

Abstract
Reovirus infection is a well-characterized experimental system for the study of viral pathogenesis and antiviral immunity within the central nervous system (CNS). We have previously shown that c-Jun N-terminal kinase (JNK) and the Fas death receptor each play a role in neuronal apoptosis occurring in reovirus-infected brains. Death-associated protein 6 (Daxx) is a cellular protein that mechanistically links Fas signaling to JNK signaling in several models of apoptosis. In the present study, we demonstrate that Daxx is upregulated in reovirus-infected brain tissue through a type I interferon-mediated mechanism. Daxx upregulation is limited to brain regions that undergo reovirus-induced apoptosis and occurs in the cytoplasm and nucleus of neurons. Cytoplasmic Daxx is present in Fas-expressing cells during reovirus encephalitis, suggesting a role for Daxx in Fas-mediated apoptosis following reovirus infection. Further, in vitro expression of a dominant negative form of Daxx (DN-Daxx), which binds to Fas but which does not transmit downstream signaling, inhibits apoptosis of reovirus-infected cells. In contrast, in vitro depletion of Daxx results in increased expression of caspase 3 and apoptosis, suggesting that Daxx plays an antiapoptotic role in the nucleus. Overall, these data imply a regulatory role for Daxx in reovirus-induced apoptosis, depending on its location in the nucleus or cytoplasm.
INTRODUCTION
Viral encephalitis is an important worldwide cause of morbidity and mortality (1). Available antiviral therapies (e.g., acyclovir treatment of herpes simplex virus encephalitis) are suboptimal, and infection remains associated with significant death and disability (2, 3). More efficacious treatment strategies are desperately needed and should ideally be developed based upon an understanding of the pathological and immunologic events that occur in the virus-infected central nervous system (CNS).
Viral encephalitis can be modeled experimentally by inoculating murine brain tissue (in vivo or ex vivo) with serotype 3 reoviruses Abney (T3A) and Dearing (T3D) (4–9). These strains specifically infect neurons and induce neurologic injury via induction of caspase 3-dependent apoptosis (4–7, 9–12). Several members of the death receptor family, including TRAIL and Fas, are known to mediate reovirus-induced apoptosis in both in vitro and in vivo settings (7, 13–15). This occurs, at least in part, by activation of the initiator caspase, caspase 8 (via the adaptor protein FADD) (13). The Fas/FasL signaling pathway is particularly important for induction of apoptosis in reovirus-infected neurons (7, 15). Serotype 3 reovirus infection results in upregulation of both Fas and Fas ligand (FasL) within brain regions susceptible to reoviral injury (15). Furthermore, blocking Fas signaling with soluble Fas (Fc:Fas) results in inhibition of reovirus-induced apoptosis in primary neuronal cultures (7). These data suggest that reovirus-induced Fas signaling results in neuropathogenesis.
c-Jun N-terminal kinase (JNK) protein is a member of the mitogen-activated protein kinase (MAPK) family and, more specifically, the stress-activated protein kinase (SAPK) family, so named for having a distinct role in proapoptotic signaling in response to cellular stress. We have previously shown that JNK activation correlates strongly with reovirus-induced apoptosis (10, 16, 17). Notably, pharmacologic JNK inhibition decreases neuronal apoptosis and enhances survival of reovirus-infected mice (10).
Daxx was originally identified, through yeast two-hybrid screening and glutathione S-transferase (GST) pulldown assay, as a protein that binds to the intracellular death domain of the Fas receptor (18). Subsequent investigations have demonstrated that Daxx participates in FADD-independent apoptosis by serving as an adaptor between the Fas death receptor and MAPK signaling cascades, including those involving JNK (18–23). Upon binding the intracellular region of activated Fas, Daxx physically interacts with Ask1 (MAPK kinase kinase [MAP3K]) (19, 24). Daxx-bound Ask1 undergoes conformational change resulting in autophosphorylation and acquirement of kinase activity (19, 25). Ask1 then phosphorylates MKK4 (MAPKK), which in turn phosphorylates proapoptotic JNK (26, 27).
Exogenous Daxx overexpression can sensitize nonneuronal cells to multiple apoptotic stimuli (23, 28, 29), including Fas-mediated apoptosis (18, 30–32). Conversely, some studies have shown that RNA interference knockdown of Daxx (21, 33) or expression of dominant negative Daxx (DN-Daxx) (23) confers protection against some apoptotic stimuli. Expression of DN-Daxx prevents apoptotic death of Fas-stimulated motor neurons (34, 35) and vesicular stomatitis virus (VSV)-infected fibroblasts (36). In contrast, other studies of Daxx knockdown (37, 38) and knockout (39) have implicated that Daxx is an antiapoptotic protein, and consensus has not been reached regarding its specific role in apoptosis.
Our observations of Fas/FasL death receptor signaling and activation of JNK in the reovirus-infected brain (7, 10, 15–17) led us to hypothesize that Daxx plays an important role in reovirus-induced neuronal apoptosis. We now demonstrate that Daxx mRNA and protein are upregulated in reovirus-infected brain tissue. We show that Daxx upregulation is secondary to activation of the interferon (IFN) arm of the innate immune response. Newly synthesized Daxx protein localizes to reovirus-infected brain regions and, particularly, to the cytoplasm of infected neurons, where it colocalizes with Fas. Our demonstration that the stable expression of DN-Daxx inhibits reovirus-induced apoptosis in L929 cells suggests that cytoplasmic Daxx promotes apoptosis following reovirus infection. However, small interfering RNA (siRNA) knockdown of Daxx was proapoptotic and was associated with increased expression of caspase 3. Overall, our results suggest that Daxx may play a regulatory role in reovirus-induced apoptosis and has different functions when present in the cytoplasm or nucleus.
MATERIALS AND METHODS
Viral stocks.
Reoviruses serotype 3 strain Abney (T3A) and serotype 3 strain Dearing (T3D) are laboratory stocks derived via plaque purification and double passage in L929 cells. Viruses were further purified via high-speed cesium chloride density gradient centrifugation.
In vivo studies.
Swiss Webster outbred mice were obtained from Harlan Laboratories (Indianapolis, IN). Breeder pairs of type I interferon receptor null mice (IFNAR−/−) were generously provided by Ross Kedl (National Jewish Health, Denver, CO), and congenic C57BL/6J mice (B6wt) were purchased from the Jackson Laboratory (Bar Harbor, ME). All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) and performed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facility.
Two-day-old mice were intracranially (i.c.) inoculated with T3A (1,000 PFU) or T3D (1,000 PFU) diluted in a 10-μl volume of phosphate-buffered saline (PBS). Mock-infected mice were i.c. injected with PBS only at an equal volume.
Organotypic brain slice culture studies.
Brain slice cultures (BSCs) were prepared from 2- to 3-day-old mice as previously described (40). Briefly, four 400-μm coronal sections of the cerebrum (containing hippocampi and thalamus) were made from a single animal by using a vibrating blade microtome (VT1000S; Leica, Bannockburn, IL). Slices were maintained in a humidified incubator (36.5°C with 5% CO2), on a semiporous membrane insert (PICMORG50; Millipore, Billerica, MA) and in 35-mm tissue culture wells containing 1.2 ml of serum-containing medium (neurobasal supplemented with 10 mM HEPES, 1× B-27, 10% fetal bovine serum (FBS), 400 μM l-glutamine, 600 μM GlutaMAX, 60 U/ml penicillin, 60 μg/ml streptomycin, 6 U/ml nystatin). Immediately after plating, slices were infected by dropwise addition of 106 PFU T3A (diluted in 20 μl PBS) to each slice. Mock infections were performed in a similar manner with vehicle PBS alone. Medium was refreshed with 5% FBS-containing medium approximately 12 h after slicing, and subsequent medium changes were made with serum-free medium every 2 days thereafter.
Cell lines.
Neuro-2a cells were maintained in Eagle's minimum essential medium (MEM) supplemented with 10% FBS, 60 U/ml penicillin, and 60 μg/ml streptomycin. L929 fibroblasts were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS, 60 U/ml penicillin, and 60 μg/ml streptomycin. L929 cells stably expressing DN-Daxx or empty vector pcDNA3.1 (EV) were generously provided by Douglas Lyles (Wake Forest University, Winston-Salem, NC) (36). These lines were maintained in DMEM, with 4 μg/ml puromycin added for selection.
RNA purification from whole brains and BSCs.
