

Abstract
The human gastrointestinal tract, in particular the colon, hosts a vast number of commensal microorganisms. Representatives of the genus Bacteroides are among the most abundant bacterial species in the human colon. Bacteroidetes diverged from the common line of eubacterial descent before other eubacterial groups. As a result, they employ unique transcription initiation signals and, because of this uniqueness, they require specific genetic tools. Although some tools exist, they are not optimal for studying the roles and functions of these bacteria in the human gastrointestinal tract. Focusing on translation initiation signals in Bacteroides, we created a series of expression vectors allowing for different levels of protein expression in this genus, and we describe the use of pepI from Lactobacillus delbrueckii subsp. lactis as a novel reporter gene for Bacteroides. Furthermore, we report the identification of the 3′ end of the 16S rRNA of Bacteroides ovatus and analyze in detail its ribosomal binding site, thus defining a core region necessary for efficient translation, which we have incorporated into the design of our expression vectors. Based on the sequence logo information from the 5′ untranslated region of other Bacteroidales ribosomal protein genes, we conclude that our findings are relevant to all members of this order.
INTRODUCTION
The human gastrointestinal tract hosts a plethora of commensal microorganisms, the majority of which are located in the colon, where they reach cell densities of 1011 per gram of content (1). It has been shown that these bacteria contain representatives of hundreds of different phylotypes (2, 3). Although the composition of the microbiota of individuals is unique and variable, a dominant phylogenetic core set has been described (4), with members of the phyla Firmicutes and Bacteroidetes constituting up to 90% of the microbiota resident in the colon of all human populations (2, 5).
The intestinal microbiome contributes considerably to the health of the human host by, for example, providing essential vitamins and breaking down and fermenting dietary fiber into short-chain fatty acids, which, besides supplying energy, have a wider physiological impact on the host (6). The microbiome also plays a key role in the development of the immune system (7), and its dysbiosis has been implicated in the development of chronic inflammatory disorders, such as Crohn's disease and ulcerative colitis (8), and may contribute to obesity (9). Among members of the Bacteroidetes phylum, representatives of the genus Bacteroides are some of the most abundant bacterial species in the human colon (10). Consequently, there is considerable interest in studying these organisms that diverged from the common line of eubacterial descent before the major eubacterial groups, including Gram-positive bacteria, and are distinct from the other major Gram-negative phylum, the Proteobacteria (11). Their membranes contain sphingolipids (12), and the structure of their promoters is different from those of proteobacteria (13), which is reflected in the fact that Bacteroides species possess a unique primary sigma factor (14) and which explains the failure to express genes from other Gram-negative bacteria (e.g., Escherichia coli) in this genus (15). Functional analyses of these bacteria require, therefore, tools specific to this order of bacteria.
To address this need, we have focused on translation initiation, the next step in protein expression, and the sequence involved in translation initiation, the ribosomal binding site (RBS) or the Shine-Dalgarno (SD) sequence (16). The available information on Bacteroides Shine-Dalgarno sequences is inconclusive. Mastropaolo et al. (17) used the SD sequence (5′-AGAAAGGAG-3′) suggested by Tribble et al. (18) in their study of 16S rRNA expression signals. By exchanging transcription and translation initiation signals between E. coli and Bacteroides thetaiotaomicron, they concluded that the RBS sequence and spacing recognition by the ribosome in Bacteroides are more selective than those in E. coli (17). This is seemingly akin to Gram-positive bacteria such as Bacillus subtilis, which have more-stringent criteria for their translation initiation signals and require a higher degree of complementarity to the 3′ end of the 16S rRNA (19). Recently, Accetto and Avgustin (20) noted from the genome sequences of several representatives of the phylum Bacteroidetes that there is enrichment in adenine and thymine in the 5′ untranslated region (UTR) of genes. Furthermore, they reported an inability of Prevotella bryantii (a member of the order Bacteroidales) to form a functional Shine-Dalgarno interaction, although it was somewhat surprising that the best-performing 5′ UTR contained a perfect core SD sequence (5′-GGAGG-3′).
Proline iminopeptidases (EC 3.4.11.5) cleave N-terminal proline residues from peptides, thereby making them more accessible to the enzymatic activities of other aminopeptidases with broader substrate specificity (21). General aminopeptidases cannot cleave the Pro-X peptide bond efficiently due to structural constraints on the peptide chain conferred by the proline residue (22). Here, we report a novel reporter gene, pepI from Lactobacillus delbrueckii subsp. lactis (21), for Bacteroides and describe in detail the Bacteroides ribosomal binding site. Furthermore, we present a set of expression vectors allowing for high, medium, and low levels of constitutive protein expression.
MATERIALS AND METHODS
Media, growth conditions, and transformations.
Escherichia coli strains were grown in Luria-Bertani medium at 37°C. Bacteroides ovatus V975 and derivative strains were grown under anaerobic conditions at 37°C in brain heart infusion (BHI) medium (Oxoid, United Kingdom) supplemented with 0.001% hemin. Lactococcus lactis UKLc10 and derivative strains were grown at 30°C in M17 medium (Oxoid) supplemented with 5 g/liter glucose. Antibiotics, ampicillin at 200 μg/ml and tetracycline, erythromycin, and chloramphenicol at 5 μg/ml, were added as selective agents when appropriate. E. coli JM109 and L. lactis UKLc10 were transformed by electroporation using a Gene Pulser II (Bio-Rad, United Kingdom). The construction of most plasmids described below was performed using E. coli JM109 as the host strain. For constructs relating to pUK200I, the host strain L. lactis UKLc10 was used. The E. coli strain J53/R751 was supplemented with 200 μg/ml trimethoprim when grown for 18 h. Plasmids were mobilized from E. coli strain J53/R751 into B. ovatus V975 using a triparental filter mating protocol (23).
Calculation of codon adaptation indices.
Codon adaptation indices (CAIs) were calculated using the EMBOSS software package (24). First, codon usage tables were created on the basis of all coding sequences (excluding pseudogenes) of the organism using the EMBOSS cusp algorithm, and then the codon adaptation index for each ribosomal gene was computed using the respective codon usage table and the EMBOSS cai algorithm.
Construction of plasmids. (i) Expression vector pFI2716.
Initially, a 1,883-bp region carrying a β-d-glucuronidase gene (gusA) was removed from the Bacteroides plasmid pMJF1 (25) through restriction digestion with BamHI and SacI, T4 polymerase treatment, and subsequent religation of the larger fragment to create pFI2708. Next, the primer pair tetQ_5′ and tetQ_3′ (general primers are listed in Table S1 in the supplemental material) was used to amplify a 2,181-bp region carrying tetQ from the Bacteroides plasmid pBT-2 (26). The tetQ fragment was cloned into the blunted EcoRI site of pFI2708 to create pFI2714. The P1 promoter from the 16S rRNA gene of Bacteroides thetaiotaomicron VPI 5482 was cloned using the primer pair BAC_P1_f and BAC_P1_r to generate a 228-bp PCR product that was digested with BamHI and PstI and ligated into the same sites in pFI2714 to create pFI2715. Finally, the primer pair BglII_pUK200-linker_5′ and BglII_pUK200-linker_3′ was used to amplify a 239-bp polylinker region from plasmid pUK200 (27) comprising an SD sequence, unique cloning sites for NcoI (to be used for translational fusions), BamHI, and SmaI, and the Tbrnq terminator. The resulting linker fragment was digested with BglII and cloned into the BamHI site of pFI2715 to create pFI2716.
