

Abstract
Human papillomavirus type 16 (HPV16) is the primary etiologic agent for cervical cancer. The infectious entry of HPV16 into cells occurs via a so-far poorly characterized clathrin- and caveolin-independent endocytic pathway, which involves tetraspanin proteins and actin. In this study, we investigated the specific role of the tetraspanin CD151 in the early steps of HPV16 infection. We show that surface-bound HPV16 moves together with CD151 within the plane of the membrane before they cointernalize into endosomes. Depletion of endogenous CD151 did not affect binding of viral particles to cells but resulted in reduction of HPV16 endocytosis. HPV16 uptake is dependent on the C-terminal cytoplasmic region of CD151 but does not require its tyrosine-based sorting motif. Reexpression of the wild-type CD151 but not mutants affecting integrin functions restored virus internalization in CD151-depleted cells. Accordingly, short interfering RNA (siRNA) gene knockdown experiments confirmed that CD151-associated integrins (i.e., α3β1 and α6β1/4) are involved in HPV16 infection. Furthermore, palmitoylation-deficient CD151 did not support HPV16 cell entry. These data show that complex formation of CD151 with laminin-binding integrins and integration of the complex into tetraspanin-enriched microdomains are critical for HPV16 endocytosis.
INTRODUCTION
Human papillomaviruses (HPVs) are nonenveloped viruses with an icosahedral capsid containing 360 copies of the major capsid protein L1, up to 72 copies of the minor capsid protein L2 (1–5), and a double-stranded circular DNA genome (6). There are over 150 different types of HPV, infecting epithelial cells and mainly causing benign epithelial warts on skin and mucosa, but there are also high-risk types, especially HPV16, which have been identified as primary etiologic agents for cervical cancer and other anogenital malignancies (7–9).
Cellular entry of HPVs is a multistep process. The first viral contact occurs through interaction with glycosaminoglycans (GAGs), mostly heparan sulfate proteoglycans (HSPGs) (10–13), or with non-HSPG components of the extracellular matrix (ECM), such as laminin-332 (previously named laminin-5) (14, 15). These interactions cause conformational changes in the capsid, followed by partial externalization and cleavage of the inner virion protein L2 by cyclophilin B and furin, respectively (11, 16–20).
It has been proposed that, subsequent to cell surface binding, the virus is transferred from the primary receptor (e.g., proteoglycans) to a secondary receptor molecule (e.g., α6-integrins or epidermal growth factor receptor [EGFR]) (21–25), which may be linked to tetraspanin microdomains (26). Subsequent internalization of the virus into the endosomal system of the host cell occurs via clathrin-, caveolin-, and dynamin-independent mechanisms (26, 27). This uptake is an asynchronous and prolonged process and involves tyrosine kinases and actin (27). After endocytosis and vesicle acidification, capsid disassembly occurs within the endosome, thereby releasing a complex of viral genome and the L2 protein. The minor capsid protein mediates all further steps essential for establishing infection (20, 28–33).
Our recent results suggest that viral entry and postentry trafficking of HPV16 also involve tetraspanin CD151 (26). Tetraspanins constitute a conserved superfamily of four-span membrane proteins that are widely expressed at the cell surface and in intracellular membranes in multicellular organisms. Like other tetraspanin proteins, CD151 is recruited to tetraspanin-enriched microdomains (TEM or TERM) and may play a critical role in TERM-associated functions, including regulation of cellular responses to external stimuli which control cell motility and proliferation (34–39). Importantly, CD151 forms stoichiometric complexes with laminin-binding integrins (e.g., α3β1 and α6β1/4), and most of its known functions are intricately linked to these integrins (40, 41). TERM are used by various viruses, such as hepatitis C virus (HCV), HIV-1, and influenza virus as molecular platforms for their entry and postentry stages during infection (42–45). One of the first discoveries was the identification of tetraspanin CD81 as a ligand for HCV. Further experiments have shown that HCV internalization relies on additional interactions with other cell surface molecules, thus suggesting that CD81 functions as a coreceptor in a multistep internalization process (46, 47). HIV-1 assembles at and buds off TERM, but the role for tetraspanins in viral infection seems to be cell type specific (44, 45).
In our previous report, we detected a strong colocalization of HPV viral particles with CD151 and CD63 on the surface of infected HeLa cells. Furthermore, the association of HPV16 particles with CD151 on the plasma membrane was confirmed by immune-electron microscopy (20, 26). Importantly, inhibition of HPV16 infection by tetraspanin-specific antibodies and short interfering RNA (siRNA) suggested that TERM act as molecular platforms for papillomavirus entry (26).
In this study, we investigated in detail how CD151 regulates HPV16 endocytosis. Using total internal reflection (TIRF) microscopy for live-cell imaging, we observed lateral movement and internalization of HPV16 in association with CD151. Infection assays and the quantification of cell surface-bound, internalized, and disassembled particles in cells expressing different CD151 mutants revealed the importance of palmitoylation, glycosylation, and interaction with integrins for CD151-dependent HPV endocytosis and infection. Furthermore, we were able to demonstrate that CD151-interacting integrins (α3β1 and α6β1/4) are necessary for efficient papillomavirus infection.
MATERIALS AND METHODS
Cell lines and pseudovirions (PsV).
The human cervical carcinoma cell line HeLa was purchased from the German Resource Centre for Biological Material (DSMZ). HaCaT cells (human nonvirally transformed keratinocytes) were obtained from Cell Lines Services (CLS), Eppelheim, Germany. The cells were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FCS), 1% Glutamax I (Invitrogen), 1% modified Eagle's medium nonessential amino acids, and antibiotics. Normal human epidermal keratinocytes (NHEK) were purchased from PromoCell and cultivated according to the manufacturer's instructions.
HPV16 PsV were prepared as previously described (48). Briefly, expression plasmids carrying codon-optimized HPV16 L1 and L2 cDNA were cotransfected with a pcDNA3.1/Luciferase reporter plasmid into 293TT cells (30). Forty-eight hours posttransfection, the cells were processed by lysis and nuclease digestion. The PsV were purified from the cell lysates by Optiprep gradient centrifugation. Codon-optimized L1 and L2 expression vectors pUF3/hu16L1 and pUF3/hu16L2 for HPV16 were kindly provided by Martin Müller, Heidelberg, Germany (49). Quantification of pcDNA3.1/luciferase-positive PsV was performed by quantitative PCR in an AB 7300 reverse transcription-PCR (RT-PCR) system with a luciferase forward primer (5′-CGCGGAGGAGTTGTGTTTGT-3′), a luciferase reverse primer (5′-TTTTCTTGCGTCGAGTTTTCC-3′), and a luciferase TaqMan probe (5′-AAGTACCGAAAGGTCTTAC-3′). For this, the marker plasmid was extracted from PsV using the High Pure viral nucleic acid kit (Roche) according to the manufacturer's instructions.
For HPV16 PsV labeling, purified HPV16 PsV were incubated for 1 h at room temperature in phosphate-buffered saline (PBS) with a 10-fold molar excess of Alexa Fluor 488 succinimidylester (Molecular Probes) over that of the major capsid protein L1 as described previously (50). PsV were separated from the labeling reagent by size-exclusion chromatography using NAP5 columns (GE Healthcare) and stored at 4°C. Infectivity after labeling was measured using a luciferase infection assay.
Plasmids and antibodies.
