

Abstract
The anaerobic acetogenic bacterium Acetobacterium woodii couples reduction of caffeate with electrons derived from molecular hydrogen to the synthesis of ATP by a chemiosmotic mechanism with sodium ions as coupling ions. Caffeate is activated to caffeyl coenzyme A (caffeyl-CoA) prior to its reduction, and the caffeate reduction operon encodes an ATP-dependent caffeyl-CoA synthetase that is thought to catalyze the initial caffeate activation. The operon also encodes a potential CoA transferase, the product of carA, which was thought to be involved in subsequent ATP-independent caffeate activation. To prove the proposed function of carA, we overproduced its protein in Escherichia coli and then purified it. Purified CarA drives the formation of caffeyl-CoA from caffeate with hydrocaffeyl-CoA as the CoA donor. The dependence of the reaction on caffeate and hydrocaffeyl-CoA followed Michaelis-Menten kinetics, with apparent Km values of 75 ± 5 μM for caffeate and 8 ± 2 μM for hydrocaffeyl-CoA. The enzyme activity had broad ranges of pH and temperature optima. In addition to being able to use caffeate, CarA could use p-coumarate and ferulate but not cinnamate, sinapate, or p-hydroxybenzoate as a CoA acceptor. Neither acetyl-CoA nor butyryl-CoA served as the CoA donor for CarA. The enzyme uses a ping-pong mechanism for CoA transfer and is the first classified member of a new subclass of family I CoA transferases that has two catalytic domains on one polypeptide chain. Apparently, CarA catalyzes an energy-saving CoA loop for caffeate activation in the steady state of caffeate respiration.
INTRODUCTION
The acetogenic bacterium Acetobacterium woodii grows by the oxidation of various electron donors, such as molecular hydrogen, C1 compounds like methanol and formate, or sugars (1). Carbon dioxide serves as an electron acceptor and is reduced to acetate in the Wood-Ljungdahl pathway (2–5). This pathway combines acetyl coenzyme A (acetyl-CoA) formation from carbon dioxide with the synthesis of ATP via a transmembrane sodium ion gradient (6–10). In addition, A. woodii uses phenyl acrylates, such as caffeate, ferulate, sinapate, or trimethoxy-cinnamate, as electron acceptors (11, 12). Reduction of the carbon-carbon double bond of phenyl acrylates such as caffeate was shown to be energy conserving in A. woodii (11) and is also coupled to the generation of a transmembrane electrochemical Na+ gradient, which is then used for ATP synthesis (9, 13–15). Hence, this type of respiration has been termed caffeate respiration.
Caffeate respiration involves an electron transfer branch in which electrons are channeled to the electron carrier ferredoxin. This is done either by way of the pyruvate-ferredoxin oxidoreductase during heterotrophic growth or via an electron-bifurcating hydrogenase during lithotrophic metabolism when hydrogen is the electron donor (16). Reduced ferredoxin then fuels the sodium-motive ferredoxin-NAD+ oxidoreductase (Rnf) that establishes the sodium ion potential across the cytoplasmic membrane used for ATP synthesis (10, 17). NADH is the actual electron donor for the reduction of the electron acceptor and is thought to be catalyzed by a soluble complex that contains the caffeyl-CoA reductase and an electron transfer protein (CarCDE) (15, 18). In analogy to the reduction of crotonyl-CoA by Clostridium kluyveri, the exergonic reduction of caffeyl-CoA might drive the endergonic reduction of ferredoxin via electron bifurcation (19, 20).
Based on thermodynamic calculations, the sodium-motive ferredoxin-NAD+ oxidoreductase may translocate a maximum of two Na+ per two electrons. Since the ATP synthase probably translocates 3.3 Na+ per ATP (21, 22), only a fraction of an ATP per mole of caffeate is reduced. In addition to the low ATP yield during its respiration, caffeate has to be activated to caffeyl-CoA prior to its reduction (15). Activation is carried out by an ATP-dependent acyl-CoA synthetase, encoded by a gene of the car operon, carB (18). This enzyme uses two energy-rich phosphodiester bonds for caffeate activation. Although some energy is saved by a sodium-motive pyrophosphatase (23), two ATP equivalents are required for activation of the electron acceptor and therefore net ATP formation cannot occur during caffeate respiration. An energy-saving alternative for caffeate activation might be a CoA loop catalyzed by CarA, encoded by the first gene of the operon that has similarities to CoA transferases (18). We have purified and characterized CarA from A. woodii and present evidence that it is indeed a CoA transferase. The role of CarA in caffeate respiration is discussed.
