

Abstract
Clostridium difficile infections are a major cause of antibiotic-associated diarrhea in hospital and care facility patients. In spite of the availability of effective antibiotic treatments, C. difficile infection (CDI) is still a major cause of patient suffering, death, and substantial health care costs. Clostridium difficile exerts its major pathological effects through the actions of two protein exotoxins, TcdA and TcdB, which bind to and disrupt gut tissue. Antibiotics target the infecting bacteria but not the exotoxins. Administering neutralizing antibodies against TcdA and TcdB to patients receiving antibiotic treatment might modulate the effects of the exotoxins directly. We have developed a mixture of three humanized IgG1 monoclonal antibodies (MAbs) which neutralize TcdA and TcdB to address three clinical needs: reduction of the severity and duration of diarrhea, reduction of death rates, and reduction of the rate of recurrence. The UCB MAb mixture showed higher potency in a variety of in vitro binding and neutralization assays (∼10-fold improvements), higher levels of protection in a hamster model of CDI (82% versus 18% at 28 days), and higher valencies of toxin binding (12 versus 2 for TcdA and 3 versus 2 for TcdB) than other agents in clinical development. Comparisons of the MAb properties also offered some insight into the potential relative importance of TcdA and TcdB in the disease process.
INTRODUCTION
Clostridium difficile infection (CDI) is a global problem that especially affects patients in hospitals and long-term health care facilities. CDI can also be community acquired. Prior antibiotic usage, being older than 65 years, and having comorbidities are considered to be major risk factors (1). The symptoms of CDI include diarrhea, fever, and inflammation of the bowel, including pseudomembranous colitis (PMC) and fulminant colitis (megacolon). Diarrhea can be prolific (∼3 to 15 bowel movements per day) and prolonged (up to 30 days) (2). The direct effects of CDI include patient suffering, prolonged hospital stays, clinical complications requiring transfer to intensive care units, surgical colon removal, and death (3). The indirect effects of CDI can include reduced ward occupancy levels as a result of “outbreak management” procedures, including increased testing and cleaning. These factors collectively result in substantially higher health care costs, variably estimated to be up to $3.2 billion for U.S. hospitals alone (4, 5), principally due to hospital stays of, on average, 6 days longer than for patients without CDI (6).
The symptoms of CDI are predominantly caused by the activity of two large exotoxins, TcdA and TcdB. Nontoxigenic strains do not cause disease in hamsters or humans, and they have been administered to human volunteers without causing diarrhea (7–9). Strains encoding either TcdA or TcdB alone cause CDI in hamsters, but the presence of both toxins was found to be more potent than the presence of either toxin alone (7). Indeed, a minority (typically ≤5%) of strains causing CDI in humans are TcdA negative and TcdB positive (A− B+), suggesting that TcdB alone is capable of causing symptoms in susceptible humans (1). The clinical effect of A− B+ strains appears to conflict with data from studies in which purified TcdA or TcdB was administered via oral gavage to animals or injected into ligated gut loops of animals. In these situations, TcdB was unable to cause any symptoms unless accompanied by TcdA or unless TcdB had been administered after mechanical tissue damage (10, 11). In addition, neutralizing monoclonal antibodies (MAbs) against TcdA alone were sufficient to protect mice from death caused by A+ B+ strains; the concentration of TcdA in the gut was diminished in surviving animals, while that for TcdB was unchanged (12). TcdA alone has been shown to cause symptoms in animals (10). These kinds of studies resulted in an understanding that TcdA alone might be required to cause CDI in humans. In contrast, lethality was observed only in mice infected with strains producing measurable amounts of both TcdA and TcdB, not TcdA alone (13). These apparent experimental differences might be explained in part by the differing susceptibilities of species to toxins due to interspecies toxin receptor variation and differences in the effectiveness of mechanically administered purified toxin protein versus toxin delivered by a bacterial strain which might potentially also encode additional pathogenicity traits. The details of human susceptibilities to toxins/strains remain to be established.
Analyses of the activities of TcdA and TcdB in vitro show that TcdB is 1,000 times more potent on a molar basis than TcdA and that both toxins are capable of causing loss of cell form and hence death (14). However, important qualitative and quantitative differences between TcdA and TcdB have been observed (15). TcdA caused a larger, more diffuse, and more edematous lesion in guinea pig skin than TcdB (16). Both toxins cause loss of transepithelial electrical resistance (TEER), but TcdA was substantially more capable (faster and more complete) in causing TEER loss than TcdB at equivalent toxin concentrations (17). TcdA has been shown to facilitate the penetration of TcdB into Caco-2 monolayers, whereas TcdB does not facilitate the penetration of TcdA (18). Finally, TcdA was found to cause a 5-times-greater fluid loss from xenotransplanted human gut tissue than TcdB (19). The loss of TEER is initiated by a loss of tight junction function, which is a likely early step in the physiology of the diarrhea and fluid loss with CDI (17, 20).
Collectively, the published research is consistent with the concept that both TcdA and TcdB are important in the disease process and is suggestive of the importance of the relative and total amounts of functional toxin used during in vivo studies. It is possible that TcdA is better evolved to catalyze early events in the disease process, including loss of tight junctions and loss of cell form, leading to both diarrhea and enhanced penetration of TcdB. TcdB appears to be better evolved to catalyze the more long-lasting inflammatory events in the disease process by killing human gut and immune cells and eliciting or subverting local inflammatory responses by causing the local production of interleukin 1β (IL-1β), tumor necrosis factor alpha (TNF-α), IL-6, and IL-8 and being involved in systemic disease in a piglet model (21–23).
Some strains also produce a third protein toxin called binary toxin or CDT. The in vivo function of CDT remains to be fully elucidated, but it is clear that A+ B+ CDT− and A− B+ CDT− strains are capable of causing CDI symptoms in humans. The influence and importance of additional virulence factors cannot be discounted. Notwithstanding uncertainties around the clinical significance of minor virulence factors, TcdA and/or TcdB are completely associated with disease in humans. That typically 85% to 95% of clinical isolates are of the A+ B+ (rather than A− B+) phenotype also supports the idea of an important role in C. difficile pathogenesis and/or survival for both toxins. Hence, in order to maximize the clinical impact of mixtures of antitoxin MAbs, one might prefer to neutralize both TcdA and TcdB.
Neutralizing and protective antibodies against TcdA and TcdB have been generated by both active and passive vaccination of animals (24, 25). Vaccination offers the potential advantage of generating a polyclonal response, but the disadvantages relate to the timing of vaccine administration relative to the perceived risk of infection and variability in the strength and speed of a patient's immune response (26–29). Parenteral administration of purified antibodies offers control over both timing and dose. The manufacture of colostrum or serum polyclonal antibodies from cow, sheep, chicken, and alpaca or other species for therapeutic use can have production quality difficulties in controlling the variability of batch-to-batch antitoxin activities (titer and specificity) and the total production capacity, and there can be immunogenicity concerns (24, 25, 30–34). The use of mixtures of monoclonal antibodies offers an advantage over polyclonal sera in these respects. However, previous efforts have generally not generated MAbs with high affinities, in vitro potencies, or very high levels of protection against infection in animal models (35–37).
Two MAbs discovered by Babcock et al. (referred to as CDA1 and MDX-1388) (35) conferred incomplete and nondurable protection against the death of hamsters in infection studies but demonstrated a statistically significant reduction (38% versus 7%, P = 0.006) in disease recurrence in a phase II clinical trial. The MAb mixture did not improve the duration or severity of diarrhea or the death rates, although this study was not powered to demonstrate these (38). Hence, there remain unmet clinical needs which might be treatable with more efficacious MAbs. Improvements in these areas might positively influence future patient outcomes and health care costs. We aimed to discover a mixture of MAbs with high affinities and in vitro toxin neutralization activities and hence high levels of protection of hamsters in an infection study. We compared a mixture of 3 MAbs against a mixture of CDA1 and MDX-1388 (35).
MATERIALS AND METHODS
Design, cloning, expression, and purification of toxin subdomains.
A range of different immunogens and screening reagents were either commercially sourced or produced in-house using conventional Escherichia coli expression techniques in order to provide a diverse and broad immune response and to facilitate the identification and characterization of monoclonal antibodies (see Table S1 in the supplemental material). The E. coli expression and subdomain purification methods used are described in Text S1 in the supplemental material. Toxoid A was purchased from List Labs (Quadratech, United Kingdom). Where recombinant proteins or peptides were generated, the sequences were based on that of ribotype 027 (UniProt accession numbers C9YJ37 [TcdA] and C9YJ35 [TcdB]).
Sequence and structural analyses were performed in order to define functional binding subdomains which might represent practicable expression domains using the crystal structure of Protein Data Bank accession number 2G7C and sequence alignments as guides. For simplicity, herein we called each functional domain boundary a combined repetitive oligopeptide repeat (CROP) and numbered them sequentially from the N terminus to the C terminus. Our analysis showed that TcdA comprised seven CROPs, while TcdB comprised four CROPs. Each CROP was defined by a central beta-hairpin which contained a putative sugar binding site and then extended by three additional beta-hairpins to both its N terminus and its C terminus. Thus, each expression CROP contained seven beta-hairpins, each overlapping with its immediate neighbors by one beta-hairpin on either side. We also divided the C-terminal regions of TcdA and TcdB into larger subunits: TcdA was divided into CROPs 1 to 3 and 4 to 6, annotated as TcdA123 and TcdA456 (CROP 7 was observed to be shorter than the other six CROPS and hence was excluded), while the entire C terminus of TcdB was expressed (TcdB1234).