Mock- and T3A-infected brains were harvested from Swiss Webster mice at appropriate time points and immediately placed into RNAlater (Ambion, Foster City, CA). Total RNA was purified by Dounce homogenization of the whole brain into 5 ml of Qiazol. This emulsion was transferred into a clean Falcon tube and set aside for at least 5 min before the addition of 1 ml chloroform. The mixture was shaken vigorously for 15 s and set aside for 5 min before centrifugation at 5,000 × g for 15 min at 4°C. The upper 2 ml of the aqueous phase was then transferred into a new tube containing 2 ml of 70% ethanol (prepared with diethyl pyrocarbonate-treated water). The solution was mixed and transferred onto an RNeasy Midi spin column (Qiagen, Germantown, MD), and RNA was purified according to the manufacturer's specifications. To prevent degradation, RNase inhibitor was added, and the sample was stored at −80°C.
For RNA purification from BSCs, four experimentally similar slices were washed three times in PBS and triturated in 600 μl RLT buffer (Qiagen, Germantown, MD) containing 1% β-mercaptoethanol. Samples were stored at −80°C until lysate was processed through a QIAshredder (Qiagen, Germantown, MD) and loaded onto an RNeasy minispin column (Qiagen, Germantown, MD) for RNA purification, according to the manufacturer's protocol. RNA samples were stored at −80°C until reverse transcription-quantitative PCR (RT-qPCR) analysis.
RT-qPCR quantification of gene transcripts.
RT-qPCR was utilized to quantify reovirus transcript in total RNA samples. Two primers designated RV-3 (5′-CAT ATG ACT ACC ACT TTC CCG-3′) and RV-4 (5′-GCT ATG TCA TAT TTC CAT CCG-3′) were synthesized (Invitrogen, San Diego, CA) to amplify a 298-bp segment of the reovirus L1 gene (41). Daxx primers (PPM03086E) were purchased from SABiosciences (Frederick, MD). Determination of L1 or Daxx gene expression, relative to a housekeeping gene, was achieved for each sample by concurrent amplification of mouse β-actin (primer PPM02945A; SABiosciences, Frederick, MD). Purified RNA template, primers, RT-qPCR master mix, and reverse transcriptase (iScript One-Step RT-qPCR kit with SYBR green; Bio-Rad, Hercules, CA) were mixed into a total volume of 20 μl. Forty cycles of PCR amplification were performed in triplicate on a Bio-Rad CFX96 thermocycler (Hercules, CA) as follows: cDNA synthesis at 50°C for 10 min, reverse transcriptase inactivation at 95°C for 5 min, denaturation at 95°C for 10 s, and annealing/extension at 60°C for 30 s. Melt curve analysis confirmed the absence of nonspecific products and primer dimers. Threshold cycle values were converted to relative expression values by using Bio-Rad CFX Manager Analysis software.
Western blot analysis.
Immediately upon harvesting, whole brains were triturated in lysis buffer (1% Triton-X, 10 mM triethanolamine-HCl, 150 mM NaCl, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1× Halt protease, and phosphatase inhibitor cocktail [Thermo Scientific, Rockford, IL]). Brain slices from a single well were rinsed three times with PBS and triturated in lysis buffer. Cell nuclear and cytoplasmic fractions were prepared using a nuclear/cytosol fractionation kit (Biovision, Mountain View, CA) according to the manufacturer's protocol. Lysates were cleared, electrophoresed through 10% polyacrylamide gels at a constant 65 V, and transferred to Hybond-C nitrocellulose membranes (Amersham Biosciences, Pittsburgh, PA). Immunoblotting was performed with anti-Daxx antibody (catalog number 4533; Cell Signaling Technology, Danvers, MA) and anti-β-actin antibody (Calbiochem, Sunnyvale, CA). Following washes and 1 h of incubation with appropriate horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), images were obtained on a FluorochemQ MultiImage III imaging workstation (Cell Biosciences, Santa Clara, CA), and densitometry was performed with the manufacturer's software (Alphaview v3.0).
Histological studies.
Freshly harvested brain tissue was immersed in 10% buffered formalin for no less than 12 h, embedded in paraffin, serially sectioned into 6-μm-thick sections, and mounted onto slides. Sections were desiccated at 50°C for 15 min and then rehydrated in PBS over 30 min. Tissue was subjected to antigen retrieval (antigen unmasking solution; Vector Laboratories, Burlingame, CA) and permeabilization/blocking (5% fetal bovine serum and 5% normal goat serum in 0.3% Triton–PBS) prior to incubation with primary antibodies: monoclonal reovirus σ3 antibody (4F2; 1:100), polyclonal reovirus antibody (1:500), mouse anti-neuronal nuclear antibody (NeuN; 1:100; Millipore, Billerica, MA), and/or rabbit anti-Daxx antibody (1:100; ab2017; Abcam, Cambridge, MA). Sections were then incubated with secondary antibodies: biotinylated goat anti-rabbit antibody (1:100; Invitrogen, Carlsbad CA) followed by detection by diaminobenzidine reaction (DAB; Trevigen, Gaithersburg, MD), or Alexa Fluor 488-conjugated IgG (1:200; Invitrogen, Carlsbad, CA) and Cy3-conjugated AffiniPure IgG (1:300; Jackson ImmunoResearch, West Grove, PA). Nuclei were Hoechst stained (10 μg/ml; Immunochemistry Technology, Bloomington, MN) for immunofluorescence imaging. Sections were mounted with VectorShield mounting medium (Vector Laboratories, Burlingame, CA), and slides images were obtained using a Marianas fluorescence imaging workstation (Intelligent Imaging Innovation Inc., Denver, CO) based on a Zeiss 300 M inverted microscope and CoolSnap HQ2 charge-coupled-device camera, all controlled by Slidebook 4.2 software (Intelligent Imaging Innovation). The images presented were obtained using Slidebook without changing the “gamma” settings and without cropping.
Primary hippocampal neurons.
Primary hippocampal neurons were prepared from newborn (0- to 1-day-old) Swiss Webster mice as previously described (42). Neurons were plated on BioCoat poly-d-lysine–laminin–coated coverslips (BD Biosciences, Franklin Lakes, NJ) and infected with T3A (multiplicity of infection [MOI] of 100) at 5 days in vitro. At 7 days postinfection (dpi), neurons were processed for immunocytochemistry.
Immunocytochemistry.
Primary hippocampal neurons were fixed in 4% paraformaldehyde, permeabilized with 0.1% saponin, and antigen blocked with 8% normal goat serum. Floating and trypsinized L929 cells were harvested using Shandon cytospin cartridges, fixed, permeabilized, and stained according to previously described methods (43). Neurons and L929 cells were incubated with monoclonal anti-σ3 reovirus antibody (4F2 laboratory stock; 1:100) and rabbit anti-Daxx antibody (10 μg/ml; ab2017; Abcam, Cambridge, MA) or rabbit anti-cleaved caspase 3 (1:400; catalog number 9661; Cell Signaling, Danvers, MA). Secondary antibodies of choice were Alexa Fluor 488-conjugated IgG (1:200; Invitrogen, Carlsbad, CA) and Cy3-conjugated AffiniPure IgG (1:300; Jackson ImmunoResearch, West Grove, PA). Hoechst staining, slide mounting, and imaging were performed as described for paraffin-embedded animal tissue.
Interferon treatment of cells and BSCs.
L929 fibroblasts and Neuro-2a cells were treated with 1,000 U/ml recombinant IFN-β (PBL InterferonSource, Piscataway, NJ), and at specified time points whole-cell lysate was prepared for Western blotting detection of Daxx protein. Brain slices prepared from 2- to 3-day-old Swiss Webster mice were treated with 50 μg/ml cycloheximide and 1,000 U/ml universal type I IFN (PBL InterferonSource, Piscataway, NJ) on the fifth day of culture. At 1 and 2 days posttreatment, BSCs were harvested for total RNA purification and RT-qPCR quantification of Daxx and β-actin transcripts.
MTT quantification assay.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), a yellow tetrazolium salt, is cleaved to purple formazan crystals by metabolically active cells. L929 fibroblasts were T3A infected (MOI, 20) or PBS (mock) treated. MTT (0.5 mg/ml, final concentration; Roche Applied Science, Boulder, CO) was added to cell medium at specified time points, and cells were detergent lysed 12 h later. Relative cell number was quantified on a microplate reader (Emax; Molecular Devices, Sunnyvale, CA) as a function of the optical density (OD) measured at a wavelength of 590 nm.
Caspase 3/7 FLICA assay.