(ii) Cloning of the pepI reporter gene.
An 882-bp fragment was amplified from pUK200I plasmid DNA (27) using the primer pair pepI_5′ and pepI_3′. The pepI fragment was cloned into the NcoI (blunted) and SmaI sites of pFI2716 to create pGH001. In pGH001, the distance between the SD sequence (5′-AAGGAGG-3′) and the ATG start codon is 8 nucleotides. To move the SD sequence closer to the ATG start codon, the spacing was adjusted by either 2 or 6 nucleotides. To this end, primers phosphorylated at the 5′ end were employed to amplify the 8,876-bp pGH001 plasmid. The primer pair RBS-F and RBS_R_dis_6 was used to generate an 8,874-bp product (Δ2nt), and RBS-F and RBS_R_dis_2 were used to generate an 8,870-bp product (Δ6nt); each was then religated, creating plasmids pGH004 and pGH006, respectively.
For the generation of splice overlap extension PCR products, the details of the primers and templates are listed in Table S2 in the supplemental material. In each case, two PCR products (A and B) were amplified, and the amplified fragments from each of these reactions were then used as templates for the final splice reaction involving the forward primer from reaction A and the reverse primer from reaction B.
(iii) Cloning of the 5′ UTR upstream of orf1.
To exchange the SD sequence in pFI2716 with the 5′ UTR of orf1 in the B. ovatus V975 xylan utilization operon, splice overlap extension PCR was employed. A 365-bp SphI- and NcoI-digested fragment of the splice PCR product was then used to replace the corresponding 257-bp fragment in the SD sequence-related Bacteroides expression vector pFI2716, creating pGH009. To reduce the upstream sequences between the transcription initiation site (TIS) and the ATG start codon in pGH009 to either 20 or 30 nucleotides (nt), splice overlap extension PCR was used. A 169-bp or 179-bp SphI- and NcoI-digested fragment of the splice PCR products was then used to replace the corresponding 257-bp fragment of pFI2716, creating pGH020 and pGH021, respectively. The pepI reporter gene was added to pGH009, pGH020, and pGH021 as described for pGH001 to create pGH010, pGH022, and pGH023, respectively.
(iv) Removal of RNase E site from pFI2716.
To remove a region containing the RNase E site upstream of the SD sequence in pFI2716, splice overlap extension PCR was employed. A 329-bp SphI- and NotI-digested fragment of the splice product was used to replace the corresponding fragment of pGH022 to create pGH078.
(v) Replacing tetQ with ermF.
The primer pair ccr_amont2 and ccr_aval2 was used to amplify a 1,500-bp region carrying ermF from the plasmid pFD516 (28). The ermF fragment was digested with NdeI and cloned into NdeI-digested (blunted) and NsiI-digested pFI2716, pGH020, and pGH021 to replace the existing 1,839-bp fragment of each, creating pGH043, pGH044, and pGH090, respectively.
(vi) Altered 5′ UTR lengths.
To reduce the length of the 5′ UTR in pGH023 to 24 nt, splice overlap extension PCR was used. A 333-bp SphI- and NcoI-digested fragment of the splice PCR product was then used to replace the corresponding 329-bp fragment of pGH022 to create pGH079. To remove the entire 5′ UTR between the TIS and the start codon or to reduce the length of the 5′ UTR in pGH022 to 10, 16, or 18 nt, splice overlap extension PCR was employed. A 309-bp (0-nt 5′ UTR), 319-bp (10-nt 5′ UTR), 325-bp (16-nt 5′ UTR), or 327-bp (18-nt 5′ UTR) SphI- and NotI-digested fragment of each splice PCR product was then used to replace the corresponding 329-bp fragment of pGH022 to create pGH039, pGH040, pGH041, and pGH042, respectively.
(vii) Exchanging individual bases within the 5′ UTR.
Splice overlap extension PCR was used to exchange bases in pGH022 at positions −11 (replacing a guanosine with a cytidine), −10 (replacing an adenosine with a cytidine), and −9 (replacing an adenosine with a cytidine). A 329-bp SphI- and NotI-digested fragment of each splice PCR product was then used to replace the corresponding 329-bp fragment of pGH022 to create pGH045, pGH046, and pGH047, respectively.
To move the RBS closer to the ATG start codon in pGH022, the spacing was adjusted by either 2 or 4 nucleotides using splice overlap extension PCR. A 327-bp (Δ2nt) or 325-bp (Δ4nt) SphI- and NotI-digested fragment of each splice PCR product was then used to replace the corresponding 329-bp fragment of pGH022 to create pGH048 and pGH049, respectively.
(viii) Comparing proline iminopeptidase I expression levels in a Gram-positive host.
The 20-nucleotide lactococcal SD sequence upstream of pepI in pUK200I was exchanged for the 20 nt of the 5′ UTR from pGH022 using splice overlap extension PCR. A 362-bp BglII- and NotI-digested fragment of the splice PCR product was then used to replace the corresponding restriction fragment of pUK200I to create pGH068.
(ix) Cloning of the gusA reporter gene.
A 1,806-bp fragment was amplified using the primer pair GUS_5′ and GUS_3′, with pBI101 plasmid DNA (Clontech/TaKaRa Bio Europe, France) as the template. The resulting PCR fragment was then cloned into NcoI-digested (blunted) and SmaI-digested pFI2716 to create pGH075. The SD region present in pGH075 was replaced with the 5′ UTR from pGH020 by way of splice overlap extension PCR. A 555-bp SphI- and SnaBI-digested fragment of the splice PCR product was then used to replace the corresponding 643-bp fragment of pGH075 to create pGH076.
The insert regions for all plasmids constructed as described above were sequenced in full using the BigDye Terminator v3.1 cycle sequencing kit (Life Technologies Ltd., United Kingdom). Table S3 in the supplemental material summarizes all plasmid constructs.
Preparation of cell extracts.
A 20-ml culture of B. ovatus or E. coli was grown to an optical density at 600 nm (OD600) of 1.0. Cells from two independent cultures were harvested by centrifugation at 10,000 × g for 25 min at 4°C. Cell pellets were washed once in 0.2 M Tris-HCl (pH 7.2) before freezing at −20°C. Each thawed cell pellet was resuspended in 250 μl of 0.2 M Tris-HCl (pH 7.2), and cell disruption via sonication was performed using eight 10-s pulses (amplitude, 6 micrometers), with 30-s pauses on ice between each pulse. Cell extracts were obtained in the form of supernatants after centrifugation at 14,000 × g for 30 min at 4°C. For L. lactis, cultures were grown to an OD600 of 0.5 before induction with 1 ng/ml nisin A (Aplin & Barret Ltd., Beaminster, United Kingdom) in order to switch on the nisA promoter. Following a 2-h induction period, cells from two independent cultures were collected by centrifugation at 10,000 × g for 15 min at 4°C using sample volumes equivalent to 37 OD600 units. Cell extracts were prepared principally as described by Wegmann et al. (27).