Animal immunizations.
Sprague-Dawley rats and half-lop rabbits were immunized with either synthetic peptides (coupled to keyhole limpet hemocyanin, ovalbumin, or bovine serum albumin) mapping to regions common to both TcdA and TcdB full-length toxins, formaldehyde-inactivated toxoid A, binding domain fragments (TcdA123, TcdA456, or TcdB1234), or, in some cases, a combination of all of the (nonpeptide) immunogens. The first doses were formulated in complete Freund's adjuvant, and all subsequent doses were formulated in incomplete Freund's adjuvant. Following 2 to 6 immunizations, the animals were sacrificed, and peripheral blood mononuclear cells (PBMCs), spleen, and bone marrow were harvested. Sera were monitored for binding to TcdA domains, TcdB domains, toxin, or toxoid by enzyme-linked immunosorbent assays (ELISAs) and for toxin neutralization ability using a Caco-2 proliferation assay.
Antibody discovery and humanization.
B cell cultures from immunized animals were prepared using a method similar to that described by Lightwood et al. (see reference 39 and references therein).
Antibody variable-region genes were cloned as mouse IgG1(κ) full-length antibodies for rat variable regions and rabbit IgG(κ) full-length antibodies for rabbit variable regions. Antibodies were reexpressed in a HEK-293 transient-expression system. Recombinant antibodies were retested for their ability to neutralize toxin in cell-based assays and screened by surface plasmon resonance (SPR) to determine the affinity for a given toxin domain and to determine the specificity and approximate the number of binding events of antibody to toxin. Lead candidates were selected for humanization. Complementarity-determining regions (CDRs) from rabbit and rodent sequences were transferred onto human germ line V region sequences selected for their high levels of sequence similarity to donor V regions. Successful humanization was determined as a <0.5-log change in affinity relative to that of chimeric rat-human or rabbit-human antibodies in a surface plasmon resonance binding assay. Unless otherwise stated, all the data provided herein were generated using humanized antibodies.
Toxin neutralization assays. (i) Caco-2 proliferation assay (methylene blue).
Caco-2 cells were maintained and screened in minimal essential medium (MEM) plus 20% fetal calf serum, 2 mM glutamine, and nonessential amino acids in 96-well polystyrene plates at 37°C. All antibody combinations were of equal molar ratios unless stated otherwise. Caco-2 cells (HTB-37; ATCC) were plated at 3,000 cells per well in 50 μl medium and incubated for 24 h at 37°C. Antibody and toxin A or toxin B (TGC Labs) (typically VPI10463 in origin, used at an 80% lethal dose [LD80]) were premixed for 1 h at 37°C in sterile 96-well round-bottom polypropylene plates before 50 μl of this mixture was transferred to the cell-containing plates. The MAb-toxin mixture and cells were incubated for an additional 96 h. Medium was aspirated from all wells of the assay plates before 50 μl methylene blue (0.5% [wt/vol] dissolved in 50% [vol/vol] ethanol) was added to the cell culture. The cells were incubated for 1 h at room temperature before being washed gently with tap water (to remove excess stain) and air dried. The cells were then lysed by adding 100 μl 1% (vol/vol) N-lauryl-sarcosine and incubated on a shaker for 15 min at room temperature. The cell biomass was determined by measuring the absorbance of each well on a BioTek Synergy 2 plate reader at 405 nm. Toxin activity and working LD80 concentrations were defined empirically in preliminary experiments and for each individual batch/lot of toxin used.
(ii) TEER assay.
Cell monolayer integrity assays were performed using the Becton Dickinson Caco-2 BioCoat HTS plate system (catalog no. 354802; BD Biosciences). All cells were cultured and incubated in a humidified incubator at 37°C and 5% CO2. Caco-2 cells were seeded at 2 × 105/ml per well of the plate insert in 500 μl of basal seeding medium, and 35 ml of basal seeding medium was added to the feeder tray. The cells were incubated for 24 h before replacement of the basal seeding medium from the inserts and feeder tray with Entero-STIM differentiation medium, 500 μl of which was added per well insert and 35 ml of which was added to the feeder tray. The cells were incubated for another 72 h. Antibodies and TcdA (VPI10463) were preincubated together in polypropylene wells for 1 h, while 1 ml of Caco-2 growth medium (MEM plus 20% fetal calf serum, 2 mM glutamine, and nonessential amino acids) was added to each well of a standard 24-well tissue culture plate. The BioCoat insert plate was transferred to the 24-well tissue culture plate, and the Entero-STIM medium from the inserts was replaced with 400 μl of the toxin-Ab mixture. The cells were incubated with this mixture for 4 h. After this time, the integrity of each monolayer was analyzed by measuring the resistance between the upper and lower compartments of the plate.
Antibody expression, purification, and preparation for hamster study.
Antibodies were transiently expressed in HEK or CHO cells and purified using standard methods to produce high-purity protein which was low in aggregate and endotoxin (see Text S2 in the supplemental material). The variable region sequences for the MAbs, CDA1 and MDX-1388, were obtained from a published patent application. Gene synthesis and restriction/ligation were used to produce human IgG1 (hIgG1) expression plasmids. CA997, CA1125, and CA1151 and the control MAbs CDA1 and MDX-1388 were transiently expressed in CHO cells purified to homogeneity and low % of aggregate levels (1.0%, 2.6%, and 1.3% for CA997, CA1125, and CA1151, respectively) and concentrated to 20 mg/ml in phosphate-buffered saline (PBS). Each of the MAbs destined to form part of a combination (i.e., animal group) were aliquoted individually into coded tubes within UCB laboratories before being shipped to Ricerca Biosciences laboratories in order to “blind” the study. Separate aliquots for each coded MAb were shipped for each of the four dosing days along with an empty tube for mixing. The contents of the three tubes for each animal group were mixed immediately prior to dosing of the hamsters on each day. Preliminary studies had shown no significant change in aggregate levels over 1 month (intended to mimic the shipping and study lead time) and also that no changes in protein quantity or quality (visual inspection at A280 with size exclusion high-pressure liquid chromatography [SE-HPLC]) occurred during or after mixing (data not shown).
Antibody affinity measurement and valency of MAb binding to toxin subdomains. (i) Kinetic binding constants for the interaction of anti-C. difficile antibodies.
TcdA and TcdB antibodies were determined by surface plasmon resonance (SPR) conducted on a BIAcore 3000 using CM5 sensor chips. All experiments were performed at 25°C. AffiniPure F(ab′)2 fragment goat anti-human IgG, Fc fragment specific (Jackson), was immobilized on a CM5 sensor chip (GE) via amine-coupling chemistry to a capture level of ∼7,000 response units (RUs). HBS-EP buffer (10 mM HEPES [pH 7.4], 0.15 M NaCl, 3 mM EDTA, 0.005% surfactant P20 [BIAcore AB]) was used as the running buffer with a flow rate of 10 μl/min. A 10-μl injection of each purified antibody at a concentration of ≤1 μg/ml was used for capture by the immobilized anti-human IgG Fc fragment. TcdA123, TcdA456, or TcdB1234 was titrated over captured purified antibodies at doubling dilutions from 12.5 nM at a 30-μl/min flow rate. Kinetics were calculated from two independent MAb captures and kinetic measurements. Double-referenced background-subtracted binding curves were analyzed using the BIA evaluation software (version 3.2) following standard procedures. Kinetic parameters were determined from the fitting algorithm.
(ii) Relative molar binding events of anti-C. difficile antibodies.
Molar binding equivalents of neutralizing antibodies to TcdA123, TcdA456, and TcdB1234 were studied using streptavidin immobilization on a CM5 sensor chip to a level of ∼4,000 RU. Biotinylated TcdA123 was bound to one flow cell and TcdA456 was bound to a different flow cell with a response of ∼500 RU. TcdB1234 was bound at ∼400 RU in independent experiments. Two 30-μl injections of the same anti-TcdA antibody at 1 μM were injected over both flow cells at 10 μl/min, and the saturating binding response was recorded. Two 20-μl injections of the same anti-TcdB antibody mixtures (final concentration of each antibody was 500 nM) were injected over this surface at 10 μl/min, and the saturating binding response was recorded. All the data were corrected for background binding using the response to the streptavidin-only reference flow cell.
Antibody biophysical characterization. (i) Thermal stability (Tm).
The midpoint unfolding temperature (Tm) of each purified antibody was determined with a Thermofluor assay using methods described previously (40).
(ii) Isoelectric point.
The pI of each purified antibody was determined using capillary isoelectric focusing after antibody samples at 2 mg/ml were mixed with 0.35% methylcellulose, 4% pH 3 to 10 ampholytes (Pharmalyte), and synthetic pI markers (4.65 and 9.77) and run on an iCE280 isoelectric focusing system (prefocusing at 1,500 V for 1 min followed by focusing at 3,000 V for 6 min).
(iii) Shaking aggregation assay (stressed stability).
Purified antibodies were subjected to stress by vortex mixing 250 μl at 1 mg/ml (three replicates per MAb) in 1.5-ml conical capped plastic tubes in PBS (pH 7.4) at 1,400 rpm and 25°C. Aggregation was monitored photometrically by absorbance at 340 nm and/or 595 nm.
Hamster infection study.