Nontoxic, cell-permeable FLICA (fluorescent inhibitor of caspases reagent) binds irreversibly to activated caspases to make apoptotic cells readily detectable by flow cytometry. Empty vector and dominant negative Daxx L929 cells were plated at low confluence (∼20%). Then, the cells were either inoculated at serial time points with T3A (MOI, 20) or treated with 1 μM staurosporine. At the time of the assay, medium (containing detached cells) was pipetted into Falcon tubes. Attached cells were trypsinized and added directly into the corresponding Falcon tube. Cells were centrifuged at 300 × g for 5 min, and medium was aspirated. Cells were resuspended in medium containing FLICA reagent (Immunochemistry Technologies, Bloomington, MN), and incubation, with frequent gentle mixing, took place for 30 to 60 min at 37°C in the dark. Cells were then washed with the manufacturer's apoptosis wash buffer to allow unbound FLICA to diffuse out of cells. Cells were then incubated with the manufacturer's supplied propidium iodide (PI) for 5 min at 37°C in the dark. After a final spin down and rinse, a suspension of cells in apoptosis wash buffer was analyzed on a flow cytometer (cyan ADP analyzer; Beckman Coulter, Brea, CA) with 488-nm laser excitation and measurements of 6-carboxyfluorescein on FL1 channel and PI on FL2 channel. For each sample, no less than 15,000 cells were quantified.
Transfection of siRNA.
ON-TARGETplus mouse Daxx siRNA smart pool (siDaxx; L-044803-00-0005; Dharmacon, CO) and nontargeting control pool (siControl; D-001810-10-05; Dharmacon, CO) were transiently transfected according to the manufacturer's protocols. L929 (5 × 104 cells) were seeded in 24-well plates. siRNA at 30 nM was transfected using the DharmaFECT transfection reagent 1 24 h after cell plating. Total RNA was isolated 48 h after transfection, and total cell lysates were prepared at 72 h posttransfection to determine efficiency of Daxx knockdown. To examine the effect of siDaxx on T3A-induced apoptosis, cells were inoculated with T3A (MOI, 20) at 48 h posttransfection.
Statistical analysis.
All bar graphs are presented as means ± standard deviations (SD). Numbers of independent experiments are indicated by the N value. All statistical analyses were performed using Instat and Prism software programs (GraphPad Software Inc., San Diego, CA). Statistical comparisons between two groups (e.g., mock versus infected) were made using a two-tailed, unpaired t test with Welch correction. Significant differences (P < 0.05) are denoted in figures by asterisks.
RESULTS
Brain expression of Daxx mRNA and protein is upregulated during reovirus encephalitis.
Oligonucleotide microarray analysis led to the identification of Daxx as a cellular gene that was increased in expression within the brains of mice infected with T3D (44). To confirm this observation and determine the kinetics of reovirus-elicited Daxx upregulation, 2-day-old Swiss Webster mice were inoculated i.c. with the highly encephalitogenic reovirus strain T3A (1,000 PFU) or PBS only (mock) prior to total brain RNA isolation and RT-qPCR quantification of Daxx transcript. Infection with T3A resulted in significant (P ≤ 0.02) upregulation of Daxx mRNA in comparison to mock-infected animals at both 6 and 8 dpi (Fig. 1a). The magnitude of Daxx transcript upregulation at 8 days post-T3A infection (5.2-fold over mock; P = 0.003) was comparable to that previously seen 8 days post-T3D infection (4.1-fold over mock; P = 0.000059) by microarray analysis (44).
Fig 1.
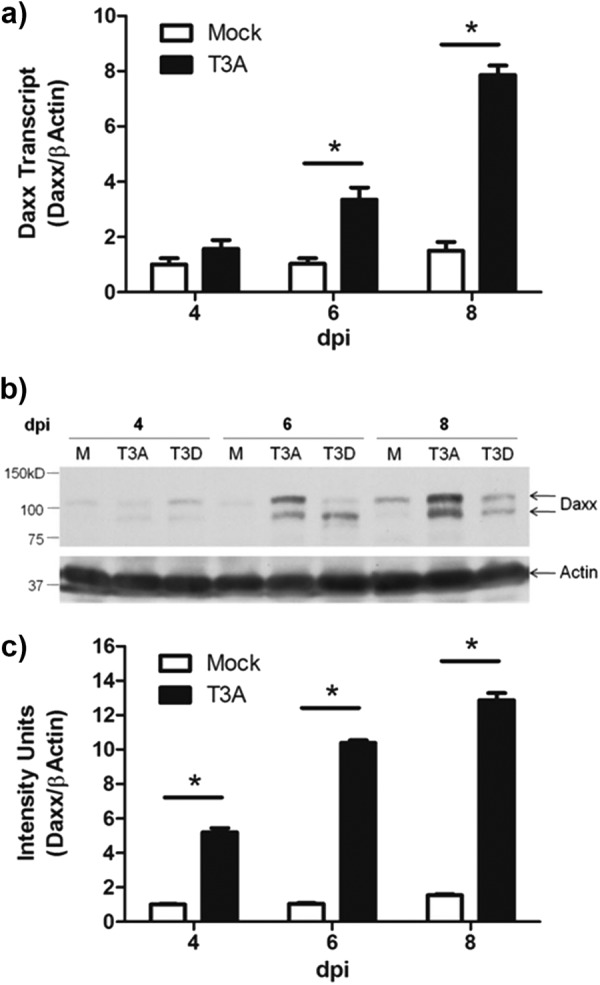
Daxx is upregulated in the reovirus-infected brain. Two- to 3-day-old Swiss Webster mice were inoculated i.c. with 1,000 PFU T3A, T3D, or an equal volume of PBS (mock). Brains were harvested for total RNA purification or protein lysate preparation at 4, 6, or 8 dpi. (a) RT-qPCR quantification of Daxx transcript relative to actin transcript for individual brains (n = 3) revealed progressive Daxx mRNA upregulation during T3A infection. (b) Representative Western blot demonstrating reovirus-induced Daxx protein upregulation. (c) Western blot densitometry results with T3A-infected brains (n = 3), showing progressive Daxx upregulation during reovirus infection.
To determine whether increased transcript is translated into more abundant Daxx protein, Western blot assays were performed on total brain lysates prepared from T3A-, T3D-, and mock-infected mice. Hollenbach et al. (45) reported three distinctly migrating Daxx isoforms with apparent molecular masses of 70 kDa, 97 kDa, and 120 kDa. Although the 120-kDa form was shown to result from posttranslational phosphorylation, the functional significance of such phosphorylation remains unclear. We found here that the 97-kDa and 120-kDa isoforms are expressed in the mouse brain and are increased as a result of T3A and T3D infection, with T3A inducing the most pronounced upregulation (Fig. 1b). Densitometry analysis of the grouped isoforms in T3A-infected and mock-infected brains demonstrated a significant (P ≤ 0.001; n = 3) virus-induced protein upregulation at 4, 6, and 8 dpi, with maximal 8.4-fold (P = 0.0005) upregulation at 8 dpi (Fig. 1c). Taken together, these findings indicate that infection with T3 reoviruses initiates the progressive transcription and translation of Daxx in the host brain.
Type I interferon signaling mediates Daxx expression during reovirus infection.
Upregulation of Daxx was previously reported in bone marrow cells following type I IFN exposure (46, 47). We previously showed that interferon signaling is initiated in the reovirus-infected brain and serves to limit local viral tropism and systemic viral spread (11, 40, 44). Thus, we hypothesized that IFN was driving Daxx upregulation during reovirus infection. In vitro IFN-β (1,000 U/ml) treatment elicited a rapid and sustained upregulation of Daxx in both L929 fibroblasts (Fig. 2a) and Neuro-2A cells (Fig. 2b). Our laboratory has recently developed an ex vivo culture system to study reovirus infection that allows for direct application of pharmacologic agents to live brain tissue (9). In the presence of cycloheximide, universal type I interferon (1,000 U/ml) or the PBS control was applied to BSCs prepared from wild-type Swiss Webster mice. RT-qPCR of Daxx transcript showed rapid IFN-induced upregulation, which was sustained for at least 2 days (Fig. 2c). IFN treatment did not induce overt cytotoxicity in slices or cell lines (data not shown). These data demonstrate that type I interferon signaling is capable of initiating Daxx transcription in the CNS.
Fig 2.