MATERIALS AND METHODS
Cloning of carA.
The carA gene was amplified from A. woodii chromosomal DNA by PCR with the primers (5′ → 3′) AGCATATGGCTAAATTTATTTCAGCAAAAG and AAGCGGCCGCCTTGCCCATTATTGTATCAAG. The PCR was performed in an Eppendorf mastercycler gradient (Eppendorf, Hamburg, Germany) with the following program: 95°C for 2 min, 30 cycles of 95°C for 1 min, 59°C for 1 min, and 72°C for 1 h 40 min. PCR products were cleaved with the appropriate restriction enzymes and ligated into the overexpression vector pET21a (Novagen, Darmstadt, Germany). The ligation mixture was transformed into chemically competent Escherichia coli DH5α. The resulting transformants were screened for plasmids of the appropriate size. Inserts were sequenced to ensure gene integrity. The final plasmid used in this study was named pET21a-carA. The plasmid contained a sequence encoding a hexahistidine tag at the C terminus for purification of the recombinant protein.
Protein production and purification.
The chemically competent E. coli strain BL21(DE3) was transformed with pET21a-carA and grown on Luria-Bertani (LB) plates, supplemented with 100 μg/ml ampicillin, and incubated overnight at 37°C. Three liters of LB medium, containing 100 μg/ml ampicillin, was inoculated with 80 ml of an overnight culture of E. coli BL21(DE3) containing pET21a-carA. Cells were grown aerobically to an optical density at 600 nm (OD600) of 1.0 at 37°C; gene expression was induced by the addition of IPTG (isopropyl-β-d-thiogalactopyranoside) (Roth, Karlsruhe, Germany) to a final concentration of 0.66 mM. After 3 h at 37°C, the cultures were harvested, and cells were washed in 50 ml lysis buffer (300 mM NaCl, 50 mM NaH2PO4, 10 mM imidazole [pH 8]). Cells were frozen at −20°C until use.
To purify the CarA protein, cells were resuspended in 25 ml lysis buffer containing 1 mg/ml DNase I (Roche) and then disrupted by three passages through a French press (65 MPa). Cell debris and whole cells were removed by two centrifugation steps (24,000 × g, 30 min, 4°C). The cell extract was separated into the cytoplasmic and membrane fractions by ultracentrifugation (150,000 × g, 2 h, 4°C). The supernatant was incubated for 40 min at 4°C with nickel-nitrilotriacetic acid (Ni2+-NTA) resin (Qiagen, Hilden, Germany). Nonspecifically bound protein was removed by a washing step with lysis buffer containing 60 mM imidazole. Protein was eluted from the resin with lysis buffer containing 150 mM imidazole. To determine the size of CarA, the Ni2+-NTA fraction was further applied to a Superose 6 column, equilibrated with 50 mM NaPi buffer (pH 8) containing 300 mM NaCl.
Assay of CoA transferase activity.
Standard assays were performed at 40°C. One milliliter of 100 mM KPi buffer (pH 7.5) was filled into a quartz cuvette and supplemented with purified CarA-His6 and 250 μM caffeate. The reaction was started with the addition of 45 μM hydrocaffeyl-CoA. Caffeyl-CoA formation was followed spectrophotometrically at 346 nm (ε = 18 mM−1 · cm−1) (24). For determination of the substrate specificity of CarA, the following extinction coefficients (mM−1 · cm−1) were used to determine specific activity: ε346 = 19 (ferulyl-CoA) (24), ε333 = 21 (ρ-coumaryl-CoA) (24), ε308 = 26.5 (cinnamyl-CoA) (25), ε352 = 20(sinapyl-CoA) (26), and ε330 = 24 (ρ-hydroxybenzyl-CoA) (27).