The hamster infection study was performed by Ricerca Biosciences LLC (Cleveland, OH). The study protocol was approved by the Ricerca IACUC. Active and control components (composition and dose) were blinded to the Ricerca staff until after completion of the planned 28-day study period. Male golden Syrian hamsters (weight, 82 to 103 g; age, 54 days) were individually housed in HEPA-filtered disposable cages and fed nonsterilized Teklad Global Diet 2016 and tap water ad libitum. After being acclimated for 1 week, the hamsters were predosed by intraperitoneal (i.p.) inoculation with MAb mixtures or PBS (vehicle control) once per day for 4 days (days −3, −2, −1, and 0, relative to infection on day 0). Two doses of MAb were investigated: 50 mg/kg of each anti-TcdA and anti-TcdB component (high dose) and 5 mg/kg of each anti-TcdA and anti-TcdB component (low dose). Hamsters were sensitized (day −1) with 50 mg/kg of clindamycin phosphate in PBS administered subcutaneously (s.c.) before being challenged 1 day later (day 0) with 3.4 × 106 CFU of vegetative cells from strain ATCC 43596 (ribotype 012, strain 545). One group of animals was dosed perorally (p.o.) with 5 mg/kg of vancomycin twice per day for 5 days (on days 1, 2, 3, 4, and 5). This dose was chosen because it carried a greater probability that the hamsters dosed would succumb to spontaneous reinfection during the 28-day study. The high-dose regimen was designed to mimic that of the primary challenge model used by Babcock et al. (35) in order to enable comparison with published data. All groups contained 11 animals, except the vancomycin control group, which contained 5 animals.
Viability checks were performed on the animals twice per day, and animals found to be in extremis were euthanized and recorded as dead. Body weights were recorded on each day of dosing and then twice weekly and before euthanizing the survivors. Three animals from each of the 5 treatment groups were selected randomly by the Ricerca Biosciences staff for blood sampling through the retroorbital sinus on days 1 and 6; thus, some animals were bled once and others twice. The blood was processed to collect plasma, and plasma samples were frozen at −70°C or lower. All animals surviving to the end of the study were also bled. Gross necropsy was performed on all animals; the status and color of the ilea and ceca were scored, and any anogenital staining (“wet tail”) was noted. Survival curves, created using the method of Kaplan and Meier, were analyzed using the P value from the log-rank test compared to the Bonferroni-corrected threshold of P = 0.005. The vancomycin-treated group was not included in the analysis. All statistical tests were done with Prism v5.04 software.
Pharmacokinetics and biodistribution of humanized MAbs in noninfected hamsters by ELISA.
The pharmacokinetics and distribution to the gut of a hIgG1 MAb was studied in “normal” (noninfected) golden Syrian hamsters. Purified MAb was administered to male hamsters (120 to 135 g) by CARE Research LLC (Fort Collins, CO), and samples were assayed by UCB Pharma. The study was approved by the CARE IACUC. Eight animals each received a single dose of 20 mg/kg of IgG1; four were dosed i.p., and four were dosed s.c. Blood was collected at 1, 3, 8, 24, 48, 72, 103, and 168 h after dose administration, and serum was separated before storage at −80°C. Blood was also taken from two untreated hamsters in order to provide assay controls. Following euthanasia, a 2-cm length of colon was cut from the cecum junction onward from each hamster. The colon section was flushed with wash buffer (50% [vol/vol] PBS containing 50% [vol/vol] Sigma protease inhibitor cocktail [P2714]) before being opened for the separation and removal of the mucosa from the underlying muscle. Mucosal samples were placed in 0.5 ml of wash buffer, homogenized until visually uniform, and stored at 4°C before immediate shipping on wet ice. Samples were assayed by ELISA as described in Text S3 of the supplemental material.
RESULTS
Expression and purification of toxin subdomains.
Our aims were to express and purify the entire polypeptide sequences of TcdA and TcdB from a ribotype 027 strain as a series of contiguous or overlapping subdomains to use as immunogens and screening reagents. Using an E. coli cytoplasmic His-tagged expression and purification system, we found that the majority of toxin subdomains were either poorly expressed or insoluble upon purification. A list of the subdomains successfully expressed is shown in Table S1 of the supplemental material. The major C-terminal subdomains (TcdA123, TcdA456, and TcdB1234) were well expressed and soluble. In order to confirm that these domains were correctly folded, we performed a thermal unfolding experiment. All three of these subdomains demonstrated clear unfolding transitions (see Fig. S1 in the supplemental material) and Tms of 42.9°C, 45.2°C, and 50.2°C for TcdA123, TcdA456, and TcdB1234, respectively, consistent with each of them being in a folded state.
Immunization, serum screening, MAb identification, and humanization.
Rabbits and rats were immunized with different combinations and serial orders of peptides, toxoids, and subdomains. Sera from immunized animals were tested for (polyclonal) toxin neutralization ability in order to best identify individual animals suitable for detailed evaluation using UCB's antibody discovery process to generate toxin-neutralizing MAbs. Following B cell culture from selected animals, approximately 12,000 toxin-specific positives were identified in the primary toxin binding assay from a total of 4,050 (96-well)-plate B cell culture experiments (3% hit rate). The anti-TcdA MAbs described were derived from rabbits immunized with 4 doses of toxoid A, while the anti-TcdB MAbs described were derived from rats immunized with 4 doses of TcdB1234.
Many approaches were taken in an attempt to identify cross-neutralizing (i.e., neutralizing both TcdA and TcdB) MAbs. These included cycling the immunogens used between subdomains derived from TcdA and then TcdB, immunizing with one toxin-derived sequence and screening on the other, immunizing with CROP domains which had the highest levels of intertoxin similarity, and using peptides with very high levels of sequence similarity and identity between TcdA and TcdB (see Table S2 in the supplemental material). Although animals immunized with peptides produced high serum titers (typically 1:100,000), neutralizing antibodies could not be isolated. Animals which had been cross-toxin immunized and screened produced a small number of cross-reactive MAbs, none of which demonstrated neutralizing activity, consistent with previous observations (34, 41). Together with the results of analysis of sequence alignments, these data suggested that it was not possible to identify MAbs which potently neutralized both TcdA and TcdB.
We assessed more than 20 MAbs with neutralizing potential against TcdA and more than 50 MAbs with neutralizing potential against TcdB. A more-detailed evaluation focused on five anti-TcdA MAbs and four anti-TcdB MAbs for their ability to function alone and together as mixtures. Antibodies were first expressed and tested as chimeric (rabbit-mouse IgG1, rat-mouse IgG1) antibodies before being humanized and then characterized in detail.
Characterization of anti-TcdA MAbs.
Five humanized rabbit MAbs were evaluated in a range of in vitro assays to test their suitability for in vivo testing and potential clinical development. An antibody with the same sequence as CDA1 (35) was included in some assays. The MAbs were tested for their ability to neutralize the activity of TcdA derived from strain VPI10463 (ribotype 003) in a 4-day Caco-2 proliferation assay. TcdA was used at different lethal doses (LD80, LD90, LD95, and LDmax) in order to evaluate how each MAb might perform in the presence of high levels of toxin, such as might be encountered during the early stages of active infection and diarrhea. The neutralization curves in Fig. 1a to d show that high levels (80 to 100%) of neutralization were achieved with all of the MAbs when TcdA was used at an LD80, with EC50s ranging from 0.3 to 35 ng/ml. The performance characteristics, the degree of increase in the EC50 (reduction in potency) and decrease in maximum neutralization, were notably different in the presence of higher toxin concentrations. Overall, CA922 had the lowest EC50 (highest potency) and the broadest range of TcdA concentrations (0.27 ng/ml to 1.2 ng/ml at LD80–95, respectively). CA922 and CA997 had the highest levels of maximum neutralization (100 to 80% at LD80–95, respectively), but CA997 demonstrated a tendency toward higher maximal neutralization at the higher TcdA concentrations. Of note was that four of the MAbs were significantly more potent than CDA1 at an LD80–95 and that CDA1 demonstrated no determinable TcdA neutralization at the LDmax tested.
Fig 1.

(a to d) Neutralization of TcdA from ribotype 003 (VPI10463) by humanized MAbs. Each data point represents the mean of two independent experiments each of three assay replicates.
Mixtures of two to five of the humanized MAbs were tested in order to determine if even greater potencies were achievable. It was conceivable that MAbs binding to different and nonoverlapping neutralizing epitopes might demonstrate significant synergies. No increases in potency were observed when comparing equimolar total amounts of any of the mixtures against the two most potent single antibodies (see Table S3 in the supplemental material). Repeat studies where just CA922 and CA997 were combined confirmed these observations (data not shown). In light of these data, subsequent studies focused on single MAbs.
A therapeutic MAb should be broadly neutralizing, i.e., capable of neutralizing toxins with different primary sequences/derived from different strains. To that end, each MAb demonstrated a good ability to neutralize TcdA produced by strains of ribotypes 027 and 078. EC50s for the three most potent MAbs (CA922, CA997, and CA1000) are shown in Table 1. Activities ranging from 0.11 to 2.25 ng/ml were observed using TcdA at an LD80. It was evident that CDA1 was largely impotent against TcdA derived from these strains, even at the lower LD80, and hence the EC50s could not be calculated (see Figure S2a to d in the supplemental material), data which are consistent with the observations of others (42). CA922 and CA997 demonstrated maximal neutralization abilities of ∼90 and 100%, respectively, at the highest TcdA concentration tested, LDmax (see Fig. S2b and d in the supplemental material). Collectively, these data suggest a ranking of CA922 ≈ CA997 > CA1000 > CA995 > CA923 with respect to TcdA neutralization, but with CA997 tending toward higher maximal neutralization at higher TcdA concentrations.