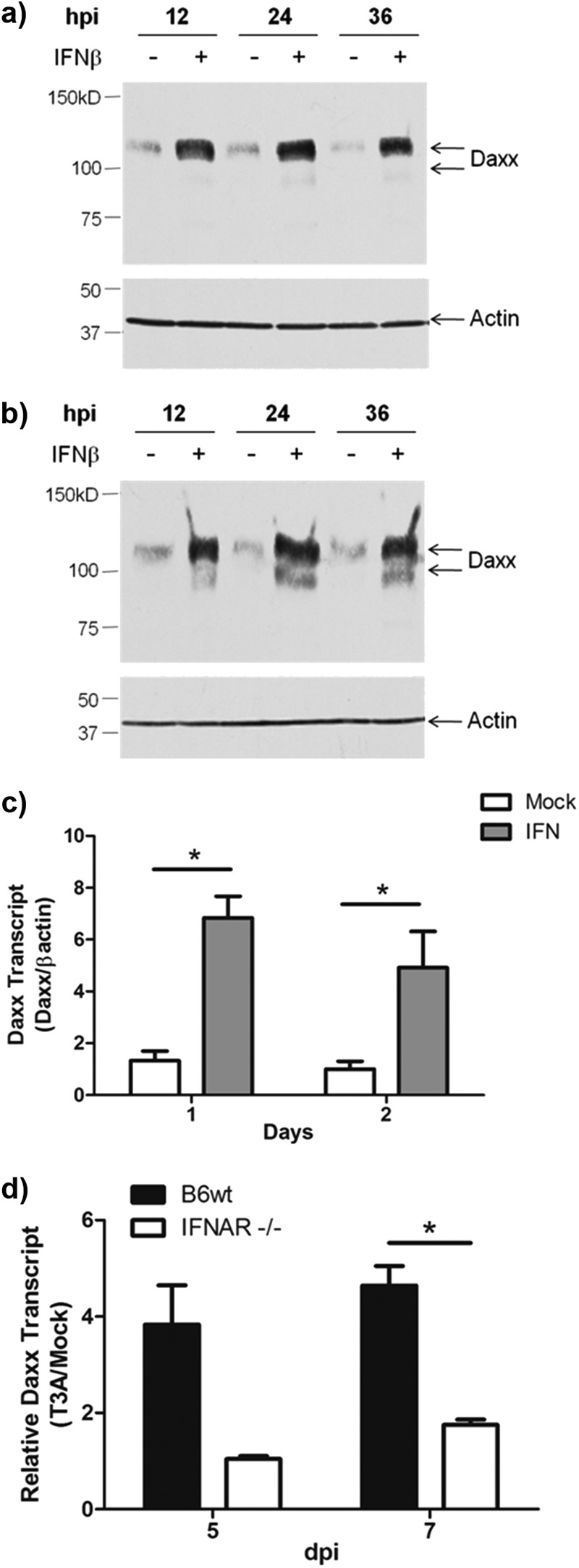
Reovirus-induced Daxx expression is type I interferon dependent. (a and b) L929 fibroblasts (a) or Neuro-2a cells (b) were treated with 1,000 U/ml IFN-β or PBS control, and whole-cell lysates were prepared at 12, 24, and 36 h for Western blotting detection of Daxx. (c) BSCs, prepared from 2- to 3-day-old Swiss Webster mice, were cultured for 5 days prior to exposure to cycloheximide and 1,000 U/ml universal type I IFN. Slices were harvested, and total RNA was purified at 1 and 2 days after treatment. RT-qPCR quantification of Daxx transcript relative to actin transcript for individual cultures (n = 5) revealed type I interferon-induced Daxx expression. (d) BSCs, prepared from 2- to 3-day-old type I interferon knockout mice (IFNAR−/−) and congenic wild-type mice (B6wt) were infected with T3A immediately upon plating. RT-qPCR quantification of Daxx transcript relative to actin transcript for individual cultures (n = 3) showed reovirus-induced Daxx mRNA upregulation infection, which was blocked in the absence of type I interferon signaling.
Next, we sought to assess the possibility that IFN drives Daxx expression specifically in the context of reovirus infection. To accomplish this, we prepared BSCs from mice that lacked the type I interferon receptor (IFNAR−/−) and congenic C57BL/6J wild-type (B6wt) mice. The respective BSCs were T3A or mock infected ex vivo, and total RNA was isolated at 5 and 7 dpi. RT-qPCR revealed that Daxx was upregulated in reovirus-infected B6wt slices (Fig. 2d), to a degree similar to that seen in vivo. Such upregulation was largely blocked in slices prepared from IFNAR−/− mice. These IFNAR−/− brain slices were previously shown to produce higher viral titers than B6wt slices (40); thus, minimal Daxx transcription in IFNAR−/− brain slices cannot be attributed to viral absence. These findings suggest that reovirus-infected brain tissue, in isolation from systemic and cell-mediated immune responses, produces IFN at levels sufficient to drive Daxx expression.
Daxx upregulation occurs in reovirus-infected brain regions and cells.
Having shown that Daxx is upregulated in the reovirus-infected brain as a result of type I interferon, we next sought to understand the potential functionality of Daxx in this context by characterizing its regional and cellular localization. Daxx upregulation occurred within brain regions known to be infected by reovirus (cingulate, hippocampus, and thalamus), but not within brain regions that are typically uninfected (e.g., lateral parietal cortex) (Fig. 3a). Fluorescent dual labeling of Daxx and reovirus antigen σ3 demonstrated that Daxx upregulation occurs predominantly within reovirus-infected cells (Fig. 3b and c). Cell counts revealed that 51% (760/1,491) of reovirus antigen-positive cells were also Daxx positive (Fig. 3d), and 89% (606/681) of Daxx-positive cells were also reovirus antigen positive (Fig. 3e). These data indicated that Daxx upregulation occurs within a subpopulation of reovirus-infected cells and in a limited number of noninfected cells.
Fig 3.

Daxx is upregulated in reovirus-infected brain regions and cells. Two- to 3-day-old Swiss Webster mice were inoculated i.c. with 1,000 PFU T3A, T3D, or an equal volume of PBS (mock). Brains were harvested at 8 dpi for histological processing. (a) Immunohistochemical staining of Daxx protein specifically in brain regions that were characteristically infected by reovirus (cingulate, hippocampus, and thalamus). The parietal cortex served as an internal negative control, as it is not infected by reovirus to any significant degree. (b) Following T3A infection, fluorescently labeled Daxx (red) was found largely within cells that were positively labeled for reovirus antigen σ3 (green). Magnification, ×630. Bar, 10 μm. (c) An isolated cell in the thalamus (inset) is enlarged by an additional 10× for demonstration of Daxx and reovirus in the same cell. (d) The percentage of cells that expressed high levels of Daxx within populations of either reovirus antigen-negative (-) or positive (+) cells. (e) The percentage of cells that expressed reovirus antigen within populations of cells expressing either low (-) or high (+) levels of Daxx. For both panels d and e, cells were quantified from 2 different fields obtained from the hippocampi of 2 individual reovirus-infected mice.
Daxx protein is upregulated in the cytoplasm of reovirus-infected neurons in vivo and reovirus-infected L929 cells in vitro.
Given that T3 reoviruses are neuronotropic and Daxx upregulation was seen predominantly in reovirus-infected cells, it was likely that Daxx is specifically increased in neurons. To confirm this, we examined Daxx expression in reovirus-infected primary hippocampal neurons prepared from Swiss Webster mice. Dual staining of these neurons for reovirus σ3 antigen and Daxx demonstrated robust Daxx upregulation specifically within reovirus-infected neurons (Fig. 4a). Furthermore, the staining appeared to localize to the neuronal cytoplasm. In comparison, neighboring neurons had detectable nuclear expression of Daxx but no evidence of reovirus antigen or cytoplasmic Daxx upregulation.
Fig 4.

Upregulated Daxx protein is localized to the cytoplasm of reovirus-infected neurons and L929 cells. (a) Primary hippocampal neurons were prepared from Swiss Webster mouse pups at the age of 0 to 1 day. Neurons were infected with T3A at 5 days in vitro and processed for immunocytochemistry at 7 dpi. Labeled Daxx (red) was found in neurons that were reovirus antigen σ3 positive (Reo; green). Magnification, ×400. Bar, 10 μm. (b) Two- to 3-day-old Swiss Webster mice were inoculated i.c. with 1,000 PFU T3A or an equal volume of PBS (mock). Brains were harvested at 4 to 8 dpi for immunohistochemical labeling of Daxx (red) and NeuN (green), a neuron-specific marker. Magnification, ×1,000. Bar, 10 μm. (c) An isolated cell in the thalamus (inset) is enlarged an additional 10× for demonstration of Daxx in NeuN-positive neurons. (d) Cytoplasmic and nuclear fractions prepared from brains extracted at 8 dpi from Swiss Webster mice that had been intracerebrally inoculated with 100 PFU T3A or an equal volume of PBS (mock). The cytoplasmic and nuclear extracts were analyzed by Western blotting for detection of Daxx and lamin B nuclear marker. (e) L929 fibroblasts were treated with T3A reovirus at an MOI 20 or PBS (mock), and cells were harvested at 2 dpi for preparation of cytoplasmic (Cyto) and nuclear (Nuc) fractions. Western blotting was performed for detection of Daxx and the lamin B nuclear marker.