Synthesis and purification of hydrocaffeyl-CoA.
Hydrocaffeyl-CoA was synthesized by a modification (28) of the Kawaguchi method (29). Briefly, 50 μmol of hydrocaffeic acid dissolved in about 1 ml dry acetonitrile was reacted with 45 μmol of 1.1′-carbonyl-diimidazole (CDI). The mixture was quickly added to 40 μmol of free CoA dissolved in 1 ml of 1 M NaHCO3. The progress of the reaction was monitored by dot blotting with Ellman's reagent [5,5′-dithiobis(2-nitrobenzoate)]. After 30 to 60 min, the reaction was complete. The mixture was diluted to 10 ml with distilled water and acidified to pH 2 with 5 M HCl. Hydrocaffeyl-CoA was purified by reverse-phase chromatography through C18 Sep-Pak columns (Waters, MA) washed with methanol and equilibrated with 0.1% trifluoroacetic acid (TFA) (vol/vol). After being loaded, the column was washed with 3 volumes of the same solution. Elution was performed with 0.1% TFA containing 50% acetonitrile (vol/vol). The eluted hydrocaffeyl-CoA was freed from acetonitrile by freezing and centrifuging on a Speed-Vac concentrator (Bachofer, Reutlingen, Germany). It was then refrozen and vacuum dried on a lyophilizer (Alpha1-4, Christ, Osterode am Harz, Germany). The lyophilized powder was stored at −80°C till further use. Hydrocaffeyl-CoA was then analyzed by UV-visible spectroscopy. The concentration of hydrocaffeyl-CoA was determined in a single assay. A sample of hydrocaffeyl-CoA was added to the reaction mixture of the standard assay with 1 mM caffeate. The reaction was started with the addition of CarA. The resulting increase in absorbance at 346 nm was used to calculate the amount of caffeyl-CoA formed. As caffeate was present in excess, the initial amount of hydrocaffeyl-CoA in the assay was proportional to the amount of caffeyl-CoA formed.
RESULTS AND DISCUSSION
Heterologous production and purification of CarA from A. woodii.
To analyze the function of the carA gene product, the gene was cloned in an expression vector and overexpressed in E. coli. The protein had an engineered His6 tag at the C terminus that was used to purify the overproduced protein via affinity chromatography. After chromatography on Ni2+-NTA, the preparation contained a major protein of 60 kDa, which fits well to the predicted mass of 57.5 kDa (including the His6 tag) (Fig. 1). In addition, anti-His6 antibodies reacted with the 60-kDa protein. Analytical gel filtration experiments revealed a mass of ∼130 kDa for the protein, indicating that it might be a homodimer.
Fig 1.
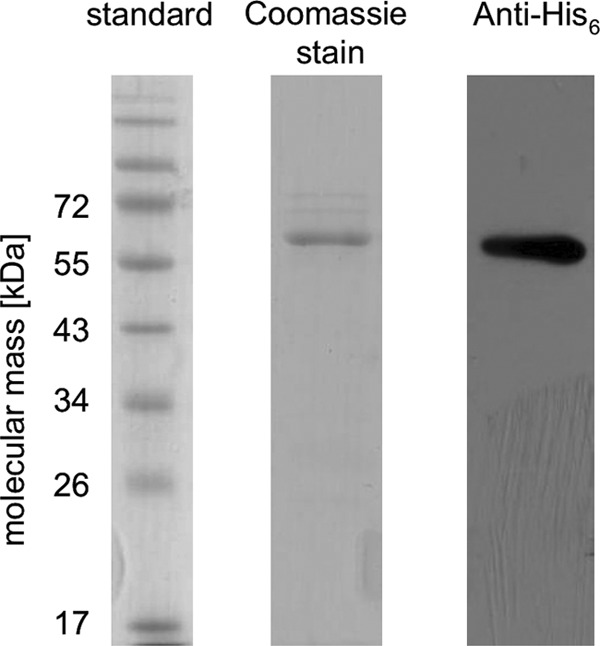
Purification of C-terminally His6-tagged hydrocaffeyl-CoA:caffeate CoA transferase CarA of A. woodii. E. coli pET21a-carA was cultured at 37°C in LB medium. CarA was purified by affinity chromatography on Ni2+-NTA. Samples were separated on SDS-PAGE; proteins were stained with Coomassie blue or the recombinant His6 tag was detected via Western blotting.