Table 1.
Neutralization of TcdA derived from ribotypes 027 and 078
| MAb | Neutralization EC50 (ng/ml) of TcdA from: |
|||||||
|---|---|---|---|---|---|---|---|---|
| Ribotype 027 |
Ribotype 078 |
|||||||
| TcdA LD80 | TcdA LD90 | TcdA LD95 | TcdA LDmax | TcdA LD80 | TcdA LD90 | TcdA LD95 | TcdA LDmax | |
| CA922 | 0.19a | 0.25 | 0.41 | 1.46 | 0.11 | 0.12 | 0.25 | 0.68 |
| CA997 | 0.92 | 1.27 | 1.75 | 7.19 | 0.33 | 1.11 | 1.11 | 2.57 |
| CA1000 | 2.25 | 2.49 | 3.52 | 16.32 | 2.04 | 5.03 | 5.03 | 14.16 |
| CDA1 | NDb | ND | ND | ND | ND | ND | ND | ND |
The data represent means of two independent experiments each of three assay replicates.
ND, not determined.
We wanted to select antibodies which had the highest potential to ameliorate diarrhea; hence, we tested our MAbs in a cell monolayer TEER assay, measuring the loss of transepithial electrical resistance. Resistance readings from the 4-h time point are shown in Fig. 2. Large differences were observed in the abilities of the MAbs in this assay which would not have been predicted by the neutralization assay performance. Of particular note was that CA922 had the poorest performance in the TEER assay, whereas it was the most potent in the neutralization assay. CA997 had a substantial potential to neutralize TEER loss with maximal inhibition approaching 80% and an EC50 of approximately 80 ng/ml. In contrast, CDA1 demonstrated little ability to neutralize TEER loss in terms of both extent of inhibition (percent protection of TEER loss) and potency (EC50).
Fig 2.
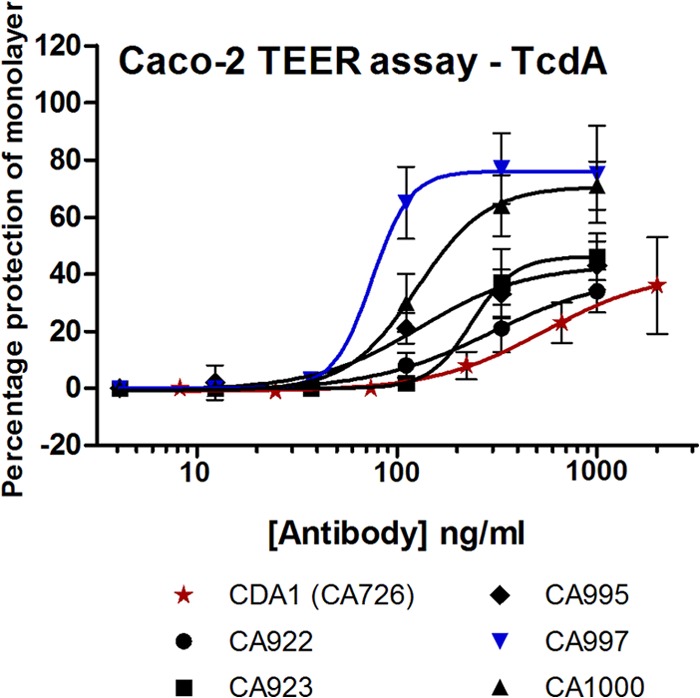
Neutralization of TEER loss by humanized MAbs. Each data point represents the mean and standard deviation of four independent experiments (except CA997, n = 3) each of three assay replicates.
The affinities of the five humanized MAbs were determined by binding to the subdomains of TcdA123 and Tcd456 using surface plasmon resonance. The results in Table 2 show that all of the humanized MAbs had substantially subnanomolar affinities against the C-terminal subdomains of TcdA derived from the ribotype 027 sequence. CA922 had the highest affinity (∼2 to 4 pM). CA995 did not bind at all to the three most N-terminal domains of the cell binding region (TcdA123). CDA1 was observed to have more moderate affinities against the subdomains (∼0.5 to 0.9 nM), data which are broadly consistent with the published affinity of 1.4 nM (43). We next attempted to estimate the number of times that each antibody was able to bind to TcdA by comparing the relative binding signals using the TcdA123 and TcdA456 subdomains during surface plasmon resonance. Others have observed that MAbs to TcdA can bind multiple times, probably due to the unusual multidomain and repetitive primary sequence structure of the cell binding domains (36). Our data (Table 2) show the valencies of binding ranging from 1 to approximately 16 for TcdA. Of note, CA922, CA923, and CA997 appear to be capable of binding TcdA >12 times each, CA1000 binding twice, and CA995 binding only once. CDA1 also appeared to be capable of binding TcdA twice, once each within the TcdA123 and TcdA456 regions. No obvious correlations were observed between neutralization ability, affinity, and valency of binding. The most stark comparison was between CA922 and CA1000, both of which were potent neutralizers of TcdA but which had considerably different affinities (2 to 4 pM versus 73 to 84 pM), valencies of binding (∼16 versus 2), and relative TEER activities (modest versus high), respectively. Hence, such multiple-binding events might comprise a mixture of neutralizing/nonneutralizing and higher-/lower-affinity epitopes.
Table 2.
Biophysical properties of humanized anti-TcdA MAbs
| MAb | Affinity (pM) |
Valency of binding (n) |
pI | Fab Tm (°C) | Resistance to shaking aggregation | ||
|---|---|---|---|---|---|---|---|
| TcdA123 | TcdA456 | TcdA123 | TcdA456 | ||||
| CA922 | 4.06 | 2.59 | ∼10 | 6 | 8.8 | 81 | Very good |
| CA923 | 64.7 | 312 | 7 to 8 | 5 | 9.2 | 79 | Good |
| CA995 | 0 | 119 | 0 | 1 | 8.5 | 71 | Good |
| CA997 | 132 | 66.8 | 7 to 8 | 5 | 8.3 | 79.2 | Good |
| CA1000 | 73.3 | 84.1 | 1 | 1 | 7.7 | 70.5 | Moderate |
| CDA1 | 871 | 557 | 1 | 1 | 8.8 | 66.2 | Moderate |
The physical stability of MAbs destined for human use is of interest, especially where mixtures of MAbs are being contemplated. We analyzed the thermostability (Tm) of each humanized MAb. The Tm is an estimate of global stability and, hence, bioprocessing and storage stability risk. The CH2 and CH3 domain Tms were found to be typical of those expected for IgG1 (data not shown) (44), and so only the differentiating Fab arm Tms are shown in Table 2. All five humanized MAbs were observed to have high Tms, with three (CA922, CA923, and CA997) being particularly high (81, 79, and 79°C, respectively). CDA1 was observed to have a Tm of 66°C. Since we were contemplating creating an antibody mixture, we also performed a “shaking aggregation” assay. All of the MAbs tested demonstrated adequate stability (Table 2), but CA1000 had the highest rate, and most substantial extent, of aggregation under the test conditions.
CA997 was chosen to continue to the hamster infection studies. It demonstrated very potent neutralization of TcdA at a wide range of toxin concentrations and from three different sequence types (ribotypes 003, 027, and 078). It had high affinity and functionally oligoclonal toxin binding/neutralization, and it demonstrated high levels of physical stability. Of particular interest was the strong potency of CA997 in the TEER assay, since MAbs for intervention in CDI have shown little clinical impact on diarrheal symptoms to date.
Characterization of anti-TcdB MAbs.
The screening of anti-TcdB MAbs failed to identify single MAbs with both highly potent and substantial neutralizing activities. Hence, MAbs with evident neutralizing activities were arrayed in pairs in order to identify combinations which offered significant activities. During these studies, it quickly became apparent that one MAb, CA1125, worked well in a significant number of pairings. Hence, CA1125 became a “partner of choice,” which enabled a focused screening on identification of its optimal partner(s). Three key partners for CA1125 (CA1134, CA1151, and CA1153) were identified and tested for their ability, together with CA1125, to neutralize TcdB. Three concentrations of TcdB which were shown to be equivalent to LD60, LD77, and LD85 were tested. The relative ratios of each MAb within a pair were also varied (without affecting the total amount of antibody) in order to evaluate if the total potency was stable if the MAbs were subjected to subtle changes in relative concentrations, such as might be experienced in vivo. The data for CA1125 combined with CA1134, CA1151, and CA1153 are shown in Table 3. An antibody with the same amino acid sequence as MDX-1388 (35) was included as a comparator. In short, the combination of CA1125 and CA1151 was selected for additional in vivo study. It had the lowest EC50 of all three TcdB concentrations at equimolar ratios and was robustly neutralizing at altered relative MAb ratios. The CA1125-plus-CA1151 combination was additionally investigated in more complex (three or more) MAb mixtures. No significant increases in the EC50s were observed using such combinations (data not shown), and hence these two MAbs were studied further for their affinities, valencies of toxin binding, Tms, and resistance to aggregation. The data in Table 4 show affinities for TcdB ranging from ∼50 to 1,820 pM for the antibodies described. In contrast to the anti-TcdA MAbs, the valencies of binding to TcdB were much lower (either 1 or 2). The MDX-1388 MAb had a 1,570 pM affinity, a binding valency of 2, and a Tm of 68°C. The CA1125 and CA1151 MAbs demonstrated high stabilities, both thermal (79.5°C and 80.8°C, respectively) and resistance to aggregation. They bound to noncompetitive epitopes (data not shown) and as a combination were capable of binding TcdB three times.