It has been suggested that the presence of Daxx in the cytoplasm is critically important to its proapoptotic functionality (18, 19, 24, 25, 48–50). To better understand where Daxx is localized on a subcellular level during reovirus infection in vivo, Swiss Webster mice were injected i.c. with 1,000 PFU T3A or PBS (mock), and brains were harvested at 8 dpi for dual labeling of Daxx and neuronal nuclei with NeuN antibody. High-magnification microscopy was utilized to demonstrate discrete subnuclear structures containing Daxx in noninfected neurons (Fig. 4b). These structures were previously identified as promyelocytic leukemia-nuclear bodies (PML-NBs) (51, 52). Again, Daxx staining was not observed in the cytoplasm of noninfected neurons. In contrast, in reovirus-infected brains, Daxx was markedly upregulated within the neuronal cytoplasm. The inset of Fig. 4b is enlarged to show a representative neuron with evidence of Daxx upregulation in the cytoplasmic compartment (Fig. 4c).
To confirm that reovirus upregulation of Daxx occurs largely in the cytoplasm, brains from infected mice were subjected to subcellular fractionation and Western blotting for Daxx protein (Fig. 4d). Daxx was indeed upregulated in the cytoplasmic (Lamin B negative) fractions at both 6 and 8 dpi, compared to mock-infected controls. Similarly, upregulation of Daxx was observed in the cytoplasmic fractions of infected L929 cells (Fig. 4e). Nuclear Daxx was only modestly upregulated in infected brains (Fig. 4d) and L929 cells (Fig. 4e). Overall, these studies clearly demonstrated that reoviral infection results in accumulation of Daxx protein within the neuronal/cellular cytoplasm.
Upregulated Daxx colocalizes with Fas in reovirus-infected brains.
Finding Daxx expression in cytoplasm raises the distinct possibility that it is interacting with membrane-expressed Fas as previously described (18, 19). Upon dual staining for Fas and Daxx in reovirus-infected brains, we showed that Daxx is present in the cytoplasm of Fas-expressing cells (Fig. 5a and b). Furthermore, z-stack images revealed subcellular colocalization of Fas and Daxx (Fig. 5c), which was consistent with the previously reported binding interaction between the two proteins (18). Since we previously showed that Fas is expressed in infected cells following reovirus infection of the brain (15), the present findings suggest that upregulated Fas and Daxx colocalize in the cytoplasm of reovirus-infected cells to promote virus-induced apoptosis.
Fig 5.

Daxx colocalizes with Fas in reovirus-infected brains. Two-to 3-day-old Swiss Webster mice were intracerebrally inoculated with 1,000 PFU T3A. Brains were harvested at 8 dpi for immunohistochemical labeling of Daxx (red) and Fas (green). (a) The images show Daxx in the cytoplasm of cells expressing Fas (white arrows). Magnification, ×630. (b) The percentage of cells that expressed Fas within populations of cells expressing either low (-) or high (+) levels of cytoplasmic Daxx. Percentages were derived from 2 different fields obtained from the hippocampi of 3 individual reovirus-infected animals. (c) Subcellular colocalization of Daxx and Fas, demonstrated by digitally deconvoluted z-stacked imaging. Magnification, ×1,000. Bar, 10 μm.
Stable expression of DN-Daxx in L929 fibroblasts is associated with limited cytopathic effect following reovirus infection.
Having demonstrated Fas-Daxx colocalization, we next wished to perturb Daxx function and study the potential impact on reovirus-induced cell death. To accomplish this, we utilized L929 fibroblasts that stably express DN-Daxx (36). DN-Daxx is made up of only the C-terminal 114 amino acids of Daxx, corresponding to the Fas-binding site (Fig. 6A). Exogenous expression of this construct blocks interactions between wild-type Daxx and Fas, thereby inhibiting signaling through this pathway. L929 fibroblasts were used for these studies because these cells (i) are readily amenable to stable construct expression following standard transfection methods and (ii) undergo well-characterized reovirus-induced death receptor-mediated apoptosis that involves JNK signaling pathways (16, 17). We found that the cells stably expressing DN-Daxx, compared to those expressing EV, were remarkably resistant to cytopathic effect (CPE) upon reovirus challenge, as shown by qualitative photomicrographs (Fig. 6b), MTT assays (Fig. 6c), and propidium iodide staining followed by flow cytometry (Fig. 7c). These assays demonstrated that both cell types proliferate at a similar rate when mock infected. EV cells demonstrated characteristic cytopathic effect (CPE) in the days following reovirus infection, ultimately resulting in death of all cells. A similar time course of CPE was noted in reovirus-infected parental L929 cells (data not shown). Conversely, there was a significant delay/inhibition of CPE in cells expressing DN-Daxx.
Fig 6.

In vitro DN-Daxx expression reduces reovirus-induced cell death. (a) Schematic of the DN-Daxx construct stably expressed in L929 fibroblasts. DN-Daxx is composed solely of the C-terminal end of Daxx, corresponding to the Fas-binding region. Functionally, this mutant binds Fas but fails to recruit downstream effectors that initiate apoptosis. (b) L929 fibroblasts were T3A infected at an MOI of 20 or PBS (mock) treated. Photomicrographs were taken at 1, 2, and 3 dpi to demonstrate resistance to reovirus-induced cell death in DN-Daxx-expressing cells. Magnification, ×100. (c) DN-Daxx and EV L929 fibroblasts were T3A infected at an MOI 20. MTT was added to cell medium (n = 3) at specified time points, and the relative cell number was quantified by using a colorimetric plate reader.
Fig 7.

In vitro DN-Daxx expression reduces reovirus-induced apoptosis. L929 fibroblasts, stably expressing either EV or DN-Daxx, were T3A infected at an MOI of 20. (a) Cells were harvested, and total RNA was isolated at different time points as indicated. cDNA was synthesized from each experimental sample, and T3A cDNA standards (with known virus titers, as determined by a traditional plaque assay) were subjected to RT-qPCR amplification of the reovirus L1 gene. PFUe/ml indicates the equivalent number of PFU/ml as determined by placement of each data point on the standard curve. (b) Representative scatter plots of flow cytometry-based quantification of caspase 3/7 FLICA and PI staining at specified time points postinoculation with T3A. (c) Graphic representation of flow cytometry data (n = 4), showing the percentage of cells that positively stained with PI in T3A samples. (d) Graphic representation of flow cytometry data (n = 4), showing the percentage of cells that were positively stained with caspase 3/7 FLICA in T3A samples. (e) DN-Daxx or EV L929 fibroblasts were treated with 1 μM staurosporine. At specified time points, total cells were analyzed with flow cytometry for quantification of caspase 3/7 FLICA and PI staining. The data are representative of three experiments with similar results.
Reovirus-induced apoptosis is reduced in L929 fibroblasts stably expressing DN-Daxx.
To determine whether CPE resistance in DN-Daxx-overexpressing cells is secondary to decreased reovirus growth and/or apoptosis inhibition, we infected DN-Daxx- and EV-expressing cells with T3A (MOI, 20) and assessed viral replication and apoptosis along a time course of 3 days. Viral growth was similar between the two cell types, as demonstrated by RT-qPCR (Fig. 7a). Caspase 3/7 FLICA flow cytometry was used to quantify virus-induced apoptosis in DN-Daxx- and EV-expressing cells. As shown in Fig. 7b, reovirus infection induced significant apoptotic death in EV L929 cells at 1, 2, and 3 dpi. In comparison, DN-Daxx-expressing cells displayed phenotypic resistance to reovirus-induced apoptosis (Fig. 7b and d). There was no difference in apoptosis in DN-Daxx- and EV-expressing cells treated with staurosporine, indicating that DN-Daxx does not inhibit apoptosis mediated primarily through mitochondrial apoptotic signaling and the activation of caspase 9 (Fig. 8).
Fig 8.

In vitro DN-Daxx expression does not affect staurosporine-induced apoptosis. L929 fibroblasts, stably expressing either EV or DN-Daxx, were treated with staurosporine. At specified time points, total cells were analyzed with flow cytometry for quantification of caspase 3/7 FLICA and PI staining. The data are representative of three experiments with similar results.
Depletion of Daxx increases apoptosis in reovirus-infected cells.
We next used siDaxx to examine the effects of depleting Daxx on reovirus-induced apoptosis. siDaxx was used to successfully silence both Daxx mRNA and protein expression in L929 cells (Fig. 9a and b). The effect of siDaxx on T3A-induced caspase 3/7 activation was then investigated. T3A infection (MOI, 20) of L929 cells pretreated with control siRNA resulted in the activation of caspase 3 in around 40% of cells (Fig. 9c). Pretreatment with siDaxx resulted in an increased percentage of reovirus-infected cells that were positive for activated caspase 3/7 (Fig. 9c), which may have been a consequence of increased caspase 3 mRNA expression in siDaxx-treated cells (Fig. 9d) and was consistent with earlier reports (37, 53).
Fig 9.