CarA is a CoA transferase.
When CarA was incubated with 250 μM caffeate and 45 μM hydrocaffeyl-CoA, caffeyl-CoA was produced, as evident from the increase of absorbance at 346 nm (Fig. 2). This activity was strictly dependent on the presence of recombinant protein, caffeate, and hydrocaffeyl-CoA. These data show that CarA indeed catalyzes CoA transfer from hydrocaffeyl-CoA to caffeate.
Fig 2.
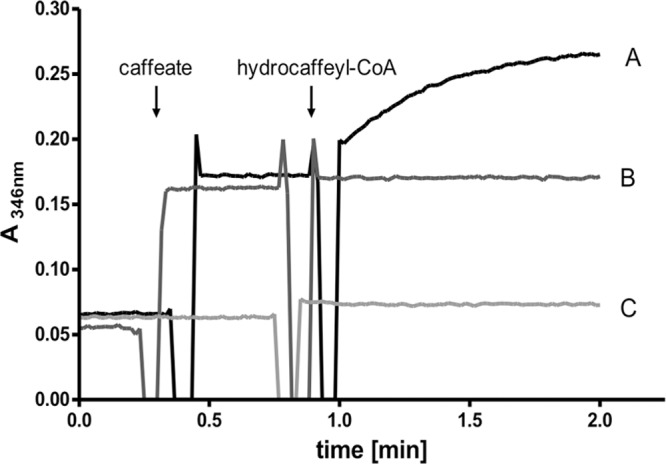
Heterologously produced and purified CarA catalyzes CoA transfer from hydrocaffeyl-CoA to caffeate. The assay mixture contained 1 ml 100 mM KPi buffer (pH 7.5) and 0.15 μg CarA. (A) 250 μM caffeate and 45 μM hydrocaffeyl-CoA were added as indicated. (B) No enzyme added. (C) No caffeate added. The formation of caffeyl-CoA was followed spectrophotometrically at 346 nm.
The dependence of the reaction on caffeate (Fig. 3) followed Michaelis-Menten kinetics. The apparent Km value was determined to be 75 ± 5 μM, the Vmax was 125 U/mg, and the kcat was 120 s−1. Figure 4 shows that the dependence on the amount of hydrocaffeyl-CoA also followed Michaelis-Menten kinetics; the Km value was determined to be 8 ± 2 μM. Double-reciprocal plots of initial velocity against the caffeate concentration gave straight parallel lines (Fig. 5), indicating a ping-pong mechanism as found in other CoA transferases (30, 31) of the class I CoA transferase family.
Fig 3.
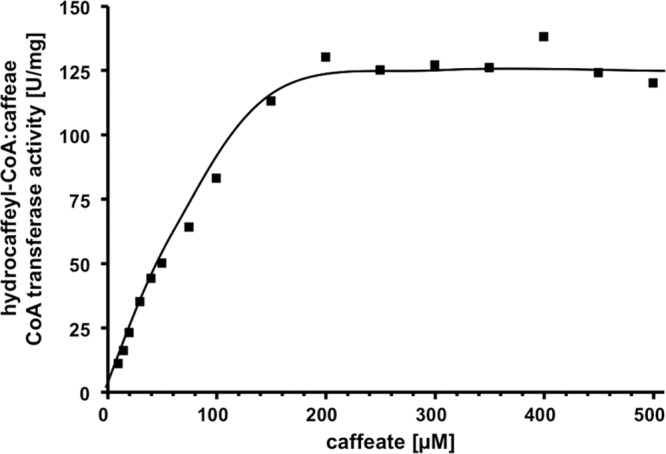
Caffeate dependence of hydrocaffeyl-CoA:caffeate CoA transferase activity of CarA. The formation of caffeyl-CoA by purified recombinant CarA (0.15 μg/ml) was monitored at 346 nm.