Table 3.
Neutralization of TcdB by humanized antibody mixtures
| MAb or combination (ratio) | Neutralization of TcdB (EC50 [ng/ml]) ata: |
||
|---|---|---|---|
| LD60 | LD77 | LD85 | |
| CA1125 + CA1134 (25:75) | <0.15 | 0.84 | 7.2 |
| CA1125 + CA1134 (50:50) | 0.48 | 1.4 | 4 |
| CA1125 + CA1134 (75:25) | 0.66 | 1.2 | 2.5 |
| CA1125 + CA1151 (25:75) | 0.73 | 1 | 2.1 |
| CA1125 + CA1151 (50:50) | 0.85 | 0.85 | 1.5 |
| CA1125 + CA1151 (75:25) | 1.4 | 1.2 | 8.3 |
| CA1125 + CA1153 (25:75) | 7 | 10 | 27 |
| CA1125 + CA1153 (50:50) | 2.7 | 5.2 | 25.2 |
| CA1125 + CA1153 (75:25) | 2.9 | 7.5 | 30 |
| MDX-1388 | 2.2 | 2.5 | 7.7 |
All data represent one experiment each of three assay replicates. The relative and absolute EC50s are representative of two additional independent assays each of three assay replicates, where the TcdB LD values differ by <20% due to interassay variability of TcdB toxicity to Caco-2 cells.
Table 4.
Biophysical properties of humanized anti-TcdB MAbs
| MAb | Affinity for TcdB1234 (pM) | Valency of binding for TcdB1234 (n) | pI | Fab Tm (°C) | Resistance to shaking aggregation |
|---|---|---|---|---|---|
| CA1125 | 170 | 1 | 9.2 | 79.5 | Good |
| CA1134 | 47 | 2 | 9.3 | 76.4 | Moderate |
| CA1151 | 1,820 | 2 (1.5) | 9.2 | 80.8 | Very Good |
| CA1153 | 260 | 1 | NDa | ND | ND |
| MDX-1388 | 1570 | 2 | 9.2 | 68.2 | Good |
ND, not determined.
Hamster infection study.
CA997, CA1125, CA1151, and the comparator MAbs CDA1 and MDX-1388 were transiently expressed in CHO cells, purified to homogeneity and low % of aggregate, and concentrated to 20 mg/ml in PBS. Each MAb was shipped to Ricerca Biosciences in a blinded manner. Specifically, we did not disclose the number, total dose, concentration, origin, or format/isotype of the MAbs or the comparator until after completion of the study.
The hamster study was designed to closely mimic that used by Babcock et al. (35) in order to enable comparison with historical data. In the Babcock et al. protocol, individual hamsters were dosed i.p. with a MAb on each of four consecutive days with 50 mg/kg of anti-TcdA and 50 mg/kg of anti-TcdB before oral challenge with C. difficile spores/vegetative cells. Hence, each animal had received a total of 200 mg/kg of both anti-TcdA and anti-TcdB. In order to maintain dosing parity with the Babcock et al. study and hence enable direct comparison against MDX-1388, CA1125 and CA1151 were each dosed at 25 mg/kg per day to maintain a total of 200 mg/kg of anti-TcdB. Additional groups of animals were dosed with 1/10 of these amounts, i.e., 5 mg/kg × 4 for each of the anti-TcdA and anti-TcdB antibodies.
The survival data in Fig. 3 show that the infectious dose elicited an early and complete symptomatic infection, with all animals (11/11) dead on day 2 or 3 in the vehicle (PBS) control group. Five days of dosing with vancomycin (5 mg/kg twice per day) protected hamsters through this period, but all the hamsters in this group (5/5) also succumbed to infection on days 10 and 11. These vancomycin controls suggested that individually housed animals had effectively been reinfected from their cage environments or had suffered a spontaneous “reactivation” of the spores remaining within their bodies, as was shown by others using a mouse model (45, 46). Animals dosed at the 50-mg/kg dose with UCB MAbs showed very high levels of protection. One hundred percent of the animals (11/11) survived to day 11, and ∼82% (9/11) survived until the end of the study on day 28. These data reflected higher levels of, and more durable, protection than those demonstrated by Babcock et al. (35), who reported protection of ≤55% beyond day 5. Medarex (MDX) sequence MAbs conferred 100% protection until day 5, but then the animals succumbed fairly steadily until day 20; only 2/11 animals survived until the end of the study on day 28. Hence, UCB MAbs provided higher levels of protection than MDX sequence MAbs. To control the experimental statistical significance level at 0.05, pairwise comparisons between the five groups were conducted using conservative Bonferroni-adjusted significance levels of 0.005 per test. Log-rank tests were used for these pairwise comparisons. Both 50-mg/kg groups were found to confer significant protection (P = 0.0001) relative to the vehicle control, both demonstrating survival beyond that of animals that received the vehicle control. The comparison between the 50-mg/kg groups almost achieved the significance threshold (P = 0.0052), and at the end of the study, the risk of death was more than four times greater in the MDX 50-mg/kg group than in the UCB 50-mg/kg group. Indeed, had we focused on these comparisons and used a Bonferroni-corrected log-rank test of these three key groups, as suggested previously by the literature (35), i.e., PBS control and both 50-mg/kg groups, then the significance threshold would have been 0.017 and the comparison of the two 50-mg/kg groups would have obtained statistical significance.
Fig 3.
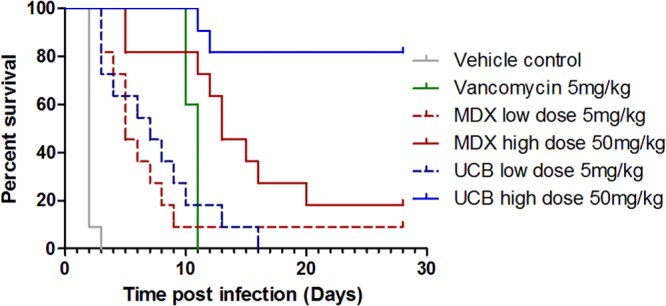
Protection of hamsters using MAb mixtures. All groups contained 11 animals except the vancomycin control group, which contained 5. Four doses of the MAb mixture for each of the anti-TcdA and anti-TcdB components were administered i.p. at the scale indicated on days −3, −2, −1, and 0 relative to infection with C. difficile.
The lower 5-mg/kg doses of both UCB and MDX MAbs were 100% protective only until day 3. Thereafter, the animals dosed with UCB MAbs succumbed steadily to infection until day 16, when there were no survivors, while those dosed with MDX sequence MAbs succumbed steadily to infection until day 9, and 1/11 animals survived thereafter until the end of the study. Both 5-mg/kg groups were also conferred significant protection (P = 0.0001) compared to those that received the vehicle control. The contrast between the 5- and 50-mg/kg × 4 dose groups was perhaps suggestive of the importance of dose early in the infection.
The body weights of the animals were monitored throughout the infection, and gross cecum pathology scores were assigned during the autopsies. The data in Fig. 4 show that animals in both the control and vancomycin-treated groups had no noticeable weight changes before they succumbed to the effects of the infection. In contrast, animals treated with the 50-mg/kg doses of antibodies typically lost weight during the first 5 to 10 days after infection. In many cases, animals which survived to the end of the 28-day study showed recovery of their weight losses. These and data of others suggest that stabilization or reversal of weight loss might be an important feature of survival in response to antibody treatment (47). More animals treated with UCB MAbs showed stabilization or reversal of weight loss than those that received MDX sequence MAbs, but differences in the numbers of survivors between the UCB and MDX sequence MAb treatment groups (9 versus 2) confounded statistical comparisons. One might speculate that protection from gut damage by MAbs might result in reduced fluid loss and earlier resumption of normal eating and digestion and hence rebound of weight loss. This is supported by the cecum pathology observed at autopsy (Table 5). All (11/11) PBS-treated animals showed visible changes to their ceca, with the majority of these (9/11) being severe changes, scored as “dark red” in appearance. Additionally, one PBS-treated animal had visible changes to its small intestine, scored as “red.” In contrast, the majority (9/11) of animals treated with UCB MAbs were scored as “normal,” with the remaining two (both nonsurvivors) showing changes at the lower end of the scoring scale. Animals treated with MDX sequence MAbs had a broader spectrum of scoring: 2/11 animals (both survivors) were judged to have normal ceca, 8/11 to have pink or red ceca, and 1/11 animals observed to have a dark-red cecum. One of the animals with red ceca also had a dark-red ileum. Wet tail was not used as a readout, since it was poorly associated with other symptoms. Few of the animals were judged to have anogenital staining (i.e., wet tail) in any of the groups, and only 1/11 in the PBS group had wet tail.
Fig 4.

Hamster body weight during infection study.
Table 5.
Pathology scores
| Group | No. of animals with: |
||||||
|---|---|---|---|---|---|---|---|
| Cecal pathology |
Small intestine pathology |
||||||
| Black | Dark red | Red | Pink | Normal | Dark red | Red | |
| PBS control | 1 | 9 | 1 | 0 | 0 | 0 | 1 |
| MDX high | 0 | 1 | 4 | 4 | 2 | 1 | 0 |
| UCB high | 0 | 0 | 1 | 1 | 9 | 0 | 0 |
Antibody pharmacokinetics and biodistribution.