In vitro Daxx knockdown potentiates reovirus-induced apoptosis. (a) L929 fibroblasts were transfected with 30 nM siControl (siCon) or siDaxx. Total RNA was isolated 48 h after transfection. The uninfected L929 (no-si) was used as an additional control. RT-qPCR quantification of Daxx transcripts relative to actin transcripts for individual cultures (n = 3) revealed siDaxx knockdown of Daxx mRNA. (b) Total cell lysates were prepared 72 h after siRNA transfection and analyzed by SDS-PAGE and Western blotting for quantitation of Daxx and tubulin. The data are representative of three experiments with similar results. (c) Two days after siRNA transfection, cells were infected with T3A reovirus at an MOI of 20 or 200. At 3 dpi, floating cells and trypsin-treated cells were harvested and stained with caspase 3/7 FLICA and PI. The quantifications of FLICA- and PI-positive cells were achieved by flow cytometry. The data are representative of three independent experiments with similar results. (d) Total RNA was isolated 48 h after siRNA transfection. RT-qPCR quantification of caspase 3 transcript relative to actin transcript for individual cultures (n = 3) revealed that siDaxx upregulated caspase 3 expression.
DISCUSSION
Reovirus encephalitis is characterized by severe neurologic injury and caspase 3-dependent neuronal apoptosis, which occur as a result of viral replication and/or the host-mediated response to viral presence (5). Daxx has been implicated in several neurologic disease processes in which apoptotic neuronal death likely plays a key pathogenic role, including amyotrophic lateral sclerosis (ALS) (34, 54), Alzheimer's disease (55, 56), and stroke/ischemia (49, 50). However, the potential role of Daxx in CNS infection has not, to our knowledge, undergone prior investigation.
It was previously reported that Daxx mRNA is upregulated in L929 fibroblasts following infection by multiple reovirus strains, with apoptotic strains inducing the greatest amount of Daxx mRNA (57). We have now demonstrated, for the first time, virus-mediated upregulation of Daxx in vivo (Fig. 1). Upregulation of Daxx is observed in reovirus-infected brains as early as 4 dpi; that is, before apoptosis can be detected by immunoblotting (data not shown) or fluorogenic assay (58) of cleaved/activated caspase 3. Daxx upregulation becomes more pronounced as disease progresses and reaches maximal levels immediately prior to animal death at 9 to 11 dpi. In addition, the degree of Daxx upregulation is greater in T3A-infected than T3D-infected brains at all time points, an observation that parallels the known differences in strain neuropathogenicity (T3A > T3D). We postulated that the correlative relationship between Daxx expression levels and the development of reovirus encephalitis might be indicative of the protein's role in reovirus-induced CNS pathogenesis. Daxx upregulation, similar to that observed in the reovirus-infected brain, is known to contribute to apoptotic injury in other systems, including in vitro oxidative stress (59) and in vivo ischemic kidney injury (60).
We previously established that i.c. inoculation of Swiss Webster mice results in type I interferon signaling, which is measurable in reovirus-infected brains as early as 4 dpi and is sustained through 8 dpi (19). We now have shown that Daxx upregulation occurs with similar kinetics in the reovirus-infected brain. Furthermore, we demonstrated that Daxx expression can be directly induced by treatment with type I interferon in multiple systems, including cell lines and brain slices (Fig. 2). To confirm that IFN signaling is the mechanism by which Daxx is upregulated during reovirus infection, we prepared brain slices from IFNAR−/− mice and congenic B6wt mice. Utilization of this ex vivo culture system was necessary, because IFNAR−/− mice succumb to systemic reovirus infection prior to Daxx upregulation, precluding in vivo investigation of neuronal Daxx expression in this mouse strain. We found that reovirus infection induced Daxx upregulation in wild-type brain slices (Fig. 2). However, Daxx was not upregulated in reovirus-infected brain slices prepared from IFNAR−/− donor mice, despite the fact that viral growth is enhanced in this context (40). This indicated that the type I interferon arm of innate immunity is the major mechanism underlying reovirus-induced Daxx upregulation. Furthermore, our findings strongly suggest that Daxx is an IFN-stimulated gene (ISG). In support of this, we identified several putative interferon stimulated response elements (ISRE) within the promoter regions of both mouse and human Daxx (by use of Genomatix software) (data not shown).
The fact that Daxx was upregulated in wild-type brain slices suggests that cell-mediated adaptive immunity is not required for Daxx upregulation in our model system. Daxx was also upregulated in infected primary hippocampal neurons (Fig. 4a), which indicates that reovirus-infected neurons do not require glial cell (e.g., astrocyte and microglia) activity for IFN-mediated Daxx upregulation. Instead, our data suggest that IFN released from a given infected neuron binds to that very neuron to mediate its expression of Daxx. In support of this autocrine signaling hypothesis, neurons have a known capacity to both produce and respond to IFN, as discovered in other models of viral encephalitis (61). Furthermore, it has been demonstrated that the profile of ISG expression in infected cells can differ significantly from that of uninfected, bystander cells residing in the same tissue (62).
To investigate the functional significance of Daxx upregulation in our system, we characterized the expression of the protein at regional, cellular, and subcellular levels. As demonstrated by dual immunostaining for the reovirus major outer capsid protein (σ3) and Daxx, upregulation of Daxx was limited to a subpopulation of reovirus-infected cells existing within brain regions known to be subject to reovirus-induced apoptotic injury (Fig. 3). Quantification of cells expressing either Daxx, reovirus σ3, both proteins clearly demonstrated that the vast majority of cells with increased Daxx expression are infected by reovirus.
Given that Daxx upregulation mainly occurred in reovirus-infected cells and that serotype 3 reovirus strains are known to specifically infect neurons in vivo, it was not surprising that reovirus-induced Daxx upregulation was neuron specific (Fig. 4). Labeling neuronal nuclei with NeuN antibody serendipitously led to the observation that reovirus-induced Daxx is largely localized to the cytoplasm. Such cytoplasmic staining of Daxx is found neither in neurons within the mock-infected brain nor in neurons within uninfected brain regions (i.e., the lateral parietal cortex) of infected animals. Other investigators have observed Daxx in the cytoplasm following in vitro Fas stimulation (24) and Ask1 overexpression (25). Still others have found rapid translocation of nuclear Daxx into the cytoplasm in association with apoptosis induced by oxidative stress (27, 63, 64), glucose deprivation (65), ischemia/reperfusion (48–50), β-amyloid peptide exposure (63), and 1-methyl-4-phenylpyridium treatment (64, 66). Daxx translocation to the cytoplasm was recently utilized as a surrogate marker of Ask1 activity (67), although the exact role of nuclear-to-cytoplasm translocation of Daxx remains a controversial issue (68). It has even been suggested that Daxx is solely expressed within the nucleus (32, 39, 68–70), although this does not seem to be the case in neurons existing within the reovirus-infected mouse brain. In our model, we observed punctate Daxx staining within neuronal nuclei, consistent with Daxx association with PML-NBs. We did not observe significant changes in nuclear Daxx expression, localization, or the staining pattern, most likely indicating that Daxx is synthesized and retained in the neuronal cytoplasm during reovirus encephalitis.
Once in the cytoplasm, Daxx proapoptotic activity is dependent upon binding molecules expressed in the cytoplasm (i.e., Ask1) and at the cell membrane (i.e., Fas) (18). Our data (Fig. 5) indicated that reovirus-induced Daxx may bind to upregulated Fas (15) in reovirus-infected neurons to propagate apoptotic signals through the Fas-Daxx-Ask1-MKK4-JNK pathway.
Experimentation in our reovirus system revealed a pronounced resistance to virus-induced cell death in cells stably expressing DN-Daxx (Fig. 6). We found that this phenotype was due to restricted apoptosis rather than altered viral replication (Fig. 7). As reported previously, DN-Daxx inhibits apoptosis by specifically blocking signaling through the Fas death receptor (18, 34, 36). Thus, mitochondrial pathways of reovirus-induced apoptosis and alternative death receptor pathways (5, 71) likely remain operative in infected DN-Daxx cells, as demonstrated by our use of staurosporine (Fig. 8), providing a possible explanation for incomplete blockage of apoptosis in this context.
Similar to exogenously expressed DN-Daxx, endogenously expressed FLICE-inhibitory protein (c-FLIP) binds to the Fas-binding domain and renders cells resistant to Fas-induced cell death (72). We have previously reported that c-FLIP is downregulated during reovirus infection as a prerequisite to reovirus-induced apoptosis (73, 74), raising the possibility that in the relative absence of c-FLIP, upregulated Daxx becomes increasingly capable of binding upregulated Fas to ultimately promote JNK-mediated apoptosis. Given that JNK mediates Fas upregulation in the reovirus-infected brain (15), we now suggest a mechanism underlying reovirus-induced apoptosis involving a positive feedback loop that connects Fas-Daxx-JNK signaling to Fas upregulation and potentiated signaling through the same death receptor-mediated pathway.