Expression plasmids encoding wild-type CD151 (CD151-wt) and various CD151 mutants, including CD151-YALA, CD151-QRD, CD151-gly(−), CD151-ΔC, and CD151-palm(−), were previously described (51, 52). Cyan fluorescent protein (CFP)-tagged CD151 (CFP-CD151) was generated by isolating the CD151 gene from pEGFP-C1/CD151 with BspEI and XmaI and inserted into pECFP-C1 (Clontech). The HPV16 L1-specific antibodies, mouse monoclonal antibodies (MAbs) L1-7 and 16L1-312F and rabbit polyclonal antibody (PAb) K75, have been described previously (19, 26, 53, 54). The rat MAb anti α6-integrin (NKI-GoH3), the α3-integrin mouse MAb (P1B5), the mouse MAb anti-CD151 (IIG5A), the mouse MAb anti-laminin-5 (laminin-332) (MAB1947), and the mouse MAb anti-β-actin (AC-15) were purchased from Abcam, Santa Cruz, Serotec, Millipore, and Sigma-Aldrich, respectively. Rabbit anti-CD151 serum was generated against the recombinant large extracellular loop of CD151 (rCD151), and the PAbs were affinity purified on the immobilized rCD151.
Immunofluorescence microscopy.
NHEK or HeLa cells were grown on coverslips. After transfection and/or infection, cells were treated with paraformaldehyde-Triton X-100 or methanol. Fixed cells were incubated with the indicated primary antibodies and with Alexa-conjugated species-specific secondary antibodies (Invitrogen). DNA was stained with Hoechst 33342 (Sigma) and is shown in blue. Coverslips were mounted onto slides using Fluoprep mounting medium (bioMérieux). Images were acquired using a laser scanning microscope (LSM) 710 (Carl Zeiss, Jena, Germany) and the LSM software ZEN 2008 or using an Zeiss Axiovert 200 M microscope equipped with a Plan-Apochromat 100× objective lens (1.4 numeric aperture [NA]) and the Axiovision deconvolution and/or colocalization software (version 4.7; Zeiss). Images were assembled into figures using Photoshop (Adobe).
Immunostaining of tissue slices.
Paraffin-embedded cervix tissue slices were deparaffinized with xylol two times for 20 min and rehydrated in decreasing concentrations of ethanol (90, 70, and 50%) for 10 min each as described previously (55). The slices were rinsed with water and boiled 3 times for 30 s in 10 mM citric buffer (pH 6.0). The staining was performed as described above.
TIRF and fluorescence microscopy.
For total internal reflection fluorescence (TIRF) microscopy, HaCaT and HeLa cells were grown on 35-mm glass-bottom dishes (MatTek Corporation, Ashland, MA). Cells expressing the CFP-CD151 plasmid were infected with Alexa Fluor 488-labeled HPV16 PsV for 2.5 h. Dynamics of viral particles in relation to CD151 were recorded every 5 s for 2 to 4 min using a Zeiss Axiovert 200 M microscope equipped with a Plan-Apochromat 100× objective lens (1.4 NA), a TIRF slider, a multiline argon laser, and a Zeiss AxioCamHR digital camera. For quantification of Alexa Fluor 488 fluorescent signal, noncolocalized and colocalized with CFP-CD151, 20 time-lapse images of two TIRF movies were analyzed. Pixel-by-pixel analysis was performed using the Axiovision colocalization module with fixed thresholds for all images.
Knockdown of CD151 and integrins.
The CD151-specific siRNAs CD151#1 (CAUGUGGCACCGUUUGCCUdTdT), obtained from Qiagen, and CD151#2, an siRNA smartpool which contains the target sequences UCACAGGACUGGCGAGACA, CCUCAAGAGUGACUACAUC, GCAAGACGGUGGUGGCUCU, and GAUCAUCGCUGGUAUCCUC, obtained from Dharmacon, all target the coding region of the CD151 gene. CD151#3 (Hs_CD151_5 [Qiagen]; CACAUACAGGUGCUCAAUAAAdTdT) targets the 3′-untranslated region (3′UTR) of the CD151 gene but does not affect expression of the protein from the transfected CD151-encoding plasmid. Integrin α3 and α6 subunits were targeted using the following siRNAs: α3-integrin #1 GCUACAUGAUUCAGCGCAA), α3-integrin #2 (GUUUGAAGGCUUGGGCAAA), α6-integrin #1 (GGAUAUGCCUCCAGGUUAA), and α6-integrin #2 (CUGUAAGGAUCCGGAAAGA), obtained from Sigma. HeLa, HaCaT, or NHEK cells were transfected with 30 nM siRNA using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Subsequent experiments were done 24 or 48 h after siRNA transfection. Knockdown efficiency was quantified by Western blotting and flow cytometry.
Pseudovirus infection assay.
Cells were grown in 24-well plates and infected with 100 (HeLa and HaCaT) or 500 (NHEK) luciferase vector-positive PsV per cell (56). Infection efficiencies of HPV PsV were assessed by quantification of luciferase expression 24 (HeLa and HaCaT) or 48 h (NHEK) postinfection (p.i.). Briefly, cells were lysed with cell culture lysis reagent (Promega), and relative luciferase activity was measured with the luciferase assay system (Promega) according to the manufacturer's instructions using the Tristar LB 941 luminometer (Berthold Technologies, Bad Wildbad, Germany). Statistical significances (P < 0.05) were calculated with the t test (two-tailed, paired).
Luciferase expression control assay.
HeLa cells were seeded on 24-well plates. Cells were cotransfected with 30 nM siRNA and 0.2 μg luciferase reporter plasmid using JetPrime (Polyplus transfections) according to the manufacturer's instructions. Forty-eight hours after transfection, cells were lysed with cell culture lysis reagent (Promega). One hundred μl of lysate was used for detection of luciferase activity. Relative luciferase activity was measured with the luciferase assay system (Promega) according to the manufacturer′s instructions using the luminometer Lumat LB 9507 (Berthold Technologies).
Cell binding assay.
HeLa, HaCaT, or NHEK cells grown in 12-well plates were transfected with siRNAs for 48 h. Subsequently, cells were detached, resuspended in serum-free DMEM, and transferred to siliconized reaction tubes. Cells were incubated with HPV16 PsV for 1 h at 37°C, washed four times with PBS, and then collected in SDS sample buffer for Western blotting. Cell-bound HPV16 particles were stained with anti-L1 antibody 312F, and β-actin was stained as an input control.
Detection of surface-bound particles by flow cytometry.
HeLa cells were transfected with the indicated siRNAs and/or plasmids and infected with HPV16 PsV for 1 h (for binding of virions to the cell surface). Subsequently, cells were extensively washed with PBS to remove unbound virions and analyzed by flow cytometry or incubated for an additional 17 h (18 h total). Cells were trypsinized with 0.25% trypsin–2.5 mM EDTA. Surface-bound particles were stained using the anti-L1 (K75) antibody and secondary anti-rabbit Alexa Fluor 488 antibody. Background staining was determined using an unspecific IgG serum. For double staining, CD151 was stained using the anti-CD151 (IIG5A) antibody and secondary anti-mouse Qdot 705 antibody. Cells were analyzed by flow cytometry (FACScan; Becton, Dickinson) and CellQuest 3.3 (Becton, Dickinson) software.
Detection of internalized particles by immunofluorescence.