It has been shown that infected animals leak serum components such as albumin and immunoglobulin into the gut lumen (11, 48). Antibodies from immunized but healthy animals have been shown to prevent the binding of cholera toxin and bovine serum albumin (BSA) to the gut lining and also that protection is substantially mediated by IgG, rather than IgA or IgM (49, 50). Babcock et al. (35) reported that levels of circulating CDA1 in hamsters were very much lower than anticipated and that 10% of the hamsters had no detectable levels of circulating CDA1. These observations left open the question of whether infection with C. difficile affects the pharmacokinetics (PK) of humanized antibodies in hamsters and hence might be in part the reason why such high levels (200 mg) of antibody are required for full protection. We could find no published data on the serum half-life of human IgG in noninfected or infected hamsters. To this end, we performed a PK and gut biodistribution study using a human IgG1 in noninfected hamsters and developed an antigen-independent assay to measure human IgG in infected hamsters.
Human IgG1 was administered to healthy (uninfected) hamsters both i.p. and s.c. using a single dose of 20 mg/kg, and the clearance from the serum was determined by ELISA. This high dose was selected in order to ensure the ability to measure the PK if it turned out to be very short while also enabling the possibility of detection of the MAb in gut mucosa. The antigen for this antibody was not expressed in hamsters, and thus there were no complications from target-mediated clearance of the antibody. The same human IgG1 (at the same dose) was also administered to mice to act as controls for potential interspecies differences. The data in Fig. 5 show that there were no substantial differences between mouse and hamster PK. Moreover, the data in Table S4 in the supplemental material show that the clearance parameters in hamsters are substantially normal for rodents. The half-lives were 6.2 days (i.p.) and 7.8 days (s.c.), rather like those reported generally for mice and rats. The i.p. route was somewhat quicker (36 h to reach maximum serum concentration) than the s.c. route (76 h), as seen by others in other species. The mucosal linings from 2-cm sections of colon at the end of the study (7 days) were homogenized and assayed using the same ELISA in order to determine if human IgG1 can penetrate the gut mucosa of uninfected hamsters. The data in Table S5 in the supplemental material show than human IgG1 was detectable in the colon mucosa in all animals dosed using both the i.p. and s.c. routes of administration (50).
Fig 5.

Serum pharmacokinetics of a human IgG1 in mice and hamsters. Each data point represents the mean and standard error of samples from four animals per group.
We attempted to determine the PK of CA997, CA1125, and CA1151 in infected hamsters. Serum immunoassays which use antigen as capture or reveal reagents have the potential to underestimate antibody concentrations when antibody:antigen complexes have preformed in vivo. Hence, we developed an antigen-independent mass spectrometry-based method. In short, serum samples were digested with trypsin, and liquid chromatography tandem mass spectrometry (LC MS-MS) (see Text S4 in the supplemental material) was used to detect a signature peptide which was specific for human IgG1 (CH2 domain). We took blood samples only from a restricted number of animals during the experiment (see Materials and Methods for explanation), and the number of samples available at the end of the 28-day experiment was necessarily constrained by the differing numbers of surviving animals in the various groups. The data in Table 6 (additionally plotted as a line graph in Fig. S3 in the supplemental material) show that human IgG was detected in the serum of animals at days 1, 6, and 28 postinfection. A crude comparison of the apparent half-life of neutralizing IgG1 in infected animals versus the calculated half-lives of a human IgG1 in uninfected animals did not suggest that target (toxin)-mediated antibody clearance was a significant feature. Critically, one can see no apparent difference between the serum concentrations of the UCB and MDX 50-mg/kg-dosed animals, showing that the different levels of survival between these two groups were not due to differences in antibody PK.
Table 6.
Detection of human IgG1 in the sera of infected hamsters by an LC MS-MS assaya
| Sampling day postinfection | Serum concn in μg/ml of human IgG for MAb group indicated (animal no.): |
|||
|---|---|---|---|---|
| UCB 5 mg/kg | UCB 50 mg/kg | MDX 5 mg/kg | MDX 50 mg/kg | |
| 1 | 280 (144) | 3,050 (182) | 335 (193) | 3,040 (160) |
| 1 | 302 (145) | 2,790 (183) | 322 (194) | 3,330 (161) |
| 1 | 182 (146) | 2,370 (184) | 260 (195) | 2,990 (162) |
| 6 | 61 (145) | 838 (182) | 103 (200) | 583 (162) |
| 6 | 71 (147) | 645 (183) | 62 (202) | 913 (163) |
| 6 | 45 (149) | 855 (184) | 79 (203) | 1,240 (164) |
| 28 | —b | 116 (182) | <2.5 (203) | 199 (164) |
| 28 | — | 65 (183) | — | 36 (165) |
| 28 | — | 66 (184) | — | — |
| 28 | — | 44 (185) | — | — |
| 28 | — | 101 (186) | — | — |
| 28 | — | 89 (187) | — | — |
| 28 | — | 27 (188) | — | — |
| 28 | — | 31 (189) | — | — |
| 28 | — | 66 (190) | — | — |
The data represent single experimental samples and assay replicates.
—, no surviving animal.
Finally, we were interested in establishing whether immune responses of the hamsters were involved in the survival process, specifically by the mounting of an IgG response against TcdA and TcdB. To that end, we developed an ELISA to detect the presence of hamster anti-TcdB IgG in the sera of infected animals (see Text S5 in the supplemental material). This approach was dependent upon the use of a mouse anti-TcdB IgG1 to create calibration curves, since no hamster anti-TcdB was available. Hence, the assay also depended upon the selection of an anti-hamster IgG which cross-reacted with the mouse anti-TcdB but not substantially against human IgG1. We were unsuccessful in the development of an assay for hamster IgG anti-TcdA using a similar approach due to a very substantial cross-reactivity issue (false positives; data not shown). The data in Table 7 show that the hamsters were capable of mounting strong anti-TcdB IgG responses. One best estimate of the baseline (i.e., false-positive serum titer) using the UCB MAb-treated group was that >7 μg/ml was likely to represent a genuine host IgG response (see Text S6 in the supplemental material). Strong responses were only found in animals which survived to day 28. The MDX-treated group had two animals which survived to day 28. One animal (no. 164) had no significant anti-TcdB IgG titer, while the other (no. 165) had a very high response. The animal with no anti-TcdB IgG response (no. 164) was the MDX MAb-treated survivor of the two which had started to recover weight loss earlier (day 9 versus 28) and more substantially (+34% versus −9% weight change) (Table 6; Fig. 4). A similar pattern of weight loss versus anti-TcdB response was observed for the survivors in the UCB MAb-treated group. All (3/3) of the animals with a strong anti-TcdB response (no. 185, 188, and 190) had net weight losses. Conversely, all (5/5) of the surviving animals (no. 182, 183, 184, 186, and 187) showing net final weight gain in the UCB MAb-treated group had no significant host anti-TcdB IgG response. One other animal (no. 189) had a net weight loss but no anti-TcdB IgG response. Hence, there appeared to be an inverse relationship between weight loss and hamster IgG anti-TcdB response. A plot of weight change versus hamster IgG anti-TcdB response is included in Fig. S4 in the supplemental material, which shows a negative correlation between the weight change and the anti-TcdB response. With all of the animals surviving until day 28 included (9 from the UCB and 2 from the MDX 50-mg/kg groups), the Pearson correlation coefficient was −0.67, and this was statistically significant at the 5% level using a two-tailed test (P = 0.03).
Table 7.
Detection of hamster anti-TcdB IgG in the sera of infected hamstersa
| MAb group and animal no. | Hamster IgG anti-TcdB (μg/ml) |
Final net wt change (% [day]) | Nadir of body wt (day) | ||
|---|---|---|---|---|---|
| Day 1 | Day 6 | Day 28 | |||
| MDX 50 mg/kg | |||||
| 160 | 2.39 | NSb | NS | −29 (12) | 9 |
| 161 | 2.57 | NS | NS | −14 (5) | 2 |
| 162 | 2.36 | 2.32 | NS | −37 (1) | 9 |
| 163 | NS | 2.18 | NS | −52 (20) | 20 |
| 164 | NS | 2.45 | 1.74 | +34 (28) | 6 |
| 165 | NS | NS | >20 | −9 (28) | 16 |
| UCB 50 mg/kg | |||||
| 182 | 9.41 | 7.67 | 5.81 | +35 (28) | 9 |
| 183 | 9.19 | 9.73 | 4.51 | +11 (28) | 9 |
| 184 | 9.73 | 8.81 | 6.68 | +21 (28) | 9 |
| 185 | NS | NS | 12.32 | −53 (28) | 28 |
| 186 | NS | NS | 4.89 | +38 (28) | 6 |
| 187 | NS | NS | 4.34 | +5 (28) | 9 |
| 188 | NS | NS | >20 | −44 (28) | 28 |
| 189 | NS | NS | 2.86 | −34 (28) | 13 |
| 190 | NS | NS | 27.32 | −49 (28) | 28 |
| Vehicle (PBS) controls | |||||
| 171 | <0.3 | NS | NS | +5 (2) | —c |
| 172 | <0.3 | NS | NS | −7 (2) | — |
| 173 | <0.3 | NS | NS | +3 (2) | — |
The data represent means of single experimental samples assayed in duplicate. The upper limit of quantitation was 20 μg/ml; sample 190 was diluted and reassayed to illustrate the scale of a strong response.
NS, not sampled for serum.
PBS-treated animals had no net weight loss.
DISCUSSION
Clostridium difficile infection still represents an important clinical problem which causes very substantial health care costs. Current approved treatments tackle only one aspect of primary infections: reduction of bacterial burden using antibiotics. Another need is to neutralize and clear the toxins that cause the symptoms. Tackling both bacteria and toxin might bring improvements in clinical outcomes, such as a reduction in the duration and severity of diarrhea, along with reduced death and recurrence rates.