Enzyme assays and protein determination.
Peptidase I activity was assayed using 50 μl of cell-free samples or dilutions thereof in 0.2 M Tris-HCl (pH 7.2) and 0.1% bovine serum albumin fraction V (Sigma, United Kingdom), with 0.7 mM l-proline p-nitroanilide trifluoroacetate salt (Sigma) as the substrate. The release of p-nitrophenol was measured at 405 nm at 32°C. β-Glucuronidase (GUS) activity was assayed using 25 μl of cell-free samples or dilutions thereof in 50 mM NaH2PO4 (pH 7.0), 10 mM β-mercaptoethanol, 1 mM EDTA, and 0.1% Triton X-100, with 1.25 mM 4-nitrophenol-β-d-glucuronide (PNPG) as the substrate. The release of p-nitrophenol was measured at A405 at 37°C. The protein concentrations of samples were measured at A600 using the Bio-Rad protein assay (Bio-Rad, United Kingdom) with bovine serum albumin (New England BioLabs UK Ltd.) as the standard. Specific enzyme activity is expressed as nanomoles of nitrophenol released from the chromogenic substrate per milligram of protein per minute. The means and standard deviations presented are based upon two biological replicates with three technical replicates each.
RNA purification.
To isolate RNA for use in quantitative reverse transcription-PCR (qRT-PCR) experiments, B. ovatus cells were grown to an OD600 of 1.0. Subsequently, two independent 1-ml culture samples were removed and added to 200 μl of phenol/ethanol (5:95), and the cell pellet was collected by centrifugation at 16,000 × g for 30 min at 4°C, after a 30-min incubation period on ice. RNA purification was carried out using the SV total RNA isolation system (Promega UK Ltd.) and following the manufacturer's instructions, with an additional DNase treatment step using a Turbo DNA-free kit (Life Technologies Ltd., United Kingdom).
qRT-PCR.
Transcript levels were assessed through qRT-PCR using a SYBR green qRT-PCR kit (Sigma, United Kingdom) in combination with the primer pair Q_pepI_5′ (5′-CGGCTATGAGTACACTGA-3′) and Q_pepI_3′ (5′-TGGCAAGTGATCGTACAT-3′) and, for the reference gene, the primer pair Q_tetQ_5′ (5′-GGCTGATTATTGGAATACGATA-3′) and Q_tetQ (5′-CAACAACTCATTGATACCGATA-3′). A one-step qRT-PCR procedure was followed according to the manufacturer's instructions, except that the reaction mixtures were scaled down to a final volume of 10 μl. The primers were used at a concentration of 400 nM. The amplifications were carried out with an ABI 7500 TaqMan system (Life Technologies Ltd., United Kingdom) with the following cycle parameters: one cycle of 44°C for 30 min, one cycle of 95°C for 10 min, and 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 35 s. After completion of the amplification cycles, melting curve data were generated to determine the amplification quality. The relative abundance of pepI mRNA was calculated by the threshold cycle (ΔΔCT) method. The means and standard deviations presented are based upon two biological replicates with three technical replicates each.
Determining 3′ ends of the 16S rRNA through 3′ rapid amplification of cDNA ends (RACE).
B. ovatus cells were grown to an OD600 of 1.0 and subsequently harvested by centrifugation. Cells were resuspended in 10 ml of RNAlater solution (Life Technologies Ltd., Paisley, United Kingdom) and stored overnight at 4°C. Cells were then harvested through centrifugation and subsequently sonicated in 1 ml lysis buffer (50 mM HEPES, 14 mM β-mercaptoethanol, 6 mM MgCl2, 0.5 mM CaCl2, 0.1 mM EDTA [pH 7.5]) to break the cells. Subsequently, 10 U of RQ1 DNase I was added, and after centrifugation at 20,000 × g for 30 min at 4°C, the cell extract was loaded into a Beckmann Ultra-Clear centrifuge tube filled with 10 ml lysis buffer containing 10% sucrose. After centrifugation in a Beckmann SW41 rotor for 5 h at 288,000 × g and 20°C, the supernatant was decanted, the ribosomal pellet was resuspended in 1 ml TRI reagent (Molecular Research Center Inc., Cincinnati, OH), and the rRNA was isolated following the manufacturer's recommendations. The purified rRNA (200 ng) was subsequently polyadenylated using E. coli poly(A) polymerase (New England BioLabs, Ipswich, MA) according to the manufacturer's protocol. After phenol-chloroform extraction, a ThermoScript RT-PCR system (Life Technologies Ltd., Paisley, United Kingdom) was used according to the manufacturer's recommendation to reverse transcribe the polyadenylated rRNA and subsequently amplify the 3′ ends of the cDNA product using the primers 5′-GCATGGTTGTCGTCAGCTCG-3′ and 5′-(TTTTT)6-3′. The resulting amplicons were gel purified on a 5% acrylamide gel, electroeluted, and subsequently used as the template for a second PCR using the primers 5′-TACCGGAAGGTGCGGCTGGAAC-3′ and 5′-(TTTTT)6-3′. Following gel purification, the fragments were cloned into pJET1.2 (Fermentas, St. Leon-Rot, Germany) and subsequently sequenced using pJET forward and reverse sequencing primers.
RESULTS
Comparative analysis of the 3′ ends of 16S rRNAs.
To identify optimal SD sequences for our expression vectors, we used the Bacteroidales genome sequences available in public databases. Figure 1A shows alignment of the 3′ ends of the 16S rRNA gene sequences extracted from the genome sequences of Bacteroides thetaiotaomicron VPI 5482 (29), Bacteroides vulgatus ATCC 8482 (30), Bacteroides ovatus V975 (U. Wegmann, unpublished data), Bacteroides fragilis NCTC 9343 (30), Prevotella denticola F0289 (CP002589), and Prevotella bryantii B14 (31) with the sequenced 3′ ends of 16S rRNA sequences from Escherichia coli (32, 33), Bacillus subtilis (34), Bacillus stearothermophilus (35) (now Geobacillus stearothermophilus), and Lactococcus lactis (36), which demonstrates the perfect sequence conservation with regard to the 3′ ends of the 16S rRNAs. In the cases of B. thetaiotaomicron, B. vulgatus, P. denticola, and P. bryantii, the 16S rRNA genes annotated in the respective genomes were extended with additional downstream sequence data on the basis that all rRNA gene operons present in the genome were followed by the same stretch of sequence that would be transcribed into the same 3′ ends as those reported for Gram-positive 16S rRNA sequences.
Fig 1.

(A) ClustalW alignment of 16S rRNA gene sequences extracted from complete genome sequences with the sequenced 3′ ends of 16S rRNAs from E. coli, B. stearothermophilus, B. subtilis, and L. lactis IL1403. Lowercase letters indicate that the annotated 16S rRNA gene sequence has been extended beyond the annotation in the genome. (B) Sequence logos of ribosomal protein gene 5′ UTRs spanning the region from positions −30 to +6 created with WebLogo 3.3 with the CG composition adjustment option turned on. The numbers of genes used to create the individual logos are as described in Table 1 or as follows: Bacillus subtilis, 60; Lactococcus lactis, 55; and Escherichia coli, 55. As the software designates the first nucleotide of the start codon as position 0, the logos are numbered from −30 to +5. Dashed line, SD core sequence (5′-GGAGG-3′).