HeLa cells were grown on coverslips and transfected with siRNAs targeting CD151. Twenty-four hours later, cells were infected with HPV16 PsV and incubated for 7 h at 37°C. Subsequently, cells were fixed with methanol and processed for staining with MAb L1-7 as described previously (26). This MAb recognizes a specific epitope located in the interior of the pseudovirion capsid and is not accessible in intact virions (53, 57). The samples were analyzed by fluorescence microscopy using a Zeiss Axiovert 200 M inverted microscope and quantified by ImageJ software. For quantification, the relative amount of internalized particles was determined based on the L1-7-positive pixels relative to the cell nucleus signal (DNA/Hoechst 33342-positive pixels) out of 100 randomly selected pictures (knockdown experiments) and 100 CD151-expressing cells (recovery experiments) from at least two independent experiments. A threshold value was set to exclude background.
Recovery experiments.
HeLa cells were treated with CD151-specific siRNA targeting the 3′UTR of the gene or control siRNA for 24 h and transfected with the indicated plasmids encoding CD151-wt and various CD151 mutants using Lipofectamine 2000 (Invitrogen) for an additional 24 h. Afterwards, the cells were infected with HPV16 PsV and an infection assay was performed.
RESULTS
HPV16 virions are closely associated with CD151 containing microdomains on the plasma membrane.
HPV virions exclusively infect the basal cells within the stratified epithelial tissue in order to establish infection (8). We studied the distribution of CD151 in cervical mucosa by immunofluorescence staining of cervical tissue sections and detected a high expression level of CD151 only within the basal cells (Fig. 1A). Confocal fluorescence microscopy of primary keratinocytes infected with HPV16 virions showed extensive colocalization of virions with CD151 in intracellular vesicles (Fig. 1B). Note that under the permeabilization conditions used, cells lost most of their surface pools of CD151. The side view of z stacks shows colocalization of the major capsid protein L1 with CD151 in different regions of the cytoplasm (Fig. 1B, X/Z axes side view). In addition, TIRF microscopy revealed prolonged colocalization of CD151 and HPV16 on the cell surface of HaCaT cells (Fig. 1C) when labeled viral particles were incubated with cells for 1 to 4 h. Importantly, knockdown of CD151 reduced expression of plasma membrane-expressed (Fig. 1D) and endogenous CD151 (Fig. 1F, upper) and inhibited HPV16 PsV infectivity of keratinocytes as tested using a standard luciferase assay (Fig. 1F, lower). In control experiments, we found that depletion of CD151 does not affect the expression level of the luciferase gene (Fig. 1E). These results support the biological relevance of the tetraspanin CD151 for HPV16 infection.
Fig 1.

Tetraspanin CD151 is important for HPV16 infection in keratinocytes. (A) Distribution of CD151 in the cervical mucosa. Human cervical tissue sections are stained with anti-CD151 (red) showing a high expression level of basal cells (upper right panel; laminin 332 staining, indicating the basement membrane, is shown in green). Nuclei are shown in blue (A and B). (B) CD151 colocalizes with HPV16-L1 in primary keratinocytes. Primary keratinocytes (NHEK) were infected with HPV16 PsV for 4 h and analyzed by confocal and deconvolution fluorescence microscopy (right panel; the lower right shows the X/Z view of the z stack). (C) TIRF microscopy of HaCaT cells transfected with CFP-CD151 and infected with Alexa Fluor 488-labeled HPV16 pseudovirions for 1.5 h. Arrows depict colocalization of HPV16 and CD151. (D, E, and F) Knockdown of CD151 by CD151 siRNAs. Cells were transfected with CD151-specific siRNAs for 48 h. (D) The knockdown of the CD151 level on the cell surface was measured by flow cytometry in HeLa and HaCaT cells. (E) CD151-specific siRNAs have no significant influence on luciferase expression. HeLa cells were cotransfected with control, luciferase-specific, or CD151-specific siRNA and with luciferase-expressing plasmid to control the influence of siRNA transfection on luciferase expression. The luciferase-specific siRNA serves as a positive control. (F) Depletion of CD151 in HeLa, HaCaT, and NHEK cells correlates with reduced infection. SDS-PAGE (upper panels) was performed under nonreducing conditions and shows the amount of CD151. The loading control is an unspecific band produced by the CD151 polyclonal rabbit antibody that serves as a control for protein input. For infection assay (lower panels), cells were transfected with siRNA as indicated for 48 h and then infected for 24 h with HPV16 PsV. Infectivity was measured by luciferase activity and normalized by lactate dehydrogenase (LDH) measurements. The control siRNA infection rate was set to 100%. *, P < 0.05 compared to the control.
To examine how CD151 controls HPV16 infection, we first analyzed the dynamics of cell-bound virions in relation to CD151-containing microdomains. In these experiments, we used TIRF microscopy for live-cell imaging to monitor movements of virions within the plane of the plasma membrane and their internalization. HeLa cells were transiently transfected with CFP-tagged CD151 and 24 h later were infected with Alexa Fluor 488-labeled HPV16 PsV. After virus binding to the cell surface, cells were incubated for 2 to 4 h at 37°C, and subsequently single particles were tracked with 10 frames per min. These experiments showed that viral particles remained associated with CD151 during their lateral movement on the cell surface (Fig. 2A; also see Movie S1 in the supplemental material). In addition, we observed disappearance of CD151-colocalized virions from the TIRF field due to internalization of viral particles as vesicles leave the evanescent field (27) (Fig. 2B; also see Movie S2 in the supplemental material). Importantly, we found that only CD151-associated virions disappeared from the TIRF field, while the CD151-free particles remained on the plasma membrane for the time of the observation (Fig. 2C). These results suggest the association of CD151 with microdomains as a precondition for HPV16 internalization.
Fig 2.

Lateral surface movement and cointernalization of HPV16-bound CD151. Shown are TIRF images of HeLa cells transfected with CFP-CD151, infected with Alexa Fluor 488 (AF488)-labeled HPV16 pseudovirions for 2.5 h, and analyzed by TIRF microscopy for 2 to 4 min. Time-lapse images show (A) lateral movement of HPV16-bound CD151 and (B) cointernalization of HPV16 and CD151. Arrows depict colocalization of HPV16 and CD151 during the process of internalization. (C) Quantitative analysis of HPV16 endocytosis. Cell surface areas with approximately 50% of the particles in colocalization with CFP-CD151 were selected, and the loss of fluorescent signal was determined over time. HPV16 Alexa Fluor 488 pixels noncolocalized (green) and colocalized with CFP-CD151 (blue) were analyzed by the Axiovision colocalization module with fixed thresholds for all images of two time-lapse sequences (20 frames every 5 s). The averages of the corresponding frames from two independent movies are shown with error bars representing the standard deviations of the pixel numbers.
CD151 is required for endocytic uptake of HPV16 virions.