Fig 4.
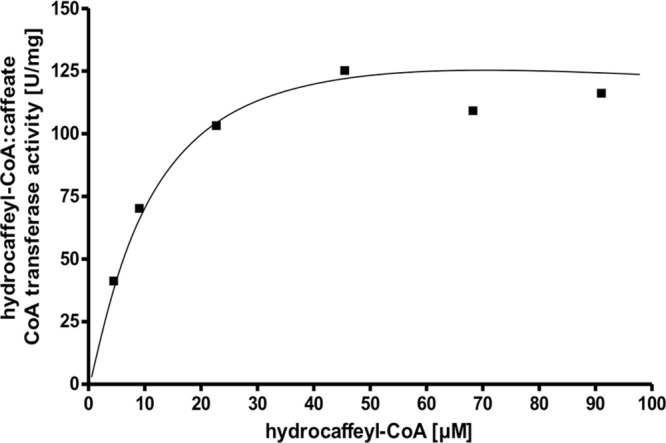
Hydrocaffeyl-CoA dependence of hydrocaffeyl-CoA:caffeate CoA transferase activity of CarA. The formation of caffeyl-CoA by purified recombinant CarA (0.15 μg/ml) was monitored at 346 nm.
Fig 5.
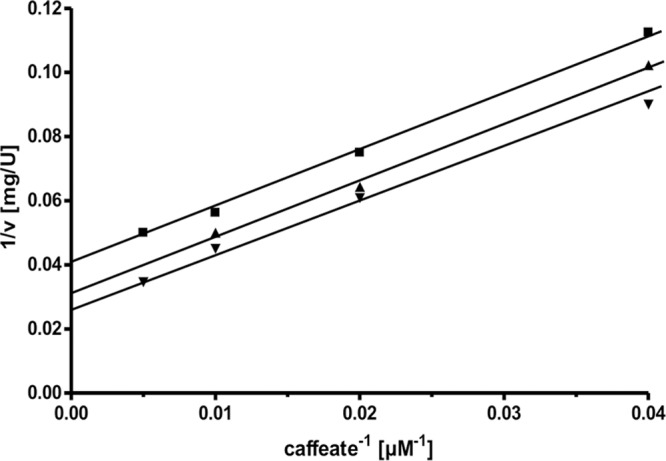
Kinetics of caffeyl-CoA formation. The assay mixture contained 1 ml 100 mM KPi buffer (pH 7.5), 0.15 μg CarA, caffeate as indicated, and hydrocaffeyl-CoA at 10 μM (■), 20 μM (▲), and 30 μM (▼).
The enzyme had a rather broad pH range for activity, ranging from 6.0 to 9.0 with an optimum at 7.5 (data not shown). It also had a rather broad temperature range for activity (20 to 50°C) with an optimum at 40°C (data not shown).
Substrate specificity of the hydrocaffeyl-CoA:caffeate CoA transferase.
Since A. woodii can reduce a number of different phenyl acrylates, we determined the specificity of CoA transfer from hydrocaffeyl-CoA to several substrates with structural similarities to caffeate (Fig. 6). Activity was highest with p-coumarate, which has one hydroxyl group less than caffeate. Caffeate and ferulate also served as substrates, with activities that were 84 and 86%, respectively, of that with p-coumarate. No activity was observed with cinnamate, sinapate, or hydroxybenzoate as the CoA acceptor or with acetyl-CoA or butyryl-CoA as the CoA donor (data not shown).
Fig 6.

Substrate specificity of the hydrocaffeyl-CoA:caffeate CoA transferase CarA. (A) Structures of phenyl acrylates and other substrates used. (B) The assay mixture contained 1 ml 100 mM KPi buffer (pH 7.5), 0.15 μg CarA, and 45 μM hydrocaffeyl-CoA. The reaction was started with the addition of 250 μM substrate. Formation of the corresponding CoA thioester was followed spectrophotometrically; 100% corresponds to 151 U/mg.