Analysis of 5′ UTRs of ribosomal protein genes.
Subsequently, we extracted the nucleotide sequences of genes encoding ribosomal proteins from positions −30 to +5 (the first base of the start codon is position 0), from the Bacteroides genome sequences mentioned above, and the genome sequences of Bacillus subtilis (37), Escherichia coli (38), and Lactococcus lactis (39). Based on codon bias, the ribosomal proteins of most bacteria are predicted to be highly expressed and, aside from optimized codon usage, this is achieved through the presence of an SD sequence that has higher affinity for the 3′ end of the 16S rRNA and occurs at closer-to-optimal spacing than the SD sequences of predicted moderately expressed genes (40). First, we inspected the Bacteroidales 5′ UTR with respect to the presence of Shine-Dalgarno-like sequences, the results of which are summarized in Table 1. In contrast to the well-documented prevalence of these sequences in the 5′ UTRs of ribosomal genes from many bacterial species, which often reaches 80% (40), the respective 5′ UTRs of the examined Bacteroidales genes contained Shine-Dalgarno-like sequences to a far lesser extent (18 to 28%). Interestingly, in comparison to the group of genes that lack such sequences, this group of genes contained an equal or greater proportion of members with a codon adaptation index (CAI) above the average calculated from the CAIs of all ribosomal genes of the organism in question, which would indicate high expression levels, also requiring efficient translation initiation.
Table 1.
Distribution of Shine-Dalgarno sequences in Bacteroides ribosomal protein genes
| Species | No. of ribosomal protein genes | % of ribosomal protein genes with an SD sequence in the 5′ UTRa | % of genes with a CAI above the mean |
|
|---|---|---|---|---|
| Genes with an SD sequence | Other genes | |||
| Bacteroides ovatus | 45 | 20.0 | ||
| Bacteroides thetaiotaomicron | 53 | 18.8 | 80 | 42 |
| Bacteroides fragilis | 52 | 18.8 | 50 | 50 |
| Bacteroides vulgatus | 51 | 17.6 | 60 | 45 |
| Prevotella denticola | 53 | 28.3 | 47 | 37 |
| Prevotella′ bryantii | 48 | 22.9 | 50 | 50 |
SD sequences are GGAGG, GGAG, and AGG. The 5′ UTR comprises positions −1 to −20.
Due to the small number of canonical SD sequences in the 5′ UTRs of Bacteroidales ribosomal genes, WebLogo 3.3 (41) was used to identify the optimal ribosomal binding site consensus sequence for the respective organism by creating sequence logos (42) of these regions without any prior alignment. The resulting sequence logos corresponded well with the 3′ ends of the 16S rRNA for B. subtilis, L. lactis (both bacilli), and E. coli (gammaproteobacteria) (Fig. 1B), and the presence of the SD core sequence (5′-GGAGG-3′) was detected (Fig. 1B, dashed lines). In contrast, the results for all Bacteroidales species revealed a dichotomy between the RBS consensus sequence logos observed for their ribosomal genes and the 16S rRNA 3′ ends predicted from their genomes, as the logos did not contain the core SD sequence (Fig. 1B). Instead, they are enriched in adenosine and, to a far lesser extent, thymidine. Positions −12 to −18 with respect to the start codon stand out because of the strong conservation of adenosine and thymidine peaks in this region. Furthermore, there is a clear dip in the sequence conservation leading up to this region, albeit at position −8 for B. vulgatus instead of position −10/11 for the other Bacteroides species. There are other positions with less striking conservation, for example, positions −1, −3, and −7, and there is a conspicuous stretch of adenosine and thymidine enrichment from positions −19 to −28, which might be important.
Determining the 3′ ends of the 16S rRNA.
Based on their unique sequence logos, we considered the possibility that the 3′ ends of the Bacteroides 16S rRNAs might be different from their counterparts in other eubacteria due to, for example, different RNase processing or posttranscriptional modification. We decided therefore to determine the 3′ end of the B. ovatus 16S rRNA through 3′ rapid amplification of cDNA ends (RACE). The corresponding sequencing results confirmed the 3′ ends predicted from the genome sequence of the organism as 5′-ACCTCCTTTCT-3′, which is identical to the sequences determined for B. subtilis and L. lactis (Fig. 1).
Proline iminopeptidase I reporter and expression vectors for Bacteroides.
We constructed a series of different expression vectors using pepI as a novel marker gene to define the Bacteroides-specific translation initiation signal in detail, to create high- and low-level protein expression vectors for Bacteroides, and to demonstrate the dynamic range of the pepI reporter. Figure 2 shows a schematic representation of the expression vectors generated. They fall into four groups. Group 1 is based on the Gram-positive expression cassette from the lactococcal expression vector pUK200 (27), allowing for translational fusion of the gene of interest with the strong SD sequence from the lactococcal nisA gene (43) under the control of the P1 promoter from the 16S rRNA gene of B. thetaiotaomicron. Group 2 is identical to group 1 apart from the region between the transcription initiation site and the start codon, which carries various derivatives of the 5′ UTR of orf1 from the xylanase operon of B. ovatus V975. Alignment of the orf1 5′ UTR with the B. ovatus ribosomal protein gene 5′ UTR logo is presented in Fig. S2 in the supplemental material. Groups 3 and 4 represent vectors from groups 1 and 2, respectively, carrying the gusA reporter gene instead of pepI.
Fig 2.

Schematic representation of the different expression vectors with the region stretching from the promoter to the start codon expanded to the nucleotide level. Tbrnq represents the terminator of the Lactobacillus delbrueckii subsp. lactis brnQ gene. Positions −33 and −7 and the transcription initiation site (position +1) of the P1 promoter (17) are in bold capital letters, ribosomal binding sites are underlined and in capital letters, and nucleotide changes are underlined and in bold lowercase letters. The 3′ end of the 16S rRNA (3′-UCUUUCCUCCA-5′) is shown for comparison. Groups 3 and 4 represent vectors from groups 1 and 2, respectively. *, pGH001 carrying the gusA reporter gene instead of pepI (i.e., pGH075); ^, pGH022 carrying the gusA reporter gene instead of pepI (i.e., pGH076).
Figure 3A shows the proline iminopeptidase I (PepI) activity present in wild-type strains and the activities obtained with the various expression vectors (the values reported were not normalized for differences in transcript levels). The first thing of note was the total lack of any PepI activity in the vector-only control (pFI2716), demonstrating the complete absence of this enzyme activity in B. ovatus. B. thetaiotaomicron VPI 5482 extracts also showed no activity, whereas extracts from B. fragilis NCTC 9343 displayed a negligible amount (2.7 nmol mg−1 min−1). The expression levels achieved in B. ovatus with the various constructs demonstrated the excellent dynamic range of the pepI gene, extending from a modest 67 nmol mg−1 min−1 with pGH006 to a medium level of 2,425 nmol mg−1 min−1 achieved with pGH078 and a high level of expression of 11,174 nmol mg−1 min−1 in the case of pGH023. For example, the expression level achieved with the xylanase operon orf1 5′ UTR (positions −1 to −20) for the pepI gene (pGH022) is high enough to visualize the corresponding protein by SDS-polyacrylamide gel electrophoresis (data not shown), something that, as far as we are aware, has not been achieved in Bacteroides with any other protein and expression system.