Overall, our data suggest that Daxx contributes to CNS disease by bridging Fas signaling to JNK activation in a type I IFN-inducible manner. While it has been well established that IFN is dispensable for reovirus-induced apoptosis (40, 75), we propose a model whereby IFN-mediated cytoplasmic Daxx expression lowers the apoptotic threshold by promoting death receptor signaling. However, our studies also demonstrated that Daxx has additional roles in the context of apoptosis, including inhibition of the expression of caspase 3 (Fig. 9). Taken together, our findings may reflect a regulatory role for Daxx in apoptosis whereby Daxx is proapoptotic when expressed in the cytoplasm and antiapoptotic when expressed in the nucleus via its role in transcriptional modification. Consequently, inhibiting the proapoptotic, Fas-associated function of Daxx (using the DN inhibitor described) inhibits reovirus-induced apoptosis, whereas inhibiting both pro- and antiapoptotic functions (using siRNA) does not prevent reovirus-induced apoptosis. Future studies will be designed to further elucidate how Daxx mechanistically translates viral sensing to the apoptotic response. Continued investigations into the molecular linkages between viral infection, innate immunity, and apoptotic signaling will potentially reveal novel therapeutic targets for the treatment of viral encephalitis.
ACKNOWLEDGMENTS
This work was supported by RO1NS076512 (K.L.T) and VA Merit Grant 1/01BX000963 (K.L.T). K.R.D. was supported by an institutional Medical Scientist Training Program training grant (T32GM008497) and a National Research Service Award for Individual Predoctoral MD/PhD Fellows (F30NS071630).
We extend our gratitude to the University of Colorado Cancer Center Histology, Flow Cytometry, and Advanced Light Microscopy Cores for their assistance with data collection. We also thank Dave Beckham and Lai Kuan Goh for editing the manuscript.
Footnotes
Published ahead of print 9 January 2013
REFERENCES
- 1. Tan K, Patel S, Gandhi N, Chow F, Rumbaugh J, Nath A. 2008. Burden of neuroinfectious diseases on the neurology service in a tertiary care center. Neurology 71:1160–1166 [DOI] [PubMed] [Google Scholar]
- 2. Tyler K. L. 2004. Herpes simplex virus infections of the central nervous system: encephalitis and meningitis, including Mollaret's. Herpes 11:57A–64A [PubMed] [Google Scholar]
- 3. Raschilas F, Wolff M, Delatour F, Chaffaut C, De Broucker T, Chevret S, Lebon P, Canton P, Rozenberg F. 2002. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clin. Infect. Dis. 35:254–260 [DOI] [PubMed] [Google Scholar]
- 4. Richardson-Burns SM, Tyler KL. 2004. Regional differences in viral growth and central nervous system injury correlate with apoptosis. J. Virol. 78:5466–5475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clarke P, Debiasi RL, Goody R, Hoyt CC, Richardson-Burns SM, Tyler KL. 2005. Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation. Viral Immunol. 18:89–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oberhaus SM, Smith RL, Clayton GH, Dermody TS, Tyler KL. 1997. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J. Virol. 71:2100–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richardson-Burns SM, Kominsky DJ, Tyler KL. 2002. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J. Neurovirol. 8:365–380 [DOI] [PubMed] [Google Scholar]
- 8. Richardson-Burns SM, Tyler KL. 2005. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp. Neurol. 192:331–339 [DOI] [PubMed] [Google Scholar]
- 9. Dionne KR, Leser JS, Lorenzen KA, Beckham JD, Tyler KL. 2011. A brain slice culture model of viral encephalitis reveals an innate CNS cytokine response profile and the therapeutic potential of caspase inhibition. Exp. Neurol. 228:222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beckham JD, Goody RJ, Clarke P, Bonny C, Tyler KL. 2007. Novel strategy for treatment of viral central nervous system infection by using a cell-permeating inhibitor of c-Jun N-terminal kinase. J. Virol. 81:6984–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goody RJ, Beckham JD, Rubtsova K, Tyler KL. 2007. JAK-STAT signaling pathways are activated in the brain following reovirus infection. J. Neurovirol. 13:373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beckham JD, Tuttle KD, Tyler KL. 2010. Caspase-3 activation is required for reovirus-induced encephalitis in vivo. J. Neurovirol. 16:306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clarke P, Meintzer SM, Gibson S, Widmann C, Garrington TP, Johnson GL, Tyler KL. 2000. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 74:8135–8139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clarke P, Meintzer SM, Spalding AC, Johnson GL, Tyler KL. 2001. Caspase 8-dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene 20:6910–6919 [DOI] [PubMed] [Google Scholar]
- 15. Clarke P, Beckham JD, Leser JS, Hoyt CC, Tyler KL. 2009. Fas-mediated apoptotic signaling in the mouse brain following reovirus infection. J. Virol. 83:6161–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clarke P, Meintzer SM, Widmann C, Johnson GL, Tyler KL. 2001. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J. Virol. 75:11275–11283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clarke P, Meintzer SM, Wang Y, Moffitt LA, Richardson-Burns SM, Johnson GL, Tyler KL. 2004. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 78:13132–13138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang XL, KhosraviFar R, Chang HY, Baltimore D. 1997. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89:1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang HY, Nishitoh H, Yang XL, Ichijo H, Baltimore D. 1998. Activation of apoptosis signal regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281:1860–1863 [DOI] [PubMed] [Google Scholar]
- 20. Chang HY, Yang XL, Baltimore D. 1999. Dissecting Fas signaling with an altered-specificity death-domain mutant: requirement of FADD binding for apoptosis but not Jun N-terminal kinase activation. Proc. Natl. Acad. Sci. U. S. A. 96:1252–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khelifi AF, D'Alcontres MS, Salomoni P. 2005. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 12:724–733 [DOI] [PubMed] [Google Scholar]
- 22. Song JJ, Lee YJ. 2004. Daxx deletion mutant (amino acids 501–625)-induced apoptosis occurs through the JNK/p38-Bax-dependent mitochondrial pathway. J. Cell. Biochem. 92:1257–1270 [DOI] [PubMed] [Google Scholar]
- 23. Wu S, Loke HN, Rehemtulla A. 2002. Ultraviolet radiation-induced apoptosis is mediated by Daxx. Neoplasia 4:486–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Charette SJ, Lavoie JN, Lambert H, Landry J. 2000. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol. Cell. Biol. 20:7602–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ko YG, Kang YS, Park H, Seol W, Kim J, Kim T, Park HS, Choi EJ, Kim S. 2001. Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (Daxx) in the cytoplasm. J. Biol. Chem. 276:39103–39106 [DOI] [PubMed] [Google Scholar]
- 26. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. 1997. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275:90–94 [DOI] [PubMed] [Google Scholar]
- 27. Song JJ, Lee YJ. 2003. Role of the ASK1-SEK1-JNK1-HIPK1 signal in Daxx trafficking and ASK1 oligomerization. J. Biol. Chem. 278:47245–47252 [DOI] [PubMed] [Google Scholar]
- 28. Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. 2001. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 3:708–714 [DOI] [PubMed] [Google Scholar]
- 29. Gostissa M, Morelli M, Mantovani F, Guida E, Piazza S, Collavin L, Brancolini C, Schneider C, Del Sal G. 2004. The transcriptional repressor hDaxx potentiates p53-dependent apoptosis. J. Biol. Chem. 279:48013–48023 [DOI] [PubMed] [Google Scholar]
- 30. Boehrer S, Nowak D, Hochmuth S, Kim SZ, Trepohl B, Afkir A, Hoelzer D, Mitrou PS, Weidmann E, Chow KU. 2005. Daxx overexpression in T-lymphoblastic Jurkat cells enhances caspase-dependent death receptor- and drug-induced apoptosis in distinct ways. Cell. Signal. 17:581–595 [DOI] [PubMed] [Google Scholar]
- 31. Muromoto R, Yamamoto T, Yumioka T, Sekine Y, Sugiyama K, Shimoda K, Oritani K, Matsuda T. 2003. Daxx enhances Fas-mediated apoptosis in a murine pro-B cell line, BAF3. FEBS Lett. 540:223–228 [DOI] [PubMed] [Google Scholar]
- 32. Torii S, Egan DA, Evans RA, Reed JC. 1999. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). Blood 94:483A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawai T, Akira S, Reed JC. 2003. ZIP kinase triggers apoptosis from nuclear PML oncogenic domains. Mol. Cell. Biol. 23:6174–6186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raoul C, Barthelemy C, Couzinet A, Hancock D, Pettmann B, Hueber AO. 2005. Expression of a dominant negative form of Daxx in vivo rescues motoneurons from Fas (CD95)-induced cell death. J. Neurobiol. 62:178–188 [DOI] [PubMed] [Google Scholar]