Classification of the hydrocaffeyl-CoA:caffeate CoA transferase.
To date, three families of CoA transferases have been distinguished based on their reaction mechanisms (32). Family I enzymes catalyze a ping-pong mechanism and, in prokaryotes, have two subunits, each having a family I coenzyme A transferase domain, PF01144 in the Pfam database (33). Family II houses the α-subunits of the citrate lyases (PF04223; citrate lyase, alpha subunit), the prototype being CitF. Family III proteins have different subunit compositions but have a characteristic PF02515 domain (CoA transferase family III in the Pfam database). A. woodii CarA shares with family I CoA transferases not only the mechanism but also the conserved domain, except that the peptide is about twice as long. This conserved domain is found in the first half of the peptide (E value, 1.5E−50 for amino acid positions 4 to 241) and in the second half, with a weaker hit to the same domain (E value, 5.2E−4 for amino acid positions 279 to 491). CoA transferases with two conserved domains on the same peptide are known to occur in eukaryotic organisms (34, 35) but have not been described in prokaryotes so far. A database search revealed the presence of genes that encode peptides with similar domain architecture in the taxa Firmicutes, Acidobacteria, Bacteroidetes, and Gamma- and Deltaproteobacteria, and especially in Alphaproteobacteria, in which they are very common.
An inspection of the genome sequences revealed that some anaerobic bacteria seem to have similar types of genes in close proximity to the CoA transferase gene. For example, closest by sequence identity to A. woodii CarA are Clostridium sp. SY8519, Pelosinus fermentans, and Holophaga foetida. In P. fermentans and H. foetida, genes for an acyl-CoA dehydrogenase, electron transfer flavoprotein, and flavin adenine dinucleotide (FAD)-dependent oxidoreductase are downstream of the CoA transferase, suggesting similar types of reactions in these anaerobes (Fig. 7).
Fig 7.

The carA gene neighborhood in A. woodii and putative similar operons in the genomes of selected anaerobic bacteria. A similar gene arrangement is found in Pelosinus and Holophaga (Firmicutes). The latter organisms and Thauera carry out the anaerobic metabolism of aromatic compounds that is known to involve a CoA transferase (36). The locus tags are shown below the gene names or annotations. Orthologs are drawn with the same pattern.
Model of caffeate respiration in A. woodii.
The current model of the enzymology and bioenergetics of caffeate respiration is shown in Fig. 8. The data presented here clearly demonstrate that CarA is a hydrocaffeyl-CoA:caffeate CoA transferase, whereas CarB has been shown to be an ATP-dependent acyl-CoA synthetase (18). It is tempting to speculate that the initial activation of caffeate is catalyzed by CarB. Once the caffeyl-CoA is produced and reduced to hydrocaffeyl-CoA, the activation reaction is maintained by the activity of CarA. This CoA loop saves energy (2 ATP equivalents) and makes the overall reaction of caffeate reduction with hydrogen as the electron donor energetically feasible. Such energy-saving CoA loops are actually found quite often in the energy metabolism of anaerobes, for example in the toluene catabolism of Thauera aromatica (36) or in the oxalate degradation in Oxalobacter formigenes (37).
Fig 8.

Model of caffeate respiration in A. woodii. The electron flow from molecular hydrogen to caffeate is shown. Ferredoxin is reduced by a bifurcating hydrogenase and reoxidized by the Rnf complex, which generates a sodium ion gradient across the cytoplasmic membrane. NADH serves as an electron donor for the caffeyl-CoA reducing complex, potentially encoded by carCDE, which might reduce another ferredoxin via electron bifurcation, fueling the Rnf complex. Caffeate is initially activated by CarB (A), whereas CarA catalyzes an energy-saving CoA loop in the steady state of caffeate respiration (B).
ACKNOWLEDGMENT
This work was supported by a grant from the Deutsche Forschungsgemeinschaft.