Close association of internalizing HPV16 with CD151-containing microdomains suggests that CD151 controls early stages of viral infection. To examine the role of CD151 in viral entry, control and CD151-depleted cells were incubated with HPV16 PsV for 1 or 18 h, and surface-associated virions were quantified by flow cytometry using polyclonal anti-L1 antibody K75 (Fig. 3A). In cells treated with the control siRNA, the amount of surface-bound PsV after 18 h of incubation decreased to 25% of the initial binding level (i.e., binding after 1 h of incubation). In contrast, the amount of PsV which remained on the surface of CD151-depleted cells after 18 h was ∼4-fold higher than the amount of PsV on control-treated cells (Fig. 3A). In additional experiments, we found that depletion of CD151 has no effect on the initial binding of HPV16 to the cell surface of HeLa, HaCaT, or primary keratinocytes (Fig. 3B). To monitor HPV16 invasion further, we examined the amount of disassembled capsids in endosomes after internalization using the specific anti-L1 MAb L1-7 (19, 26). For these experiments, siRNA-transfected cells were infected with HPV16 PsV for 7 h. As illustrated in Fig. 3C and D, CD151 knockdown resulted in a marked decrease in the number of L1-7-positive endosomes, thereby suggesting inhibition of HPV16 disassembly.
Fig 3.

CD151 is involved in HPV16 internalization but not in cell surface binding. (A) HeLa cells were treated with siRNAs for 48 h and subsequently incubated with HPV16 PsV for 1 or 18 h as indicated. The amount of surface-associated PsV was assessed by flow cytometry using anti-L1 PAb (K75). Mean fluorescence intensity for cells incubated with HPV16 PsV for 1 h was adjusted to 100%. Shown are compiled results of three independent experiments. (B) Primary binding of HPV16 to the cell surface is independent of CD151. HeLa, HaCaT, and NHEK cells were transfected with control or CD151-specific siRNAs for 48 h and subsequently incubated with HPV16 PsV for 1 h. Cell-bound PsV were detected in cell lysates by Western blotting using specific anti-L1 PAb. (C and D) HeLa cells were treated with siRNAs for 48 h and infected with HPV16 PsV for 7 h. Virus internalization was analyzed by immunofluorescence using monoclonal antibody (L1-7) recognizing an epitope that is located on the inner side of intact capsids and only accessible after internalization and capsid disassembly. Representative images are shown in panel C. (D) For quantification of virus internalization and disassembly, L1-7-positive pixels of at least 100 cells were analyzed using an ImageJ script. Shown are the results of three independent experiments normalized to control siRNA-treated and infected cells. *, P < 0.05 compared to the control.
Taken together, these data demonstrated that although CD151 is not involved in the initial binding of HPV16 to cells, it plays a critical role in endocytic uptake of the virus, leading to disassembly of the capsid.
Tyrosine-based sorting motif in CD151 is not required for HPV16 infection.
To begin characterization of molecular mechanisms underlying the activity of CD151 in HPV16 infection, we initially examined the role of the tyrosine-based sorting motif in the CD151 C-terminal cytoplasmic region (i.e., the Y245-R-S-L sequence). We have recently shown that this sequence plays an important role in the antibody-induced surface clearance of CD151 (51). Thus, it was also possible that the Y245-R-S-L motif would be important for proendocytic function of the protein in controlling HPV16 infection. To investigate the role of the Y245-R-S-L motif, we carried out infectivity assays using cells overexpressing either wild-type CD151 or a CD151 mutant with two point mutations within this sorting motif (CD151-YALA). Interestingly, overexpression of the YALA mutant resulted in a 2- to 3-fold increase of HPV16 infection in HeLa and HaCaT cells, respectively (Fig. 4A). We also noticed that the activity of the YALA mutant was slightly stronger than that of CD151-wt. As a complementary approach, we analyzed the activity of the CD151-YALA mutant using the recovery assay for HPV16 infection (Fig. 4B and C). To this end, the endogenous CD151 was depleted from cells using siRNA-CD151, which targets the 3′UTR of the mRNA; 24 h later, tetraspanin-depleted cells were transiently transfected with the constructs encoding siRNA-resistant wild-type CD151 or CD151-YALA. Reexpression of the wild-type protein increased infectivity in CD151-depleted cells from 30% in CD151 knockdown cells to ∼60% (Fig. 4C). Importantly, the activity of the YALA mutant in these experiments was comparable to that of the wild-type protein. Accordingly, the CD151-YALA mutant was as effective as the wild-type protein in recovering the virus disassembly steps (Fig. 4D and E). These experiments showed that the Y245-R-S-L sorting motif is not important for the function of CD151 in HPV16 infection.
Fig 4.

Tyrosine-based sorting motif in CD151 is not required for HPV16 internalization and infection. (A) Overexpression of CD151-wt and CD151-YALA increases HPV16 infection. HeLa and HaCaT cells were transfected with control or CD151 plasmids as indicated for 24 h and then infected for 24 h with HPV16 PsV. Infectivity was measured by luciferase activity and normalized to LDH measurements. *, P < 0.05 compared to the control). (B to E) Reexpression of CD151-wt and -YALA in CD151-depleted cells recovers HPV16 infection and virus internalization in CD151-depleted cells. HeLa cells were transfected with control or CD151-specific siRNA (CD151#3) which targets the untranslated region of the mRNA. After 24 h, cells were transfected with a control plasmid or with a plasmid encoding CD151-wt or -YALA to recover CD151 expression. (B and C) Recovery of CD151 expression in CD151-depleted cells was controlled by flow cytometry and additionally monitored using a luciferase infectivity assay. (C) Infectivity was measured by luciferase activity and normalized by LDH measurements. The control siRNA infection rate was set to 100%. *, P < 0.05 compared to the control; $, P < 0.05 compared to CD151 knockdown. (D and E) Internalization and disassembly of HPV16 PsV. HeLa cells were treated with siRNAs for 48 h and infected with HPV16 PsV for 7 h. Virus internalization was analyzed by immunofluorescence using monoclonal antibody (L1-7) recognizing an epitope that is located on the inner side of intact capsids and only accessible after internalization and capsid disassembly. Representative images are shown in panel D. (E) For quantification, at least 100 CD151-overexpressing cells were analyzed using an ImageJ script. *, P < 0.05 compared to cells transfected with control siRNA and control plasmid; $, P < 0.05 compared to cells transfected with CD151#3 and control plasmid.
The role of the CD151-integrin complexes in HPV16 infection.
CD151 controls activities of the associated integrins (41, 58). Furthermore, at least one of the CD151-associated integrins (α6β4) is proposed to serve as a receptor for HPV (22, 24). Thus, we investigated whether CD151 functions in HPV16 infection in the complex with integrins. Initially, we observed a decrease in HPV16 infectivity in cells depleted of either α3- or α6-integrin (Fig. 5B). Furthermore, in contrast to CD151-wt, overexpression of CD151 mutants which are known to affect integrin functions and localization [i.e., CD151-QRD, CD151-gly(−), CD151-ΔC, and CD151-palm(−)] (Fig. 6C) did not increase HPV16 infection (Fig. 6C). The expression levels of the CD151 constructs were controlled by Western blotting (Fig. 6B).
Fig 5.