- 35. Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. 2002. Motoneuron death triggered by a specific pathway downstream of Fas potentiation by ALS-linked SOD1 mutations. Neuron 35:1067–1083 [DOI] [PubMed] [Google Scholar]
- 36. Gaddy DF, Lyles DS. 2007. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J. Virol. 81:2792–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen LY, Chen JD. 2003. Daxx silencing sensitizes cells to multiple apoptotic pathways. Mol. Cell. Biol. 23:7108–7121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Michaelson JS, Leder P. 2003. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 116:345–352 [DOI] [PubMed] [Google Scholar]
- 39. Michaelson JS, Bader D, Kuo F, Kozak C, Leder P. 1999. Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 13:1918–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dionne KR, Galvin JM, Schittone SA, Clarke P, Tyler KL. 2011. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J. Neurovirol. 17:314–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tyler KL, Sokol RJ, Oberhaus SM, Le M, Karrer FM, Narkewicz MR, Tyson RW, Murphy JR, Low R, Brown WR. 1998. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology 27:1475–1482 [DOI] [PubMed] [Google Scholar]
- 42. Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. 2002. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J. Neurosci. 22:7027–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gown AM, Willingham MC. 2002. Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J. Histochem. Cytochem. 50:449–454 [DOI] [PubMed] [Google Scholar]
- 44. Tyler KL, Leser JS, Phang TL, Clarke P. 2010. Gene expression in the brain during reovirus encephalitis. J. Neurovirol. 16:56–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hollenbach AD, Sublett JE, McPherson CJ, Grosveld G. 1999. The Pax3-FKHR oncoprotein is unresponsive to the Pax3-associated repressor hDaxx. EMBO J. 18:3702–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gongora R, Stephan RP, Zhang Z, Cooper MD. 2001. An essential role for Daxx in the inhibition of B lymphopoiesis by type I interferons. Immunity 14:727–737 [DOI] [PubMed] [Google Scholar]
- 47. Shimoda K, Kamesaki K, Numata A, Aoki K, Matsuda T, Oritani K, Tamiya S, Kato K, Takase K, Imamura R, Yamamoto T, Miyamoto T, Nagafuji K, Gondo H, Nagafuchi S, Nakayama K, Harada M. 2002. Cutting edge: Tyk2 is required for the induction and nuclear translocation of Daxx which regulates IFN-alpha-induced suppression of B lymphocyte formation. J. Immunol. 169:4707–4711 [DOI] [PubMed] [Google Scholar]
- 48. Jung YS, Kim HY, Lee YJ, Kim E. 2007. Subcellular localization of Daxx determines its opposing functions in ischemic cell death. FEBS Lett. 581:843–852 [DOI] [PubMed] [Google Scholar]
- 49. Bi FF, Xiao B, Hu YQ, Tian FF, Wu ZG, Ding L, Zhou XF. 2008. Expression and localization of Fas-associated proteins following focal cerebral ischemia in rats. Brain Res. 1191:30–38 [DOI] [PubMed] [Google Scholar]
- 50. Niu YL, Li C, Zhang GY. 2011. Blocking Daxx trafficking attenuates neuronal cell death following ischemia/reperfusion in rat hippocampus CA1 region. Arch. Biochem. Biophys. 515:89–98 [DOI] [PubMed] [Google Scholar]
- 51. Zhang J, Cado D, Chen A, Kabra NH, Winoto A. 1998. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392:296–300 [DOI] [PubMed] [Google Scholar]
- 52. Li H, Leo C, Zhu J, Wu XY, O'Neil J, Park EJ, Chen JD. 2000. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell. Biol. 20:1784–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Croxton R, Puto LA, de Belle I, Thomas M, Torii S, Hanaii F, Cuddy M, Reed JC. 2006. Daxx represses expression of a subset of antiapoptotic genes regulated by nuclear factor-κB. Cancer Res. 66:9026–9035 [DOI] [PubMed] [Google Scholar]
- 54. Raoul C, Buhler E, Sadeghi C, Jacquier A, Aebischer P, Pettmann B, Henderson CE, Haase G. 2006. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc. Natl. Acad. Sci. U. S. A. 103:6007–6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. 2002. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 70:462–473 [DOI] [PubMed] [Google Scholar]
- 56. Lukiw WJ. 2004. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem. Res. 29:1287–1297 [DOI] [PubMed] [Google Scholar]
- 57. Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, Kaufman RJ, Bohjanen PR, Schiff LA. 2006. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80:2019–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clarke P, Leser JS, Tyler KL. 2009. Gene expression in the brain during virus-induced encephalitis, abstr. W23-2. Am. Soc. Virol. 28th Annu. Meet. Sci. Progr. Abstr. 2009 American Society for Virology, Toledo, OH [Google Scholar]
- 59. Kim KS, Hwang HA, Chae SK, Ha H, Kwon KS. 2005. Upregulation of Daxx mediates apoptosis in response to oxidative stress. J. Cell. Biochem. 96:330–338 [DOI] [PubMed] [Google Scholar]
- 60. Ma Q, Devarajan P. 2008. Induction of proapoptotic Daxx following ischemic acute kidney injury. Kidney Int. 74:310–318 [DOI] [PubMed] [Google Scholar]
- 61. Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T. 2006. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U. S. A. 103:7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Konopka JL, Penalva LO, Thompson JM, White LJ, Beard CW, Keene JD, Johnston RE. 2007. A two-phase innate host response to alphavirus infection identified by mRNP-tagging in vivo. PLoS Pathog. 3:e199 doi:10.1371/journal.ppat.0030199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Akterin S, Cowburn RF, Miranda-Vizuete A, Jimenez A, Bogdanovic N, Winblad B, Cedazo-Minguez A. 2006. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer's disease. Cell Death Differ. 13:1454–1465 [DOI] [PubMed] [Google Scholar]
- 64. Junn E, Taniguchi H, Jeong BS, Zhao X, Ichijo H, Mouradian MM. 2005. Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proc. Natl. Acad. Sci. U. S. A. 102:9691–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Song JJ, Lee YJ. 2004. Tryptophan 621 and serine 667 residues of Daxx regulate its nuclear export during glucose deprivation. J. Biol. Chem. 279:30573–30578 [DOI] [PubMed] [Google Scholar]
- 66. Karunakaran S, Diwakar L, Saeed U, Agarwal V, Ramakrishnan S, Iyengar S, Ravindranath V. 2007. Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson's disease: protection by alpha-lipoic acid. FASEB J. 21:2226–2236 [DOI] [PubMed] [Google Scholar]
- 67. Mateos L, Persson T, Kathozi S, Gil-Bea FJ, Cedazo-Minguez A. 2012. Estrogen protects against amyloid-beta toxicity by estrogen receptor alpha-mediated inhibition of Daxx translocation. Neurosci. Lett. 506:245–250 [DOI] [PubMed] [Google Scholar]
- 68. Lindsay CR, Giovinazzi S, Ishov AM. 2009. Daxx is a predominately nuclear protein that does not translocate to the cytoplasm in response to cell stress. Cell Cycle 8:1544–1551 [DOI] [PubMed] [Google Scholar]
- 69. Pluta AF, Earnshaw WC, Goldberg IG. 1998. Interphase-specific association of intrinsic centromere protein CENP-C with HDaxx, a death domain-binding protein implicated in Fas-mediated cell death. J. Cell Sci. 111:2029–2041 [DOI] [PubMed] [Google Scholar]
- 70. Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF, III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147:221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kominsky DJ, Bickel RJ, Tyler KL. 2002. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 9:926–933 [DOI] [PubMed] [Google Scholar]
- 72. Kim YY, Park BJ, Seo GJ, Lim JY, Lee SM, Kimm KC, Park C, Kim J, Park SI. 2003. Long form of cellular FLICE-inhibitory protein interacts with Daxx and prevents Fas-induced JNK activation. Biochem. Biophys. Res. Commun. 312:426–433 [DOI] [PubMed] [Google Scholar]
- 73. Clarke P, Tyler KL. 2007. Down-regulation of cFLIP following reovirus infection sensitizes human ovarian cancer cells to TRAIL-induced apoptosis. Apoptosis 12:211–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Clarke P, Debiasi RL, Meintzer SM, Robinson BA, Tyler KL. 2005. Inhibition of NF-kappa B activity and cFLIP expression contribute to viral-induced apoptosis. Apoptosis 10:513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Knowlton JJ, Dermody TS, Holm GH. 2012. Apoptosis induced by mammalian reovirus is beta interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-κB-dependent expression of Noxa. J. Virol. 86:1650–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]