We made a mixture of three antibodies which neutralized TcdA and TcdB in vitro and in vivo. These MAbs are “functionally oligoclonal,” as measured by the high valency of binding to the toxin, and hence might combine the manufacturing benefits of MAbs along with the functional benefits of a hyperimmune polyclonal serum. In order to best determine their clinical potential, we compared them against the MAbs which are the most advanced in the clinic, CDA1 and MDX-1388. These MAbs are in two phase III clinical trials (MODIFY I [identifier NCT01241552] and MODIFY II [identifier NCT01513239] [see www.clinicaltrials.gov]) as a mixture designated MK-3415A and alone as MAbs designated MK-3415 and MK-6072, respectively, for reduction in recurrence rates and as a global cure.
The UCB MAb mixture gave 100% (11/11) protection of hamsters until day 11 and ∼82% (9/11) protection at the end of the study on day 28. These were higher and more durable levels of protection than those demonstrated with CDA1 and MDX-1388 by Babcock et al. (35) and herein. One-hundred-percent protection was observed until only day 5 with the CDA1 and MDX-1388 mixture; thereafter, protection decreased until day 20, when only 2/11 (18%) of animals survived. The higher protection levels of the UCB MAbs were supported by observations made using cecum pathology scores and animal weight changes. Animals treated with the UCB MAbs had fewer moderate and severe cecum pathology scores (2/11) than MDX MAb-treated animals (9/11). A greater number of UCB MAb-treated hamsters showed rebound from weight loss than MDX MAb-treated hamsters. It is possible that weight rebound was caused directly by the toxin-neutralizing properties of the UCB MAbs or that it was a result of increased well-being of the animals due to an earlier restoration of normal diet, digestion, and, hence, normal gut microbes (45). Others have shown (42) several-log reductions in the numbers of C. difficile CFU after treatment with neutralizing antitoxin MAbs, perhaps supporting the latter suggestion. The different levels of protection between the UCB and MDX MAb treatment groups were not due to differences in antibody PK. The subtle differences between the Babcock et al. study and the one reported here, including doses and routes of administration of clindamycin (10 mg/kg i.p. versus 5 mg/kg s.c., respectively), infectious strains, and doses (140 spores of B1 versus 3.4 × 106 CFU of ATCC 43596 vegetative cells, respectively), were observed to have little or no effect on the protection test in comparison with published data (35).
The incomplete and waning protection of the lower dosing groups (5 mg/kg, total dose of 20 mg/kg), even when dosed prophylactically, might reflect at least in part complex dynamics in the model between antibody dose (biodistribution and clearance), toxin burden, and relative sensitivity of hamsters to TcdA and TcdB. Human clinical studies using therapeutic dosing suggest that a total dose of 10 mg/kg can offer significant clinical benefit (38). One other hamster infection study of neutralizing MAbs (42) used MAb doses lower than the 50 mg/kg × 4 used here, but these data are not instructive when considering the question of minimum protective dose. The dosing regimen used in our study and by Babcock et al. (35) involved MAb dosing on days −3, −2, −1, and 0, whereas Marozsan et al. (42) utilized “top-up” multidosing during the infection period, with MAb dosing on days −1, 1, 3, and 5. It is probable that the serum hIgG concentrations will differ considerably between the two regimens given the ∼6-day serum half-life of hIgG1 in (uninfected) hamsters and the potential for hIgG1 loss into the gut of infected hamsters. Perhaps in the hamster model, sufficiently high concentrations of antibody must be present during the critical first ∼5 days of infection in order for the intestine to be protected from high levels of toxin. Lower concentrations of neutralizing IgG1 might suffice thereafter for keeping the gut healthy when challenged with lower levels of toxin, such as during a recurrence. The dynamics will be quite different in human disease, not the least in that the substantially longer serum half-life of IgG1 in humans versus hamsters (∼21 versus 6 days) would ensure high drug levels during the first critical week of infection and treatment.
We have demonstrated that some hamsters which survived to day 28 were capable of mounting an anti-TcdB IgG response and, perhaps surprisingly, that there was an inverse correlation between this and hamster weight loss. It is possible that animals with persistent weight loss might have been subjected to a more persistent environmental reinfection challenge. The demonstration that surviving hamsters can mount an antitoxin response is of general interest. The mounting of an effective host antitoxin response following clearance of parenterally administered MAbs would be an excellent clinical outcome for patients in that it might confer a long-lived protection against recurrent infection.
Can we rationalize mechanisms for the higher levels of protection provided by the UCB MAbs than by the MDX sequence MAbs? Our data show that the UCB MAbs had higher affinities, higher potencies (lower EC50s), and higher valencies of toxin binding. Collectively, these were likely to result in more effective toxin neutralization and clearance from the circulation. The data also show that UCB MAbs were more stable (higher Fab Tm), which might confer some benefit for retention of MAb functionality in the gut lumen. Of special interest, however, is the observation that the CDA1 MAb was poorly neutralizing at high TcdA concentrations. In the hamster model (and unlike potential human clinical application), the MAbs were given as sole therapy rather than in addition to antibiotic therapy. The hamster intestine is likely to encounter very high levels (total amounts and local concentrations) of both TcdA and TcdB in the first few days after infection. CDA1 might have been largely ineffective in terms of TcdA activity neutralization in these circumstances, whereas CA997 will likely have retained substantial levels of neutralization. Of additional interest is the difference in protection against TEER loss in response to TcdA. A loss of tight junctions is a known first step toward fluid loss and toxin penetration and, hence, diarrhea and local inflammation (17, 20). The ability to prevent TEER loss in vitro might be considered a marker for protection against early gut damage events in vivo. CDA1 conferred low levels of protection (percent protection and EC50) against TEER loss, whereas CA997 offered a maximum protection level of ∼80% and an EC50 of ∼80 ng/ml. One might imagine that animals treated with CA997 might have suffered less fluid loss and less gut damage than those treated with CDA1. Wet tail was found to be an inaccurate disease indicator in these experiments, but the lower prevalence of cecum damage in the UCB MAb 50-mg/kg group was at least consistent with a benefit from protection against TEER loss.
The higher valencies of binding of these (human IgG1) MAbs might have more relevance in human studies than in hamster studies because of the potential for more effective engagement of Fcγ receptors (FcγRs). The clearance of toxin from the circulation is much faster (minutes rather than hours) (51, 52) when an antigen is decorated with three or more Fc fragments (53). The presence of low levels of toxin in the circulation of infected humans has been a point of some conjecture (54), but toxin was detected using a very sensitive assay in a piglet model of infection and was shown to be the cause of systemic symptoms in mice and piglets (47, 55). Neutralized MAb:antigen complexes have been demonstrated to both educate (56) and vaccinate (57) host immune systems, and antigen:Fc fusion vaccines have been shown to offer mucosal protection (58). In addition, oligoclonal MAb binding has been shown to confer neutralization advantages through altered binding kinetics (59). CA997 was estimated to bind TcdA approximately 12 times, while CDA1 was shown to bind TcdA twice. Hence, there are likely to be considerably different dynamics of toxin decoration and, hence, possibly also of toxin clearance. The anti-TcdB mixture of CA1125 plus CA1151 offers a more defined set of toxin decoration/cross-linking outcomes. CA1125 was shown to only bind to TcdB once and hence is most likely to catalyze TcdB cross-linking of two TcdB molecules. In contrast, CA1151 and MDX-1388 both appear to bind to TcdB twice and thus might be able to bind bivalently to one TcdB molecule or might cross-link two TcdB molecules. Therefore, the UCB MAb mixture is perhaps more capable of both decorating and cross-linking the Clostridium difficile toxins A and B than MDX-1388. In a piglet model of systemic disease, only anti-TcdB antibodies (including MDX-1388) were protective, whereas anti-TcdA antibodies (including CDA1) were not (23).
Of interest regarding ongoing human clinical studies of CDA1 and MDX-1388 (MK-3415A) is the observation that the CDA1 produced by us showed negligible neutralization activity against TcdA of ribotypes 027 and 078, a finding confirmed by others (42). CDA1 also showed incremental reductions of neutralization potential at the higher concentrations of all three TcdAs. These observations might have important implications in the clinic, where 15 to 50% of patients can be found to be infected with ribotype 027 strains. There also remains a need to reduce the impact on patients of the acute effects of infection (diarrhea and gut tissue damage) during the first few days of infection. Effective neutralization of TcdA might have a secondary benefit in that in vitro experiments suggest that the initial damage caused by TcdA (loss of tight junctions) can provide deeper access of the more potent TcdB to underlying tissues and hence enhance its overall inflammatory potential (18, 19, 60). UCB MAbs used proteins of ribotype 027 sequence origin as immunogens and/or screening agents, while assays were performed using that of VPI10463 (ribotype 003). Hence, UCB MAbs were likely to retain good neutralizing activity against ribotype 003 and 027. All together, the UCB MAbs have been shown to be effective against toxins produced by four different strain ribotypes: 003, 027, and 078 in vitro and 012 in vivo.