Footnotes
Published ahead of print 11 January 2013
REFERENCES
- 1. Müller V, Imkamp F, Rauwolf A, Küsel K, Drake HL. 2004. Molecular and cellular biology of acetogenic bacteria, p 251–281 In Nakano MM, Zuber P. (ed), Strict and facultative anaerobes. Medical and environmental aspects. Horizon Biosciences, Norfolk, United Kingdom [Google Scholar]
- 2. Ljungdahl LG. 1994. The acetyl-CoA pathway and the chemiosmotic generation of ATP during acetogenesis, p 63–87 In Drake HL. (ed), Acetogenesis. Chapman & Hall, New York, NY [Google Scholar]
- 3. Ljungdahl LG. 1986. The autotrophic pathway of acetate synthesis in acetogenic bacteria. Annu. Rev. Microbiol. 40:415–450 [DOI] [PubMed] [Google Scholar]
- 4. Ragsdale SW. 1997. The eastern and western branches of the Wood/Ljungdahl pathway: how the east and west were won. Biofactors 6:3–11 [DOI] [PubMed] [Google Scholar]
- 5. Ragsdale SW. 1991. Enzymology of the acetyl-CoA pathway of autotrophic CO2 fixation. Crit. Rev. Biochem. Mol. Biol. 26:261–300 [DOI] [PubMed] [Google Scholar]
- 6. Heise R, Reidlinger J, Müller V, Gottschalk G. 1991. A sodium-stimulated ATP synthase in the acetogenic bacterium Acetobacterium woodii. FEBS Lett. 295:119–122 [DOI] [PubMed] [Google Scholar]
- 7. Heise R, Müller V, Gottschalk G. 1992. Presence of a sodium-translocating ATPase in membrane vesicles of the homoacetogenic bacterium Acetobacterium woodii. Eur. J. Biochem. 206:553–557 [DOI] [PubMed] [Google Scholar]
- 8. Poehlein A, Schmidt S, Kaster Goenrich AKM, Vollmers J, Thürmer A, Bertsch J, Schuchmann K, Voigt B, Hecker M, Daniel R, Thauer RK, Gottschalk G, Müller V. 2012. An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis. PLoS One 7:e33439 doi:10.1371/journal.pone.0033439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt S, Biegel E, Müller V. 2009. The ins and outs of Na+ bioenergetics in Acetobacterium woodii. Biochim. Biophys. Acta 1787:691–696 [DOI] [PubMed] [Google Scholar]
- 10. Biegel E, Schmidt S, González JM, Müller V. 2011. Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci. 68:613–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bache R, Pfennig N. 1981. Selective isolation of Acetobacterium woodii on methoxylated aromatic acids and determination of growth yields. Arch. Microbiol. 130:255–261 [Google Scholar]
- 12. Tschech A, Pfennig N. 1984. Growth yield increase linked to caffeate reduction in Acetobacterium woodii. Arch. Microbiol. 137:163–167 [Google Scholar]
- 13. Imkamp F, Müller V. 2002. Chemiosmotic energy conservation with Na+ as the coupling ion during hydrogen-dependent caffeate reduction by Acetobacterium woodii. J. Bacteriol. 184:1947–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dilling S, Imkamp F, Schmidt S, Müller V. 2007. Regulation of caffeate respiration in the acetogenic bacterium Acetobacterium woodii. Appl. Environ. Microbiol. 73:3630–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imkamp F, Biegel E, Jayamani E, Buckel W, Müller V. 2007. Dissection of the caffeate respiratory chain in the acetogen Acetobacterium woodii: indications for a Rnf-type NADH dehydrogenase as coupling site. J. Bacteriol. 189:8145–8153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schuchmann K, Müller V. 2012. A bacterial electron bifurcating hydrogenase. J. Biol. Chem. 287:31165–31171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Biegel E, Müller V. 2010. Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U. S. A. 107:18138–18142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hess V, Vitt S, Müller V. 2011. A caffeyl-coenzyme A synthetase initiates caffeate activation prior to caffeate reduction in the acetogenic bacterium Acetobacterium woodii. J. Bacteriol. 193:971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Herrmann G, Jayamani E, Mai G, Buckel W. 2008. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190:784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, Thauer RK. 2008. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fritz M, Müller V. 2007. An intermediate step in the evolution of ATPases—the F1FO-ATPase from Acetobacterium woodii contains F-type and V-type rotor subunits and is capable of ATP synthesis. FEBS J. 274:3421–3428 [DOI] [PubMed] [Google Scholar]