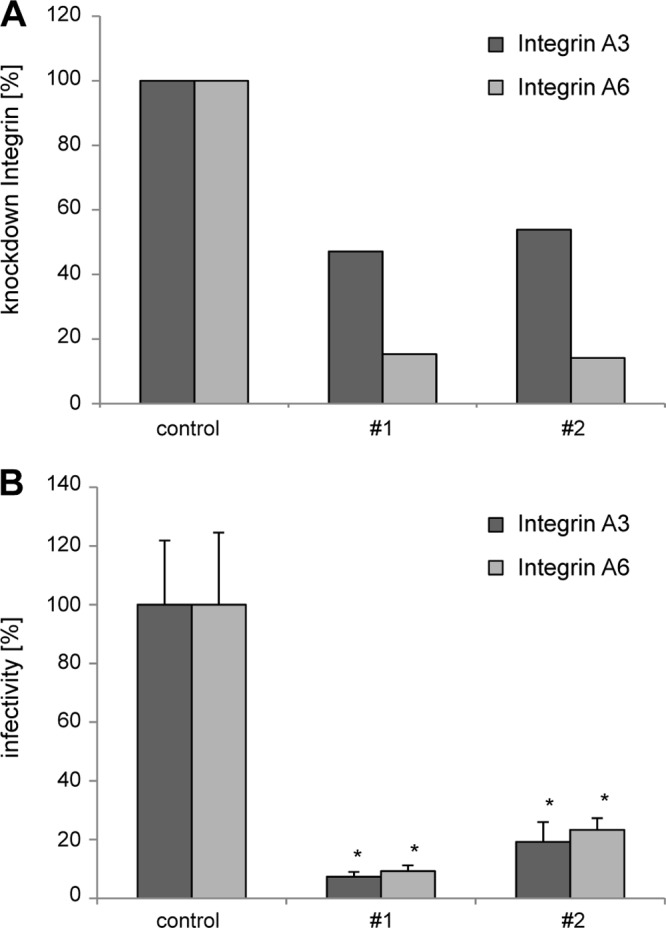
CD151-interacting integrins α3β1 and α6β1/4 are important for HPV16 infection. HeLa cells were transfected with siRNAs targeting α3β1- and α6β1/4-integrin subunits, and after 48 h cells were incubated with HPV16 PsV for 24 h. (A) The level of integrin knockdown was assessed by flow cytometry. Data are presented as percentages relative to the level expressed in cells transfected with an siRNA control. (B) Relative infectivity was measured by luciferase activity and normalized by LDH measurements. *, P < 0.05 compared to the control.
Fig 6.
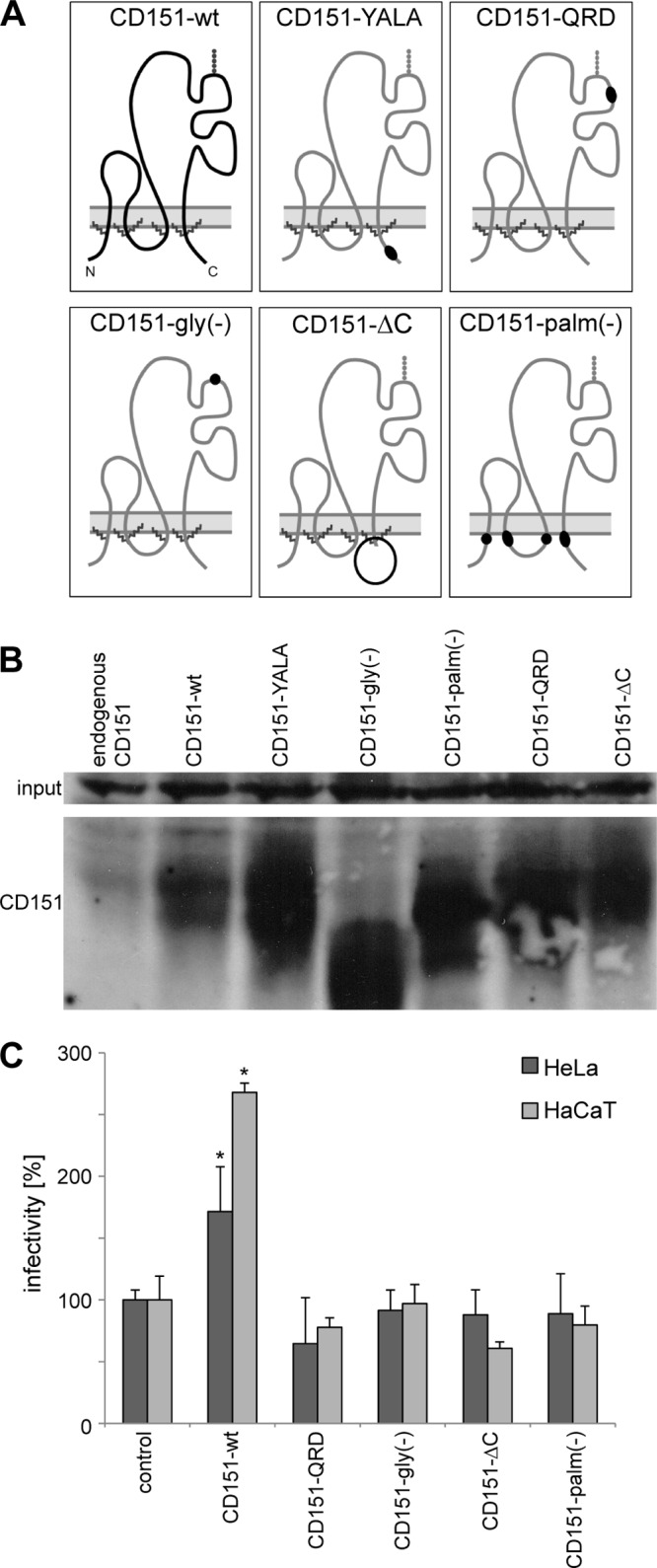
Integrin association and palmitoylation of CD151 are important for HPV16 infection. (A) Schematic diagram of CD151 mutants. (B) HeLa cells were transfected with control plasmid, CD151-wt, or CD151 mutants for 24 h. CD151 expression levels in cell lysates were analyzed by Western blotting. (C) Influence of CD151 mutants on HPV16 infection was analyzed after CD151 overexpression as described in the legend to Fig. 4A.
We next used the approach of infection and recovery of internalization/endocytosis to analyze the activity of CD151 mutants (Fig. 7). The CD151-QRD mutant is unable to associate directly with α3β1- and α6-integrins (59) and to support various integrin-dependent functions (52, 60). Notably, in agreement with the important role played by CD151-integrin complexes in HPV16 infection, the CD151-QRD mutant did not restore the activity of CD151 in internalization recovery experiments (Fig. 7B to D). The importance of CD151-integrin cooperation in HPV16 infection was further emphasized when we used the glycosylation-deficient and C terminus-deleted mutants of CD151 [CD151-gly(−) and CD151-ΔC, respectively]. We have previously reported that the CD151-gly(−) protein is unable to restore integrin-dependent migration in CD151-deficient cells (52). Others have demonstrated that the C-terminal cytoplasmic portion of CD151 is critical for the activity of the tetraspanin-associated integrins (61). Here, we found that, similar to the CD151-QRD mutant, overexpression of CD151-gly(−) or CD151-ΔC did not increase HPV16 infection (Fig. 6C). We also examined activities of CD151 mutants in the virion internalization/disassembly assay. In accordance with the results of the infectivity experiments, we found that only the wild-type protein could rescue the internalization-deficient phenotype observed in CD151-depleted cells (Fig. 7C to E). Taken together, these results indicated that CD151 functions via integrins in HPV16 infection.
Fig 7.