Fig 3.

(A, D, E, and F) Specific enzyme activities of cell extracts. (B) Relative mRNA levels determined by qRT-PCR (pGH006 was the calibrator sample, i.e., relative quantity = 1). (C) Specific enzyme activities normalized with respect to relative mRNA levels. Dashed lines separate the different construct groups. Error bars represent standard deviations based on 2 biological replicates with 3 technical replicates each.
Transcript levels.
Although all constructs carry the P1 promoter from the 16S rRNA gene of B. thetaiotaomicron, there is scope for the differences in transcript levels produced by these constructs due to the sequence variations in their 5′ UTRs. Therefore, we determined transcript levels through quantitative PCR, the results of which are shown in Fig. 3B. Most constructs produced similar levels of transcript, with the noticeable exceptions of pGH001, pGH004, and pGH006. Plasmid pGH006 showed the lowest transcript level and served as the calibrator in our qRT-PCR experiments. We identified a potential RNase E site at the 5′ end of the transcripts of these 3 vectors, explaining the stark difference in mRNA levels. Removal of this part of the sequence in plasmid pGH078 stabilized the respective mRNA (Fig. 3B). In comparison to pGH022, significantly lower levels of transcript were also determined for pGH039 and pGH040 (two-sample two-tailed t test, P < 0.05).
Defining the Bacteroides ribosomal binding site.
To define the Bacteroides ribosomal binding site, we needed to look at the translation initiation efficiency represented by PepI activity while minimizing the effects of different transcript levels. To this end, PepI expression data were normalized with respect to the qRT-PCR results (Fig. 3C). The normalized PepI expression levels, representing the translation initiation efficiency, were 198 nmol mg−1 min−1 with the lactococcal SD sequence (pGH001) and decreased markedly as the distances between the start codon and the SD sequence were progressively reduced (see pGH004 and pGH006 in Fig. 3C). The shortening of the 5′ UTR to 22 nt in pGH078 resulted in a 45% increase in activity, and exchanging the core SD sequence-containing region for the 5′ UTR of orf1 from the xylanase operon of B. ovatus V975 (pGH10) led to a slight decrease in PepI activity to 232 nmol mg−1 min−1. This was increased 3.2-fold when the stretch of nucleotides between positions +2 (transcription) and −30 (translation) was deleted (pGH023), which, with a normalized PepI activity level of 752 nmol mg−1 min−1, represents the most efficiently initiating RBS in the series. The progressive shortening of the RBS region to positions −24 (pGH079), −20 (pGH022), −18 (pGH042), and −16 (pGH041) resulted in successive decreases in PepI activity levels to 74, 67, 66, and 61%, respectively, of the peak activity. Deletion to position −10 (pGH040) reduced the peptidase activity level to 25% of the peak activity obtained with pGH023, but the activity still was twice that of the negative control (pGH039), which had only two cytidine residues in the pepI 5′ UTR. A two-sample two-tailed t test analysis was performed on the above-mentioned comparisons and, apart from the comparison of pGH006 with pGH004, all the differences were significant (P < 0.05). To define the RBS further, nucleotide exchanges were made at positions −11, −10, and −9, none of which had a substantial effect on reporter gene expression levels in comparison to the levels obtained with the equivalent plasmid pGH022. With the core RBS defined as ranging from positions −12 to −18, the spacing between the RBS and the start codon in construct pGH022 was changed. Moving the RBS closer by 2 nt (pGH048) reduced expression levels by one-third (P < 0.05), and a move of 4 nt (pGH049) resulted in the halving of expression levels (P < 0.05), demonstrating the importance of the spacing between the Bacteroides ribosomal binding site and the start codon.
To ensure that our results were not dependent on this particular reporter gene, we constructed gusA (uidA) equivalents for some of the key pepI-carrying constructs (pGH075* and pGH076^ in Fig. 2). In general, the specific enzyme activity observed with these constructs was more than 1 order of magnitude lower than that seen with the pepI reporter (Fig. 3D), demonstrating the superior performance of the pepI reporter gene. With regard to the expression levels obtained with the classical SD sequence or the Bacteroides ribosomal binding site, the findings with the pepI reporter gene were confirmed, in that employment of the latter was far more efficient and resulted in 23-fold-higher specific GusA activity.
Use of Bacteroides ribosomal binding site in other bacteria.
In light of the high expression levels achieved with the RBS originating from the B. ovatus xylanase operon orf1, we determined how it would perform in bacteria that normally employ the SD sequence for translation initiation. Two organisms were investigated: E. coli, which has less stringent SD sequence requirements and employs the ribosomal protein S1 to aid translation initiation (44), and Gram-positive L. lactis, which has more stringent requirements. The L. lactis cultures were induced with nisin. In E. coli, some expression might be derived from the Bacteroides P1 promoter, although previous research has shown that this promoter does not work very well in E. coli (17). We analyzed the upstream region of the pepI gene of the constructs in question with the SoftBerry promoter prediction program (http://linux1.softberry.com/berry.phtml), which returned 2 plausible E. coli promoters that, in our opinion, are the more likely sources of gene expression. As expected, the classical SD sequence (5′-GGAGG-3′) present in pGH078 and the lactococcal expression vector pUK200 performed best in both of these bacteria, but interestingly, the ribosomal binding site originating from Bacteroides delivered 19% of the PepI activity achieved with the SD sequence in E. coli (pGH022 in Fig. 3E) and the respective value was 30% in the case of L. lactis (pGH68 in Fig. 3F).
DISCUSSION
We have analyzed the upstream regions of ribosomal genes in Bacteroides to identify optimized ribosomal binding sites for the construction of expression vectors using a sequence logo approach. Our results, though more detailed, are in keeping with the findings by Accetto and Avgustin (20), who used the total gene contents of the organisms for their logos, containing alien as well as highly and moderately expressed genes. This explains why our logos are more extensive and detailed, as ribosomal genes are predicted to be among the most highly expressed genes and show high levels of occurrence of strong SD sequences (40), therefore providing a better starting point for identifying strong ribosomal binding sites through a sequence logo approach.
Some authors have suggested that translation initiation in the Bacteroidetes phylum is facilitated solely through ribosomal protein S1 (RPS1) (20, 45). It has been demonstrated that translational enhancers derived from the tobacco mosaic virus, whose sequence fits the binding criteria for RPS1 (no guanosine) (46), can replace the SD sequence in a variety of Gram-negative bacteria (47). However, the ability of RPS1 to initiate translation by itself has not been demonstrated (48).