- 22. Fritz M, Klyszejko AL, Morgner N, Vonck J, Brutschy B, Müller DJ, Meier T, Müller V. 2008. An intermediate step in the evolution of ATPases: a hybrid F1FO rotor in a bacterial Na+ F1FO ATP synthase. FEBS J. 275:1999–2007 [DOI] [PubMed] [Google Scholar]
- 23. Biegel E, Müller V. 2011. A Na+-translocating pyrophosphatase in the acetogenic bacterium Acetobacterium woodii. J. Biol. Chem. 286:6080–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Obel N, Scheller HV. 2000. Enzymatic synthesis and purification of caffeoyl-CoA, p-coumaroyl-CoA, and feruloyl-CoA. Anal. Biochem. 286:38–44 [DOI] [PubMed] [Google Scholar]
- 25. Yao KW, Schulz H. 1993. Specific assay of medium-chain acyl-CoA dehydrogenase based on the spectrophotometric measurement of product formation. Anal. Biochem. 214:528–534 [DOI] [PubMed] [Google Scholar]
- 26. Lee D, Meyer K, Chapple C, Douglas CJ. 1997. Antisense suppression of 4-coumarate:coenzyme A ligase activity in Arabidopsis leads to altered lignin subunit composition. Plant Cell 9:1985–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Webster LT, Jr, Mieyal JJ, Siddiqui UA. 1974. Benzoyl and hydroxybenzoyl esters of coenzyme A. Ultraviolet characterization and reaction mechanisms. J. Biol. Chem. 249:2641–2645 [PubMed] [Google Scholar]
- 28. Parthasarathy A, Buckel W, Smith DM. 2010. On the thermodynamic equilibrium between (R)-2-hydroxyacyl-CoA and 2-enoyl-CoA. FEBS J. 277:1738–1746 [DOI] [PubMed] [Google Scholar]
- 29. Kawaguchi A, Yoshimura T, Okuda S. 1981. A new method for the preparation of acyl-CoA thioesters. J. Biochem. 89:337–339 [DOI] [PubMed] [Google Scholar]
- 30. Buckel W, Dorn U, Semmler R. 1981. Glutaconate CoA-transferase from Acidaminococcus fermentans. Eur. J. Biochem. 118:315–321 [DOI] [PubMed] [Google Scholar]
- 31. Tung KK, Wood WA. 1975. Purification, new assay, and properties of coenzyme A transferase from Peptostreptococcus elsdenii. J. Bacteriol. 124:1462–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heider J. 2001. A new family of CoA-transferases. FEBS Lett. 509:345–349 [DOI] [PubMed] [Google Scholar]
- 33. Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. 2010. The Pfam protein families database. Nucleic Acids Res. 38:D211–D222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corthésy-Theulaz IE, Bergonzelli GE, Henry H, Bachmann D, Schorderet DF, Blum AL, Ornston LN. 1997. Cloning and characterization of Helicobacter pylori succinyl CoA:acetoacetate CoA-transferase, a novel prokaryotic member of the CoA-transferase family. J. Biol. Chem. 272:25659–25667 [DOI] [PubMed] [Google Scholar]
- 35. Parales RE, Harwood CS. 1992. Characterization of the genes encoding beta-ketoadipate: succinyl-coenzyme A transferase in Pseudomonas putida. J. Bacteriol. 174:4657–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leutwein C, Heider J. 2001. Succinyl-CoA:(R)-benzylsuccinate CoA-transferase: an enzyme of the anaerobic toluene catabolic pathway in denitrifying bacteria. J. Bacteriol. 183:4288–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ricagno S, Jonsson S, Richards N, Lindqvist Y. 2003. Formyl-CoA transferase encloses the CoA binding site at the interface of an interlocked dimer. EMBO J. 22:3210–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]