Recovery of endocytosis and disassembly using CD151 mutants. (A) HeLa cells were treated with siRNAs for 24 h and then transfected with plasmids as indicated. CD151 expression levels on the cell surface were analyzed by flow cytometry. (B) Primary binding of HPV16 to the cell surface is not affected by overexpression of CD151 mutants, but only CD151-wt and -YALA are able to reconstitute HPV16 endocytosis. HeLa cells were treated with CD151 siRNAs for 24 h, transfected with CD151 plasmids as indicated, and subsequently incubated with HPV16 PsV for 1 and 18 h. The amount of cell surface-bound virus particles was quantified by flow cytometry using anti-L1 pAb (K75). Mean of fluorescence intensity for cells treated with control siRNA and plasmid and incubated with HPV16 PsV for 1 h was adjusted to 100%. *, P < 0.05 compared to cells transfected with CD151#3 and control plasmid. (C) HeLa cells were treated with siRNAs and plasmids as described for panel B and infected with HPV16 PsV for 7 h. The major capsid protein L1 was detected with monoclonal anti-L1 antibody (L1-7; green) reacting with an L1 epitope that is accessible exclusively after viral entry, and CD151 was detected with monoclonal anti-CD151 antibody (red). The upper panels show the costaining of CD151 and HPV16-L1, and the lower panels show the staining of HPV16-L1 only. (D and E) Virus internalization was analyzed by immunofluorescence as described in the legend to Fig. 4D and E.
Recruitment to TERM is critical for the activity of CD151 in HPV16 infection.
Palmitoylation is critical for lateral interactions of CD151 with other tetraspanins and, therefore, has an important role in positioning associated integrins within TERMs (62–64). When analyzed in the internalization recovery experiments, palmitoylation-deficient CD151 [CD151-palm(−)] was unable to restore L1-7 reactivity in CD151-depleted cells (Fig. 7C to E) and had no effect on HPV infection after overexpression (Fig. 6C).
To exclude the possibility that loss of L1-7 reactivity is caused by inhibition of capsid binding, we examined whether expression of any of the mutants affected HPV-16 cell binding by flow cytometry of surface-bound particles. Virus binding was analyzed after cells were incubated with HPV16 PsV for 1 h. We also incubated PsV with cells for 18 h to determine the endocytic capacity of the cells. Surface-associated virions then were quantified as described above. As illustrated in Fig. 7B, none of the mutants affected surface binding of HPV16. In addition, endocytosis assay (18 h p.i.) confirmed that only the wild-type CD151 and the YALA mutant were able to restore virus internalization. Accordingly, expression of the other mutants did not rescue the HPV16 entry deficiency in CD151-depleted cells. Thus, interaction of CD151 with its integrin partners and recruitment of CD151-integrin complexes to TERM is important for endocytic uptake of HPV16.
DISCUSSION
The data presented in this study indicate that entry of HPV16 into keratinocytes is mediated by the tetraspanin CD151. Results with CD151 mutants suggest that HPV16 endocytosis is dependent on the recruitment of the tetraspanin to TERM and its association with laminin-binding integrins (i.e., α3β1 and α6β1/β4). Together, these data strongly suggest that papillomaviruses use TERM and their specific components as a platform for viral entry.
HPV virions infect the basal cells within the stratified epithelial tissue. We found that, similar to other epithelial organs (e.g., skin and mammary gland), CD151 is highly expressed in the basal layers of cervical mucosa, where epithelial cells come into direct contact with the basement membrane. Unlike HSPGs, which are expressed throughout the epithelium, expression of the CD151-associated integrins (α3β1 and α6β1/4 integrin) is largely found in basal keratinocytes (65, 66). Here, we demonstrate for the first time that the formation of integrin-CD151 complexes in keratinocytes is critical for HPV16 infection. These results strongly suggest that these molecular assemblies are used by HPV16 as entry points to infect its target cells in vivo. Previous studies implicated α6-integrins in HPV6b (22, 23) and HPV16 cell binding (24, 67). Expression of α6-integrin subunit in binding-negative B cells, DG75, caused binding of HPV6b virus-like particles (VLPs) to the cell surface (23). In addition, anti-α6-integrin antibody was able to block VLP/cell binding. In contrast to these reports, we found that depletion of CD151 or α6-integrins (not shown) had no effect on primary cell binding of HPV16 to keratinocytes. However, we discovered that α3β1- and α6-integrins in complex with CD151 function in a postbinding step(s) during viral infection, thereby playing a possible role of secondary receptors.
Our earlier studies described colocalization of viral particles with CD151 at the plasma membrane of infected cells (26). Here, we used TIRF microscopy and live-cell imaging to show that only CD151-bound HPV16 could enter the cells. Importantly, we also observed that CD151-associated virus particles underwent lateral movement within the plane of the membrane before internalization. These results add further complexity to the initial (i.e., preentry) steps in HPV infection and indicate that CD151 functions as a surface chaperon which facilitates recruitment of the virus to the sites of its entry into the cells. This model suggests that binding to CD151 (or a CD151-contaning complex) is a prerequisite for HPV internalization. It is tempting to speculate that tetraspanin-enriched microdomains represent the target sites for the moving CD151-HPV complex to initiate viral entry. Indeed, we found that surface-bound HPV16 is colocalized with other tetraspanin proteins, including CD9, CD81, CD82 (K. Scheffer, G. A. Spoden, and L. Florin, unpublished data), and CD63 (26). Furthermore, we show that palmitoylation-deficient CD151, a mutant that cannot be recruited into TERM, is unable to reconstitute HPV16 entry when expressed in CD151-depleted cells.
Our data show that in addition to its role in the preentry steps (i.e., recruitment of viral particles to TERM), CD151 also controls internalization of HPV16. Indeed, we found that CD151 is cointernalized with HPV16 particles to be found in HPV16-positive endosomes of infected primary keratinocytes. While the identity of the endocytic receptor(s) for HPV16 remains to be established, our data suggest that laminin-binding integrins (α3β1 or α6β1/β4) are involved. Binding of HPV to cells triggers integrin-dependent activation of focal adhesion kinase (FAK) and phosphotidylinositol 3-kinase (PI3K) (67, 68), both of which are important for early steps of viral entry (27, 67). Importantly, we and others have previously reported that CD151 plays an important role in regulating both of these signaling pathways (63, 69), which may explain the proendocytic function of the protein toward HPV16. Alternatively, CD151-integrin complexes may facilitate HPV16 entry via the EGF receptor, which is also a component of TERM (70) and has been shown to be important for HPV16 infection (25). In this regard, Yang and colleagues have recently found that CD151 regulated cellular responses to EGF (71).
It remains to be established whether continuing interaction with CD151-associated receptors (integrins and/or EGFR) is required for viral entry into cells. Growth factor-activated EGFR is known to endocytose via clathrin- and caveolin-dependent pathways (72). However, neither of these pathways seems to be important for HPV16 entry (26, 27). Thus, one expects CD151 to control a novel clathrin-independent endocytic pathway. Furthermore, it is feasible that CD151 controls HPV16 entry by regulating endocytosis of the associated integrins. Accordingly, we found that, like other TERM components, integrins (and growth factor receptors) are cointernalized with HPV16 into endosomes of infected keratinocytes (K. Scheffer, G. A. Spoden, and L. Florin, unpublished data). Whatever the mechanisms underlying the role of CD151 in integrin-dependent HPV infection, this pathway is unlikely to involve a tyrosine-based sorting motif, a putative binding site of the clathrin adaptor AP2, in the C-terminal cytoplasmic part of CD151 (Y245-R-S-L sequence). Indeed, our overexpression and expression recovery experiments indicate that the activities of the CD151-YALA mutant were comparable to those of wild-type CD151. Future work should clarify whether the ability of CD151 to regulate trafficking of the associated integrins is linked to integrin-dependent viral infection.