In examining regions upstream of the ribosomal genes of B. thetaiotaomicron VPI 5482, B. ovatus V975, B. fragilis NCTC 9343, B. vulgatus ATCC 8482, P. bryantii B14, and P. denticola F0289, we identified the core SD sequence in 31 of a total of 309 genes from position −1 to position −20 but not at all from position −21 to position −40. Interestingly, the presence in front of certain genes is preserved (see Table S4 in the supplemental material), for example, the genes for ribosomal proteins S18 and L6 in all 6 strains containing the core SD sequence upstream of the respective start codon. In contrast, the occurrence of 5′-GGAGG-3′ is evenly distributed between these two regions with regard to the total gene content and occurs at a rate of 2.8% in region −1 to −40 (B. ovatus not included). Strengthening the notion that these particular ribosomal genes are highly expressed is the fact that in the five members of Bacteroidales examined, an equal or greater proportion of the genes (Table 1) that possess the core SD sequence or remnants thereof in the position −1 to −20 region also display a codon adaptation index that is above the average for the ribosomal genes of the bacterial species in question. Together with the results for the reporter gene expression plasmid pGH078, these findings provide clear evidence that the core SD sequence enables efficient translation initiation in Bacteroides, although at rates somewhat lower than those for the Bacteroides ribosomal binding site defined here. It is clear from our data that B. ovatus is able to initiate translation in the absence of the SD sequence or the ribosomal binding site defined here (pGH040 in Fig. 3). Indeed, the presence of 2 nt in front of the ATG start codon (pGH039 in Fig. 3) is sufficient. This so-called leaderless translation initiation is not uncommon in bacteria (49).
The mRNA produced from constructs pGH001, pGH004, and pGH006 is very labile, most probably due to an RNase E site near the 5′ end of the molecule. Normalization of the PepI expression data with respect to the corresponding transcript data reveals that spacing is a factor affecting translation initiation efficiency for the canonical SD sequence, as described for other bacteria (50–52). A spacing effect can also be seen with regard to the Bacteroides ribosomal binding site (comparing pGH022 and pGH049). Assuming that RPS1 binds to this sequence, this is somewhat different from the situation for the translation-enhancing effect that was described by Boni et al. for RPS1 (46), where the U-rich binding sites are described as a “drifting” element of the primary mRNA structure. In a review by Kozak (53), the minimization of secondary structure in the 5′ UTR is put forward as an alternative explanation for the enhancing effects of these A/U-rich sequences. With this in mind, we compared the results of expression vector pairs that differed only in the extent of their 5′ UTRs (pGH001 with pGH078 and pGH010 with pGH023). In both cases, the efficiency of translation initiation is negatively affected by the presence of an extended 5′ UTR, leading to losses of 31 and 69% of PepI activity, respectively (Fig. 3). Both 5′ UTRs contain secondary structure elements as determined with mFold (54), with combined ΔG values of −20 and −38 kcal mol−1, respectively. Whether these predicted secondary structure elements or the sheer presence of a longer 5′ UTR is responsible for the knockdown in translation initiation cannot be elucidated from our data.
Gram-positive bacteria such as B. subtilis and L. lactis were thought to lack RPS1 (55–57). It is now known that B. subtilis has a counterpart, YpfD (58), that is not involved in translation. L. lactis also carries an rpsA gene on its chromosome (59), although Salah et al. showed that the corresponding gene product lacks the typical ribosome binding domain (domain 1), supporting the idea that the protein cannot bind to the ribosome (60). This protein is therefore unlikely to be involved in translation initiation in L. lactis. Salah et al. (60) also identified the full complement of domains in the B. fragilis RPS1 protein supporting its role in translation initiation. Together, these findings make it difficult to see how translation initiation at the Bacteroides RBS in L. lactis might also be achieved through the rpsA gene product, especially as the expression level achieved amounts to 30% of the level seen with the strong SD sequence of the nisA gene. This raises the questions of how translation initiation is achieved in L. lactis under these circumstances and whether RPS1 is the sole entry point for mRNAs of this type in Bacteroidetes unless RPS1 plays a role in L. lactis or there are two distinct systems present in these organisms.
Reporter genes are useful tools for studying the expression of genes of interest in response to extrinsic or intrinsic factors. Ideally, the gene products are easily detectable, and the measured activity is absent in the organism of interest to avoid background problems at low levels of expression. A number of reporter genes have been described for Bacteroides, although they all have limitations and suffer from high costs and complexity (chloramphenicol acetyltransferase assay), high background levels under certain conditions or in certain organisms (β-glucuronidase and xylosidase) (25, 61), relatively low levels of enzyme activity, or the need for specialized equipment to detect fluorescence or luminescence. The pepI reporter gene described here offers a simpler, more sensitive, and cheaper alternative. Measured PepI activities ranging from 102 to 104 nmol mg−1 min−1 demonstrate a greater dynamic range than the E. coli gusA gene (10 to 102 nmol mg−1 min−1) or the B. ovatus xa gene (38 to 75 nmol mg−1 min−1), measured in Bacteroides uniformis and B. thetaiotaomicron, respectively (61). Also, due to the absence of any significant constitutive PepI activity in B. thetaiotaomicron, B. ovatus, and B. fragilis, it is possible to measure low levels of expression in these organisms. The relatively small size of the pepI gene and its product (294 amino acids [aa]), in comparison with the reporter gene products GusA (602 aa) and LuxAB (683 aa), puts a smaller burden on the host cells, which can be a problem if strong promoters are examined, and is an advantage in terms of plasmid stability.
In summary, this work gives for the first time a detailed view of the unique Bacteroides ribosomal binding site. While we do not demonstrate direct binding to the ribosome, our findings are supported by extensive expression data. Based on the respective sequence logos (see Fig. S1 in the supplemental material), we predict that our findings apply to a great number of, if not all, members of the Bacteroidetes phylum. Furthermore, we show that canonical SD sequences initiate translation in B. ovatus and most probably in other Bacteroidales species as well. We have generated a set of expression vectors that allow the expression of a gene of interest at appropriate levels in these organisms. They are presented in Fig. 4 and offer different combinations of expression levels and antibiotic resistance genes.
Fig 4.
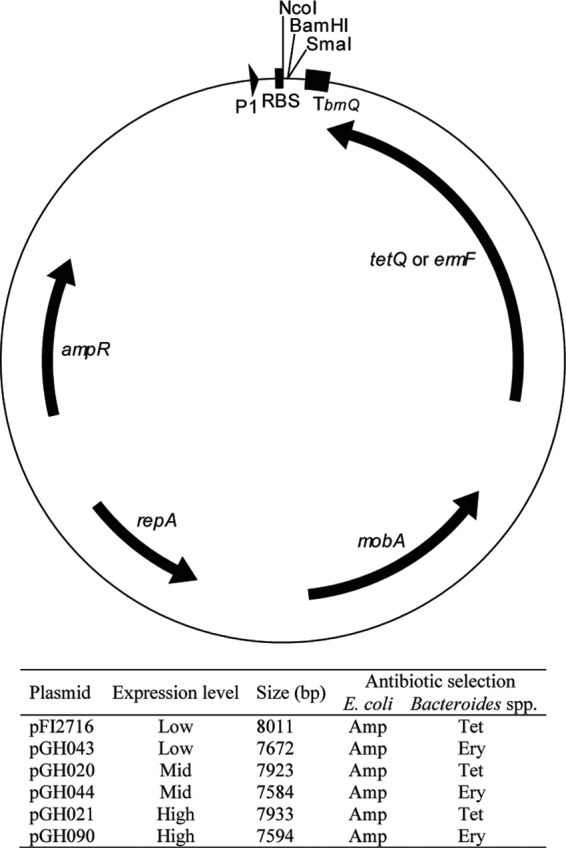
Schematic representation of expression vectors. Genes are shown as arrows on the inner circle. On the outer circle, P1 represents the B. thetaiotaomicron promoter P1, RBS represents different ribosomal binding sites, and Tbrnq represents the terminator of the L. delbrueckii subsp. lactis brnQ gene. The table summarizes the characteristics of the different expression vectors. The start codon containing an NcoI site allows for translational fusion of the genes of interest.
