]
- 23. Fehr T, Lopez-Macias C, Odermatt B, Torres RM, Schubart DB, O'Keefe TL, Matthias P, Hengartner H, Zinkernagel RM. 2000. Correlation of anti-viral B cell responses and splenic morphology with expression of B cell-specific molecules. Int. Immunol. 12:1275–1284 [DOI] [PubMed] [Google Scholar]
- 24. Gjertsson I, Nitschke L, Tarkowski A. 2004. The role of B cell CD22 expression in Staphylococcus aureus arthritis and sepsis. Microbes Infect. 6:377–382 [DOI] [PubMed] [Google Scholar]
- 25. Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M., Jr 2006. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 80:9424–9434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suthar MS, Brassil MM, Blahnik G, Gale M., Jr 2012. Infectious clones of novel lineage 1 and lineage 2 West Nile virus strains WNV-TX02 and WNV-Madagascar. J. Virol. 86:7704–7709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M., Jr 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 6:e1000757 doi:10.1371/journal.ppat.1000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Linke S, Ellerbrok H, Niedrig M, Nitsche A, Pauli G. 2007. Detection of West Nile virus lineages 1 and 2 by real-time PCR. J. Virol. Methods 146:355–358 [DOI] [PubMed] [Google Scholar]
- 29. Mehlhop E, Fuchs A, Engle M, Diamond MS. 2009. Complement modulates pathogenesis and antibody-dependent neutralization of West Nile virus infection through a C5-independent mechanism. Virology 393:11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frey A, Di Canzio J, Zurakowski D. 1998. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods 221:35–41 [DOI] [PubMed] [Google Scholar]
- 31. Purtha WE, Myers N, Mitaksov V, Sitati E, Connolly J, Fremont DH, Hansen TH, Diamond MS. 2007. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur. J. Immunol. 37:1845–1854 [DOI] [PubMed] [Google Scholar]
- 32. Brien JD, Uhrlaub JL, Nikolich-Zugich J. 2008. West Nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J. Immunol. 181:8568–8575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kasahara S, Clark EA. 2012. Dendritic cell-associated lectin 2 (DCAL2) defines a distinct CD8alpha-dendritic cell subset. J. Leukoc. Biol. 91:437–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barber DL, Wherry EJ, Ahmed R. 2003. Cutting edge: rapid in vivo killing by memory CD8 T cells. J. Immunol. 171:27–31 [DOI] [PubMed] [Google Scholar]
- 35. Bourne N, Scholle F, Silva MC, Rossi SL, Dewsbury N, Judy B, De Aguiar JB, Leon MA, Estes DM, Fayzulin R, Mason PW. 2007. Early production of type I interferon during West Nile virus infection: role for lymphoid tissues in IRF3-independent interferon production. J. Virol. 81:9100–9108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 37. Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M, Jr, Diamond MS. 2010. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol. 84:12125–12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shrestha B, Diamond MS. 2004. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 78:8312–8321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sitati EM, Diamond MS. 2006. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J. Virol. 80:12060–12069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shrestha B, Samuel MA, Diamond MS. 2006. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 80:119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shortman K, Liu YJ. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161 [DOI] [PubMed] [Google Scholar]
- 42. Shortman K, Heath WR. 2010. The CD8+ dendritic cell subset. Immunol. Rev. 234:18–31 [DOI] [PubMed] [Google Scholar]
- 43. McCandless EE, Zhang B, Diamond MS, Klein RS. 2008. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc. Natl. Acad. Sci. U. S. A. 105:11270–11275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sitati E, McCandless EE, Klein RS, Diamond MS. 2007. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J. Virol. 81:9801–9811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bréhin AC, Mouries J, Frenkiel MP, Dadaglio G, Despres P, Lafon M, Couderc T. 2008. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+ B220− BST-2+ leukocyte population. J. Immunol. 180:6760–6767 [DOI] [PubMed] [Google Scholar]
- 46. Lefrançois L, Parker CM, Olson S, Muller W, Wagner N, Schon MP, Puddington L. 1999. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response. J. Exp. Med. 189:1631–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davison AM, King NJ. 2011. Accelerated dendritic cell differentiation from migrating Ly6C(lo) bone marrow monocytes in early dermal West Nile virus infection. J. Immunol. 186:2382–2396 [DOI] [PubMed] [Google Scholar]
- 48. Lim JK, Murphy PM. 2011. Chemokine control of West Nile virus infection. Exp. Cell Res. 317:569–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luther SA, Cyster JG. 2001. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2:102–107 [DOI] [PubMed] [Google Scholar]
- 50. Masson F, Mount AM, Wilson NS, Belz GT. 2008. Dendritic cells: driving the differentiation programme of T cells in viral infections. Immunol. Cell Biol. 86:333–342 [DOI] [PubMed] [Google Scholar]
- 51. Edwards AD, Chaussabel D, Tomlinson S, Schulz O, Sher A, Reis e Sousa C. 2003. Relationships among murine CD11c(high) dendritic cell subsets as revealed by baseline gene expression patterns. J. Immunol. 171:47–60 [DOI] [PubMed] [Google Scholar]
- 52. Liu Y, Mullbacher A. 1988. Astrocytes are not susceptible to lysis by natural killer cells. J. Neuroimmunol. 19:101–110 [DOI] [PubMed] [Google Scholar]
- 53. Liu Y, Blanden RV, Mullbacher A. 1989. Identification of cytolytic lymphocytes in West Nile virus-infected murine central nervous system. J. Gen. Virol. 70(Part 3):565–573 [DOI] [PubMed] [Google Scholar]
- 54. Shrestha B, Diamond MS. 2007. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J. Virol. 81:11749–11757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nitschke L, Floyd H, Ferguson DJ, Crocker PR. 1999. Identification of CD22 ligands on bone marrow sinusoidal endothelium implicated in CD22-dependent homing of recirculating B cells. J. Exp. Med. 189:1513–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Engel P, Nojima Y, Rothstein D, Zhou LJ, Wilson GL, Kehrl JH, Tedder TF. 1993. The same epitope on CD22 of B lymphocytes mediates the adhesion of erythrocytes, T and B lymphocytes, neutrophils, and monocytes. J. Immunol. 150:4719–4732 [PubMed] [Google Scholar]
- 57. Stamenkovic I, Sgroi D, Aruffo A. 1992. CD22 binds to alpha-2,6-sialyltransferase-dependent epitopes on COS cells. Cell 68:1003–1004 [DOI] [PubMed] [Google Scholar]
- 58. Zeng J, Joo HM, Rajini B, Wrammert JP, Sangster MY, Onami TM. 2009. The generation of influenza-specific humoral responses is impaired in ST6Gal I-deficient mice. J. Immunol. 182:4721–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim CH, Broxmeyer HE. 1999. Chemokines: signal lamps for trafficking of T and B cells for development and effector function. J. Leukoc. Biol. 65:6–15 [DOI] [PubMed] [Google Scholar]
- 60. Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. 2006. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 440:890–895 [DOI] [PubMed] [Google Scholar]
- 61. Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. 2005. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 202:1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lim JK, McDermott DH, Lisco A, Foster GA, Krysztof D, Follmann D, Stramer SL, Murphy PM. 2010. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J. Infect. Dis. 201:178–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.