In summary, we propose that after papillomavirus binding to heparan sulfate proteoglycans, accompanied by structural rearrangement of the capsid and/or putative cleavage of syndecan, viral particles are transferred to secondary receptor complexes composed of CD151 and associated integrins. In turn, these complexes are responsible for virus-induced signaling and subsequent internalization of viral particles. It is also possible that GFRs are included in this multiprotein complex and contribute to the tetraspanin-dependent endocytic pathway (20, 27, 73). Further investigation is under way to establish how various components of TERM and CD151 control the multifactorial process of HPV infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant (SFB490/D2) from the German Research Foundation to L.F. and C.L. and the intern-funding program of the Johannes Gutenberg University Mainz to L.F.
We thank Teodora Nikolova, Mainz, for assistance with LSM, Fatima Boukhallouk and Kirsten Freitag, Mainz, for technical support, and Sally Roberts for critical reading of the manuscript.
Footnotes
Published ahead of print 9 January 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02906-12.
REFERENCES
- 1. Chen X, Garcea R, Goldberg I, Casini G, Harrison S. 2000. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell 5:557–567 [DOI] [PubMed] [Google Scholar]
- 2. Modis Y, Trus BL, Harrison SC. 2002. Atomic model of the papillomavirus capsid. EMBO J. 21:4754–4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baker TS, Newcomb WW, Olson NH, Cowsert LM, Olson C, Brown JC. 1991. Structures of bovine and human papillomaviruses. Analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys. J. 60:1445–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. 2008. Arrangement of L2 within the papillomavirus capsid. J. Virol. 82:5190–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Finnen RL, Erickson KD, Chen XS, Garcea RL. 2003. Interactions between papillomavirus L1 and L2 capsid proteins. J. Virol. 77:4818–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doorbar J. 2006. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 110:525–541 [DOI] [PubMed] [Google Scholar]
- 7. Longworth MS, Laimins LA. 2004. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 68:362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doorbar J. 2005. The papillomavirus life cycle. J. Clin. Virol. 32(Suppl. 1):S7–S15 [DOI] [PubMed] [Google Scholar]
- 9. Thomas M, Narayan N, Pim D, Tomaić V, Massimi P, Nagasaka K, Kranjec C, Gammoh N, Banks L. 2008. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 27:7018–7030 [DOI] [PubMed] [Google Scholar]
- 10. Joyce J, Tung J, Przysiecki C, Cook J, Lehman E, Sands J, Jansen K, Keller P. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 274:5810. [DOI] [PubMed] [Google Scholar]
- 11. Giroglou T, Florin L, Schafer F, Streeck R, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75:1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sapp M, Day PM. 2009. Structure, attachment and entry of polyoma- and papillomaviruses. Virology 384:400–409 [DOI] [PubMed] [Google Scholar]
- 13. Horvath CAJ, Boulet GAV, Renoux VM, Delvenne PO, Bogers J-PJ. 2010. Mechanisms of cell entry by human papillomaviruses: an overview. Virol. J. 7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Culp T, Budgeon L, Christensen N. 2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 347:147–159 [DOI] [PubMed] [Google Scholar]
- 15. Culp TD, Budgeon LR, Marinkovich MP, Meneguzzi G, Christensen ND. 2006. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 80:8940–8950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Selinka H-C, Giroglou T, Nowak T, Christensen ND, Sapp M. 2003. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 77:12961–12967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. U. S. A. 101:14252–14257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Day PM, Lowy DR, Schiller JT. 2008. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 82:12565–12568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bienkowska-Haba M, Patel HD, Sapp M. 2009. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 5:e1000524 doi:10.1371/journal.ppat.1000524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Florin L, Sapp M, Spoden GA. 2012. Host-cell factors involved in papillomavirus entry. Med. Microbiol. Immunol. 201:437–448 [DOI] [PubMed] [Google Scholar]
- 21. Selinka H-C, Florin L, Patel HD, Freitag K, Schmidtke M, Makarov VA, Sapp M. 2007. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J. Virol. 81:10970–10980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NA. 1997. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J. Virol. 71:2449–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McMillan N, Payne E, Frazer I, Evander M. 1999. Expression of the α6 integrin confers papillomavirus binding upon receptor-negative B-cells. Virology 261:271–279 [DOI] [PubMed] [Google Scholar]
- 24. Yoon C, Kim K, Park S, Cheong S. 2001. α6 Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem. Biophys. Res. Commun. 283:668–673 [DOI] [PubMed] [Google Scholar]
- 25. Surviladze Z, Dziduszko A, Ozbun MA. 2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 8:e1002519 doi:10.1371/journal.ppat.1002519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16–involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313 doi:10.1371/journal.pone.0003313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schelhaas M, Shah B, Holzer M, Blattmann P, Kühling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 8:e1002657 doi:10.1371/journal.ppat.1002657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kämper N, Day PM, Nowak T, Selinka H-C, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. 2006. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 80:759–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Florin L, Becker KA, Lambert C, Nowak T, Sapp C, Strand D, Streeck RE, Sapp M. 2006. Identification of a dynein interacting domain in the papillomavirus minor capsid protein l2. J. Virol. 80:6691–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneider MA, Spoden GA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol. 13:32–46 [DOI] [PubMed] [Google Scholar]
- 31. Bergant Marušič M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting Nexin 17. Traffic 13:455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the HPV16 capsid protein L1 from the L2/DNA complex following virus entry. J. Virol. 86:9875–9887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cerqueira C, Schelhaas M. 2012. Principles of polyoma- and papillomavirus uncoating. Med. Microbiol. Immunol. 201:427–436 [DOI] [PubMed] [Google Scholar]
- 34. Maecker HT, Todd SC, Levy S. 1997. The tetraspanin superfamily: molecular facilitators. FASEB J. 11:428–442 [PubMed] [Google Scholar]
- 35. Boucheix C, Rubinstein E. 2001. Tetraspanins. Cell. Mol. Life Sci. 58:1189–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yunta M, Lazo P. 2003. Tetraspanin proteins as organisers of membrane microdomains and signalling complexes. Cell. Signal. 15:559–564 [DOI] [PubMed] [Google Scholar]
- 37. Hemler ME. 2001. Specific tetraspanin functions. J. Cell Biol. 155:1103–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berditchevski F, Odintsova E. 2007. Tetraspanins as regulators of protein trafficking. Traffic 8:89–96 [DOI] [PubMed] [Google Scholar]