ACKNOWLEDGMENTS
This work was supported in part by the Biotechnology and Biological Sciences Research Council (BBSRC) in support of the Gut Health and Food Safety research program (grant BB/J004529/1).
We thank Bernhard Henrich (University of Kaiserslautern, Germany) for providing plasmid pUK200 and the pepI gene from Lactobacillus delbrueckii subsp. lactis. We are grateful to Terry Whitehead for the gift of the plasmid pMJF1.
Footnotes
Published ahead of print 18 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03086-12.
REFERENCES
- 1. Berg RD. 1996. The indigenous gastrointestinal microflora. Trends Microbiol. 4:430–435 [DOI] [PubMed] [Google Scholar]
- 2. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Dore J. 1999. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl. Environ. Microbiol. 65:4799–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, Ugarte E, Munoz-Tamayo R, Paslier DLE, Nalin R, Dore J, Leclerc M. 2009. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 11:2574–2584 [DOI] [PubMed] [Google Scholar]
- 5. Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze XL, Brown D, Stares MD, Scott P, Bergerat A, Louis P, McIntosh F, Johnstone AM, Lobley GE, Parkhill J, Flint HJ. 2011. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 5:220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flint H, Scott K, Duncan S, Louis P, Forano E. 2012. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3:289–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cebra JJ. 1999. Influences of microbiota on intestinal immune system development. Am. J. Clin. Nutr. 69:1046S–1051S [DOI] [PubMed] [Google Scholar]
- 8. Xavier RJ, Podolsky DK. 2007. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–434 [DOI] [PubMed] [Google Scholar]
- 9. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023 [DOI] [PubMed] [Google Scholar]
- 10. Qin JJ, Li RQ, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li JH, Xu JM, Li SC, Li DF, Cao JJ, Wang B, Liang HQ, Zheng HS, Xie YL, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu HM, Yu C, Li ST, Jian M, Zhou Y, Li YR, Zhang XQ, Li SG, Qin N, Yang HM, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J, MetaHIT Consortium 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woese CR. 1987. Bacterial evolution. Microbiol. Rev. 51:221–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kunsman JE, Caldwell DR. 1974. Comparison of the sphingolipid content of rumen Bacteroides species. Appl. Microbiol. 28:1088–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bayley DP, Rocha ER, Smith CJ. 2000. Analysis of cepA and other Bacteroides fragilis genes reveals a unique promoter structure. FEMS Microbiol. Lett. 193:149–154 [DOI] [PubMed] [Google Scholar]
- 14. Vingadassalom D, Kolb A, Mayer C, Rybkine T, Collatz E, Podglajen I. 2005. An unusual primary sigma factor in the Bacteroidetes phylum. Mol. Microbiol. 56:888–902 [DOI] [PubMed] [Google Scholar]
- 15. Smith CJ, Rogers MB, McKee ML. 1992. Heterologous gene expression in Bacteroides fragilis. Plasmid 27:141–154 [DOI] [PubMed] [Google Scholar]
- 16. Shine J, Dalgarno L. 1975. Determinant of cistron specificity in bacterial ribosomes. Nature 254:34–38 [DOI] [PubMed] [Google Scholar]
- 17. Mastropaolo MD, Thorson ML, Stevens AM. 2009. Comparison of Bacteroides thetaiotaomicron and Escherichia coli 16S rRNA gene expression signals. Microbiology 155:2683–2693 [DOI] [PubMed] [Google Scholar]
- 18. Tribble GD, Parker AC, Smith CJ. 1999. Genetic structure and transcriptional analysis of a mobilizable, antibiotic resistance transposon from Bacteroides. Plasmid 42:1–12 [DOI] [PubMed] [Google Scholar]
- 19. Band L, Henner DJ. 1984. Bacillus subtilis requires a “stringent” Shine-Dalgarno region for gene expression. DNA 3:17–21 [DOI] [PubMed] [Google Scholar]
- 20. Accetto T, Avgustin G. 2011. Inability of Prevotella bryantii to form a functional Shine-Dalgarno interaction reflects unique evolution of ribosome binding sites in Bacteroidetes. PLoS One 6:e22914 doi:10.1371/journal.pone.0022914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klein JR, Schmidt U, Plapp R. 1994. Cloning, heterologous expression, and sequencing of a novel proline iminopeptidase gene, pepI, from Lactobacillus delbrueckii subsp. lactis DSM 7290. Microbiology 140:1133–1139 [DOI] [PubMed] [Google Scholar]
- 22. Yaron A, Naider F. 1993. Proline-dependent structural and biological properties of peptides and proteins. Crit. Rev. Biochem. Mol. Biol. 28:31–81 [DOI] [PubMed] [Google Scholar]
- 23. Shoemaker NB, Getty C, Gardner JF, Salyers AA. 1986. Tn4351 transposes in Bacteroides spp. and mediates the integration of plasmid R751 into the Bacteroides chromosome. J. Bacteriol. 165:929–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277 [DOI] [PubMed] [Google Scholar]
- 25. Feldhaus MJ, Hwa V, Cheng Q, Salyers AA. 1991. Use of an Escherichia coli β-glucuronidase gene as a reporter gene for investigation of Bacteroides promoters. J. Bacteriol. 173:4540–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tancula E, Feldhaus MJ, Bedzyk LA, Salyers AA. 1992. Location and characterization of genes involved in binding of starch to the surface of Bacteroides thetaiotaomicron. J. Bacteriol. 174:5609–5616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wegmann U, Klein JR, Drumm I, Kuipers OP, Henrich B. 1999. Introduction of peptidase genes from Lactobacillus delbrueckii subsp. lactis into Lactococcus lactis and controlled expression. Appl. Environ. Microbiol. 65:4729–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith CJ, Rollins LA, Parker AC. 1995. Nucleotide sequence determination and genetic analysis of the Bacteroides plasmid, pBI143. Plasmid 34:211–222 [DOI] [PubMed] [Google Scholar]
- 29. Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. 2003. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299:2074–2076 [DOI] [PubMed] [Google Scholar]
- 30. Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, Henrissat B, Coutinho PM, Minx P, Latreille P, Cordum H, Van Brunt A, Kim K, Fulton RS, Fulton LA, Clifton SW, Wilson RK, Knight RD, Gordon JI. 2007. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 5:e156 doi:10.1371/journal.pbio.0050156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Purushe J, Fouts DE, Morrison M, White BA, Mackie RI, Coutinho PM, Henrissat B, Nelson KE, Bacteria NACR. 2010. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb. Ecol. 60:721–729 [DOI] [PubMed] [Google Scholar]
- 32. Brosius J, Palmer ML, Kennedy PJ, Noller HF. 1978. Complete nucleotide sequence of a 16S ribosomal RNA gene from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 75:4801–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ehresmann C, Stiegler P, Mackie GA, Zimmermann RA, Ebel JP, Fellner P. 1975. Primary sequence of the 16S ribosomal RNA of Escherichia coli. Nucleic Acids Res. 2:265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murray CL, Rabinowitz JC. 1982. Nucleotide sequences of transcription and translation initiation regions in Bacillus phage phi-29 early genes. J. Biol. Chem. 257:1053–1062 [PubMed] [Google Scholar]
- 35. Van Charldorp R, Van Kimmenade AM, Van Knippenberg PH. 1981. Sequence and secondary structure of the colicin fragment of Bacillus stearothermophilus 16S ribosomal RNA. Nucleic Acids Res. 9:4909–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiaruttini C, Milet M. 1993. Gene organization, primary structure and RNA processing analysis of a ribosomal RNA operon in Lactococcus lactis. J. Mol. Biol. 230:57–76 [DOI] [PubMed] [Google Scholar]
- 37. Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Dusterhoft A, Ehrlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim SY, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guiseppi G, Guy BJ, Haga K, Haiech J, Harwood CR, Henaut A, Hilbert H, Holsappel S, Hosono S, Hullo MF, Itaya M, Jones L, Joris B, Karamata D, Kasahara Y, Klaerr-Blanchard M, Klein C, Kobayashi Y, Koetter P, Koningstein G, Krogh S, Kumano M, Kurita K, Lapidus A, Lardinois S, Lauber J, Lazarevic V, Lee SM, Levine A, Liu H, Masuda S, Mauel C, Medigue C, Medina N, Mellado RP, Mizuno M, Moestl D, Nakai S, Noback M, Noone D, O'Reilly M, Ogawa K, Ogiwara A, Oudega B, Park SH, Parro V, Pohl TM, Portetelle D, Porwollik S, Prescott AM, Presecan E, Pujic P, Purnelle B, Rapoport G, Rey M, Reynolds S, Rieger M, Rivolta C, Rocha E, Roche B, Rose M, Sadaie Y, Sato T, Scanlan E, Schleich S, Schroeter R, Scoffone F, Sekiguchi J, Sekowska A, Seror SJ, Serror P, Shin B-S, Soldo B, Sorokin A, Tacconi E, Takagi T, Takahashi H, Takemaru K, Takeuchi M, Tamakoshi A, Tanaka T, Terpstra P, Tognoni A, Tosato V, Uchiyama S, Vandenbol M, Vannier F, Vassarotti A, Viari A, Wambutt R, Wedler E, Wedler H, Weitzenegger T, Winters P, Wipat A, Yamamoto H, Yamane K, Yasumoto K, Yata K, Yoshida K, Yoshikawa H-F, Zumstein E, Yoshikawa H, Danchin A. 1997. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249–256 [DOI] [PubMed] [Google Scholar]
- 38. Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462 [DOI] [PubMed] [Google Scholar]
- 39. Wegmann U, O'Connell-Motherway M, Zomer AL, Buist G, Shearman C, Canchaya C, Ventura M, Goesmann A, Gasson M, Kuipers O, van Sinderen D, Kok J. 2007. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 189:3256–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma J, Campbell A, Karlin S. 2002. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J. Bacteriol. 184:5733–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider TD, Stephens RM. 1990. Sequence Logos: a new way to display consensus sequences. Nucleic Acids Res. 18:6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuipers OP, Beerthuyzen MM, Siezen RJ, Devos WM. 1993. Characterization of the nisin gene cluster nisABTCIPR of Lactococcus lactis: requirement of expression of the nisA and nisI genes for development of immunity. Eur. J. Biochem. 216:281–291 [DOI] [PubMed] [Google Scholar]
- 44. Sorensen MA, Fricke J, Pedersen S. 1998. Ribosomal protein S1 is required for translation of most, if not all, natural mRNAs in Escherichia coli in vivo. J. Mol. Biol. 280:561–569 [DOI] [PubMed] [Google Scholar]
- 45. Nakagawa S, Niimura Y, Miura K, Gojobori T. 2010. Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 107:6382–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boni IV, Isaeva DM, Musychenko ML, Tzareva NV. 1991. Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res. 19:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gallie DR, Kado CI. 1989. A translational enhancer derived from tobacco mosaic virus is functionally equivalent to a Shine-Dalgarno sequence. Proc. Natl. Acad. Sci. U. S. A. 86:129–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lim K, Furuta Y, Kobayashi I. 2012. Large variations in bacterial ribosomal RNA genes. Mol. Biol. Evol. 29:2937–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moll I, Grill S, Gualerzi CO, Blasi U. 2002. Leaderless mRNAs in bacteria: surprises in ribosomal recruitment and translational control. Mol. Microbiol. 43:239–246 [DOI] [PubMed] [Google Scholar]
- 50. Chen HY, Bjerknes M, Kumar R, Jay E. 1994. Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli messenger RNAs. Nucleic Acids Res. 22:4953–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. He JL, Sakaguchi K, Suzuki T. 2012. Determination of the ribosome binding sequence and spacer length between binding site and initiation codon for efficient protein expression in Bifidobacterium longum 105-A. J. Biosci. Bioeng. 113:442–444 [DOI] [PubMed] [Google Scholar]
- 52. Vellanoweth RL, Rabinowitz JC. 1992. The influence of ribosome binding site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol. Microbiol. 6:1105–1114 [DOI] [PubMed] [Google Scholar]
- 53. Kozak M. 2005. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361:13–37 [DOI] [PubMed] [Google Scholar]
- 54. Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Higo K, Otaka E, Osawa S. 1982. Purification and characterization of 30S ribosomal proteins from Bacillus subtilis: correlation to Escherichia coli 30S proteins. Mol. Gen. Genet. 185:239–244 [DOI] [PubMed] [Google Scholar]
- 56. Kleerebezem M, Beerthuyzen MM, Vaughan E, de Vos WM, Kuipers OP. 1997. Controlled gene expression systems for lactic acid bacteria: transferable nisin-inducible expression cassettes for Lactococcus, Leuconostoc, and Lactobacillus spp. Appl. Environ. Microbiol. 63:4581–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roberts MW, Rabinowitz JC. 1989. The effect of Escherichia coli ribosomal protein S1 on the translational specificity of bacterial ribosomes. J. Biol. Chem. 264:2228–2235 [PubMed] [Google Scholar]
- 58. Sorokin A, Serror P, Pujic P, Azevedo V, Ehrlich SD. 1995. The Bacillus subtilis chromosome region encoding homologs of the Escherichia coli mssA and rpsA gene products. Microbiology 141:311–319 [DOI] [PubMed] [Google Scholar]
- 59. Bolotin A, Wincker P, Mauger S, Jaillon O, Malarme K, Weissenbach J, Ehrlich SD, Sorokin A. 2001. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp. lactis IL1403. Genome Res. 11:731–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Salah P, Bisaglia M, Aliprandi P, Uzan M, Sizun C, Bontems F. 2009. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 37:5578–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Whitehead TR. 1997. Development of a bifunctional xylosidase/arabinosidase gene as a reporter gene for the Gram-negative anaerobes Bacteroides and Porphyromonas, and Escherichia coli. Curr. Microbiol. 35:282–286 [DOI] [PubMed] [Google Scholar]