- 39. Yáñez-Mó M, Barreiro O, Gordon-Alonso M, Sala-Valdés M, Sánchez-Madrid F. 2009. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 19:434–446 [DOI] [PubMed] [Google Scholar]
- 40. Berditchevski F. 2001. Complexes of tetraspanins with integrins: more than meets the eye. J. Cell Sci. 114:4143. [DOI] [PubMed] [Google Scholar]
- 41. Stipp CS. 2010. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert Rev. Mol. Med. 12:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin F, Roth DM, Jans DA, Pouton CW, Partridge LJ, Monk PN, Moseley GW. 2005. Tetraspanins in viral infections: a fundamental role in viral biology? J. Virol. 79:10839–10851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Spriel AB, Figdor CG. 2010. The role of tetraspanins in the pathogenesis of infectious diseases. Microbes Infect. 12:106–112 [DOI] [PubMed] [Google Scholar]
- 44. Monk PN, Partridge LJ. 2012. Tetraspanins: gateways for infection. Infect. Disord. Drug Targets 12:4–17 [DOI] [PubMed] [Google Scholar]
- 45. Thali M. 2011. Tetraspanin functions during HIV-1 and influenza virus replication. Biochem. Soc. Trans. 39:529–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dubuisson J, Helle F, Cocquerel L. 2008. Early steps of the hepatitis C virus life cycle. Cell Microbiol. 10:821–827 [DOI] [PubMed] [Google Scholar]
- 47. Meredith LW, Wilson GK, Fletcher NF, McKeating JA. 2012. Hepatitis C virus entry: beyond receptors. Rev. Med. Virol. 22:182–193 [DOI] [PubMed] [Google Scholar]
- 48. Buck C, Pastrana D, Lowy D, Schiller J. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78:751–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leder C, Kleinschmidt JA, Wiethe C, Müller M. 2001. Enhancement of capsid gene expression: preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J. Virol. 75:9201–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schelhaas M, Ewers H, Rajamäki M-L, Day PM, Schiller JT, Helenius A. 2008. Human papillomavirus type 16 entry: retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 4:e1000148 doi:10.1371/journal.ppat.1000148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu L, He B, Liu WM, Zhou D, Cox JV, Zhang XA. 2007. Tetraspanin CD151 promotes cell migration by regulating integrin trafficking. J. Biol. Chem. 282:31631–31642 [DOI] [PubMed] [Google Scholar]
- 52. Baldwin G, Novitskaya V, Sadej R, Pochec E, Litynska A, Hartmann C, Williams J, Ashman L, Eble JA, Berditchevski F. 2008. Tetraspanin CD151 regulates glycosylation of (alpha)3(beta)1 integrin. J. Biol. Chem. 283:35445–35454 [DOI] [PubMed] [Google Scholar]
- 53. Sapp M, Kraus U, Volpers C, Snijders PJ, Walboomers JM, Streeck RE. 1994. Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J. Gen. Virol. 75(Pt 12):3375–3383 [DOI] [PubMed] [Google Scholar]
- 54. Volpers C, Unckell F, Schirmacher P, Streeck RE, Sapp M. 1995. Binding and internalization of human papillomavirus type 33 virus-like particles by eukaryotic cells. J. Virol. 69:3258–3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Florin L, Schäfer F, Sotlar K, Streeck RE, Sapp M. 2002. Reorganization of nuclear domain 10 induced by papillomavirus capsid protein l2. Virology 295:97–107 [DOI] [PubMed] [Google Scholar]
- 56. Spoden GA, Besold K, Krauter S, Plachter B, Hanik N, Kilbinger AFM, Lambert C, Florin L. 2012. Polyethylenimine is a strong inhibitor of human papillomavirus and cytomegalovirus infection. Antimicrob. Agents Chemother. 56:75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rommel O, Dillner J, Fligge C, Bergsdorf C, Wang X, Selinka H-C, Sapp M. 2005. Heparan sulfate proteoglycans interact exclusively with conformationally intact HPV L1 assemblies: basis for a virus-like particle ELISA. J. Med. Virol. 75:114–121 [DOI] [PubMed] [Google Scholar]
- 58. Berditchevski F, Gilbert E, Griffiths MR, Fitter S, Ashman L, Jenner SJ. 2001. Analysis of the CD151-alpha3beta1 integrin and CD151-tetraspanin interactions by mutagenesis. J. Biol. Chem. 276:41165–41174 [DOI] [PubMed] [Google Scholar]
- 59. Kazarov AR, Yang X, Stipp CS, Sehgal B, Hemler ME. 2002. An extracellular site on tetraspanin CD151 determines alpha 3 and alpha 6 integrin-dependent cellular morphology. J. Cell Biol. 158:1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sadej R, Romanska H, Kavanagh D, Baldwin G, Takahashi T, Kalia N, Berditchevski F. 2010. Tetraspanin CD151 regulates transforming growth factor beta signaling: implication in tumor metastasis. Cancer Res. 70:6059–6070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lammerding J, Kazarov AR, Huang H, Lee RT, Hemler ME. 2003. Tetraspanin CD151 regulates alpha6beta1 integrin adhesion strengthening. Proc. Natl. Acad. Sci. U. S. A. 100:7616–7621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Charrin S, Manié S, Oualid M, Billard M, Boucheix C, Rubinstein E. 2002. Differential stability of tetraspanin/tetraspanin interactions: role of palmitoylation. FEBS Lett. 516:139–144 [DOI] [PubMed] [Google Scholar]
- 63. Berditchevski F, Odintsova E, Sawada S, Gilbert E. 2002. Expression of the palmitoylation-deficient CD151 weakens the association of alpha 3 beta 1 integrin with the tetraspanin-enriched microdomains and affects integrin-dependent signaling. J. Biol. Chem. 277:36991–37000 [DOI] [PubMed] [Google Scholar]
- 64. Yang X, Kovalenko OV, Tang W, Claas C, Stipp CS, Hemler ME. 2004. Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J. Cell Biol. 167:1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sterk LMT, Geuijen CAW, van den Berg JG, Claessen N, Weening JJ, Sonnenberg A. 2002. Association of the tetraspanin CD151 with the laminin-binding integrins alpha3beta1, alpha6beta1, alpha6beta4 and alpha7beta1 in cells in culture and in vivo. J. Cell Sci. 115:1161–1173 [DOI] [PubMed] [Google Scholar]
- 66. Bailey RL, Herbert JM, Khan K, Heath VL, Bicknell R, Tomlinson MG. 2011. The emerging role of tetraspanin microdomains on endothelial cells. Biochem. Soc. Trans. 39:1667–1673 [DOI] [PubMed] [Google Scholar]
- 67. Abban CY, Meneses PI. 2010. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 403:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fothergill T, McMillan NAJ. 2006. Papillomavirus virus-like particles activate the PI3-kinase pathway via alpha-6 beta-4 integrin upon binding. Virology 352:319–328 [DOI] [PubMed] [Google Scholar]
- 69. Hong I-K, Jin Y-J, Byun H-J, Jeoung D-I, Kim Y-M, Lee H. 2006. Homophilic interactions of tetraspanin CD151 up-regulate motility and matrix metalloproteinase-9 expression of human melanoma cells through adhesion-dependent c-Jun activation signaling pathways. J. Biol. Chem. 281:24279–24292 [DOI] [PubMed] [Google Scholar]
- 70. Alexi X, Berditchevski F, Odintsova E. 2011. The effect of cell-ECM adhesion on signalling via the ErbB family of growth factor receptors. Biochem. Soc. Trans. 39:568–573 [DOI] [PubMed] [Google Scholar]
- 71. Yang XH, Richardson AL, Torres-Arzayus MI, Zhou P, Sharma C, Kazarov AR, Andzelm MM, Strominger JL, Brown M, Hemler ME. 2008. CD151 accelerates breast cancer by regulating alpha 6 integrin function, signaling, and molecular organization. Cancer Res. 68:3204–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sorkin A, Goh LK. 2009. Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 315:683–696 [DOI] [PubMed] [Google Scholar]
- 73. Bienkowska-Haba M, Sapp M. 2011. The cytoskeleton in papillomavirus infection. Viruses 3:260–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.