In a phase II study of CDA1 alone, there was no difference in the recurrence rate versus that of placebo (61), and yet a substantial reduction in the recurrence rate was observed when both CDA1 and MDX-1388 were used together in another phase II study (38). One conclusion from these studies is that TcdB is the only virulence factor which determines the risk of recurrence in humans. Indeed, the Leav et al. study (61) highlighted that the concentration of (host) anti-TcdB-neutralizing antibodies was significantly lower in subjects whose disease recurred than in those whose disease did not recur. An alternative or additional conclusion in light of the data presented here is that the anti-TcdA component of MK-3415A (CDA1) was insufficiently protective to elicit a measurable clinical outcome in humans. It is still not clear at this stage what the relative importance and specific roles of TcdA and TcdB are in human disease or what therapeutic benefits might be observed after their complete neutralization.
In summary, we show data here for humanized IgG1, which as a mixture of three MAbs provided excellent levels of protection in a hamster model of infection. This high level of protection is likely a composite of potent toxin neutralization, high affinity/avidity toxin binding, protection from loss of tight junctions, and antibody stability. Functional oligoclonality was demonstrated through high-level molar binding equivalents during surface plasmon resonance assays. Along with high-affinity toxin binding, this property might also make this MAb mixture a good agent of toxin clearance in humans. The UCB anti-TcdA MAb was capable of effective neutralization of a range of clinically relevant toxin ribotypes at high toxin concentrations and conferred significant protection from TEER loss. Finally, we show that some infected hamsters were capable of mounting their own antitoxin response after MAb-induced survival. The characteristics of these MAbs might offer advantages to patients and health care providers in terms of reducing the duration and severity of diarrhea and the incidence rates of death and recurrence.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tania Boden for immunization support; Andrew Payne and Clare Watkins of UCB for assay development discussions; Soumya Ghosh of Premas Biotech (Gurgaon, India) for reagent expression; Ann O'Leary and colleagues of Ricerca Biosciences LLC for the hamster infection model, discussions, and data analysis; Robert MacDonald (CARE) and Sara Wright for the hamster PK study; Bernie Sweeney for protein expression support; Roly Foulkes and Tim Bourne for project guidance; Ros Walley for statistical analysis; Mark Bodmer for critical reading of the manuscript; and Jon Brazier for supplying R20291 genomic DNA.
All the authors were UCB employees during the research period. K.L.T., A.R.M., D.J.L., and D.P.H. own UCB stock options. This research was wholly funded by UCB as an internal company project.
Footnotes
Published ahead of print 16 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00625-12.
REFERENCES
- 1.Bauer MP, Notermans DW, Van Benthem BHB, Brazier JS, Wilcox MH, Rupnik M, Monnet DL, Van Dissel JT, Kuijper EJ. 2011. Clostridium difficile infection in Europe: a hospital-based survey. Lancet 377:63–73 [DOI] [PubMed] [Google Scholar]
- 2.Wullt M, Odenholt I. 2004. A double-blind randomized controlled trial of fusidic acid and metronidazole for treatment of an initial episode of Clostridium difficile-associated diarrhoea. J. Antimicrob. Chemother. 54:211–216 [DOI] [PubMed] [Google Scholar]
- 3.Oake N, Taljaard M, Van Walraven C, Wilson K, Roth V, Forster AJ. 2010. The effect of hospital-acquired Clostridium difficile infection on in-hospital mortality. Arch. Intern. Med. 170:1804–1810 [DOI] [PubMed] [Google Scholar]
- 4.O'Brien JA, Lahue BJ, Caro JJ, Davidson DM. 2007. The emerging infectious challenge of Clostridium difficile-associated disease in Massachusetts hospitals: clinical and economic consequences. Infect. Control Hosp. Epidemiol. 28:1219–1227 [DOI] [PubMed] [Google Scholar]
- 5.Dubberke ER, Wertheimer AI. 2009. Review of current literature on the economic burden of Clostridium difficile infection. Infect. Control Hosp. Epidemiol. 30:57–66 [DOI] [PubMed] [Google Scholar]
- 6.Forster AJ, Taljaard M, Oake N, Wilson K, Roth V, Van Walraven C. 2012. The effect of hospital-acquired infection with Clostridium difficile on length of stay in hospital. CMAJ 184:37–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713 [DOI] [PubMed] [Google Scholar]
- 8.Seal D, Borriello SP, Barclay F, Welch A, Piper M, Bonnycastle M. 1987. Treatment of relapsing Clostridium difficile diarrhoea by administration of a non-toxigenic strain. Eur. J. Clin. Microbiol. 6:51–53 [DOI] [PubMed] [Google Scholar]
- 9.Shim JK, Johnson S, Samore MH, Bliss DZ, Gerding DN. 1998. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet 351:633–636 [DOI] [PubMed] [Google Scholar]
- 10.Lyerly DM, Saum KE, Macdonald DK, Wilkins TD. 1985. Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. 47:349–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lima AAM, Lyerly DM, Wilkins TD, Innes DJ, Guerrant RL. 1988. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect. Immun. 56:582–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corthier G, Muller MC, Wilkins TD, Lyerly D, L'Haridon R. 1991. Protection against experimental pseudomembranous colitis in gnotobiotic mice by use of monoclonal antibodies against Clostridium difficile toxin A. Infect. Immun. 59:1192–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vernet A, Corthier G, Dubos-Ramare F, Parodi AL. 1989. Relationship between levels of Clostridum difficile toxin A and toxin B and cecal lesions in gnotobiotic mice. Infect. Immun. 57:2123–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothman SW, Brown JE, Diecidue A, Foret DA. 1984. Differential cytotoxic effects of toxin A and B isolated from Clostridum difficile. Infect. Immun. 46:324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaves-Olarte E, Weidmann M, Von Eichel-Streiber C, Thelestam M. 1997. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J. Clin. Invest. 100:1734–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor NS, Thorne GM, Bartlett JG. 1981. Comparison of two toxins produced by Clostridium difficile. Infect. Immun. 34:1036–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hecht G, Koutsouris A, Pothoulakis C, Lamont JT, Madara JL. 1992. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 102:416–423 [DOI] [PubMed] [Google Scholar]
- 18.Du T, Alfa MJ. 2004. Translocation of Clostridium difficile toxin B across polarised Caco-2 monolayers is enhanced by toxin A. Can. J. Infect. Dis. 15:83–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. 2003. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 125:413–420 [DOI] [PubMed] [Google Scholar]
- 20.Nusrat A, Von Eichel-Streiber C, Turner JR, Verkade P, Madara JL, Parkos CA. 2001. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect. Immun. 69:1329–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flegel WA, Muller F, Daubener W, Fischer HG, Hadding U, Northoff H. 1991. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect. Immun. 59:3659–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linevsky JK, Pothoulakis C, Keates S, Warny M, Keates AC, Lamont JT, Kelly CP. 1997. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am. J. Physiol. 273:G1333–G1340 [DOI] [PubMed] [Google Scholar]
- 23.Steele J, Mukherjee J, Parry N, Tzipori S. 2013. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis. 207:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim PH, Iaconis JP, Rolfe RD. 1987. Immunization of adult hamsters against Clostridium difficile-associated ileocecitis and transfer of protection to infant hamsters. Infect. Immun. 55:2984–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giannasca PJ, Zhang ZX, Lei WD, Boden JA, Giel MA, Monath TP, Thomas WD. 1999. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect. Immun. 67:527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotloff KL, Wasserman SS, Losonsky GA, Thomas W, Nichols R, Edelman R, Bridwell M, Monath TP. 2001. Safety and immunogenicity of increasing doses of a Clostridium difficile toxoid vaccine administered to healthy adults. Infect. Immun. 69:988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sougioultzis S, Kyne L, Drudy D, Keates S, Maroo S, Pothoulakis C, Giannasca PJ, Lee CK, Warny M, Monath TP, Kelly CP. 2005. Clostridium difficile toxoid vaccine in recurrent C. difficile-associated diarrhea. Gastroenterology 128:764–770 [DOI] [PubMed] [Google Scholar]
- 28.Kovaiou RD, Herndler-Brandstette D, Grubeck-Loebenstein B. 2007. Age-related changes in immunity: implications for vaccination in the elderly. Expert Rev. Mol. Med. 9:1–17 [DOI] [PubMed] [Google Scholar]
- 29.Lee BY, Popovich MJ, Tian Y, Bailey RR, Ufberg PJ, Wiringa AE, Muder RR. 2010. The potential value of Clostridium difficile vaccine: an economic computer simulation model. Vaccine 28:5245–5253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyerly DM, Bostwick EF, Binion SB, Wilkins TD. 1991. Passive immunization of hamsters against disease caused by Clostridium difficile by use of bovine immunoglobulin G concentrate. Infect. Immun. 59:2215–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelly CP, Pothoulakis C, Vavva F, Castagliuolo I, Bostwick EF, O'Keane JC, Keates S, Lamont JT. 1996. Anti-Clostridium difficile bovine immunoglobulin concentrate inhibits cytotoxicity and enterotoxicity of C. difficile toxins. Antimicrob. Agents Chemother. 40:373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kink JA, Williams JA. 1998. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 66:2018–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun X, Wang H, Zhang Y, Chen K, Davis B, Feng H. 2011. Mouse relapse model of Clostridium difficile infection. Infect. Immun. 79:2856–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts A, McGlashan J, Al-Abdulla I, Ling R, Denton R, Green S, Coxon S, Landon J, Shone C. 2012. Development and evaluation of an ovine antibody-based platform for treatment of Clostridium difficile infection. Infect. Immun. 80:875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatring N, Stack AM, Lowy I, Graziano R, Molrine D, Ambrosino DM, Thomas WD., Jr 2006. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 74:6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demarest SJ, Hariharan M, Elia M, Salbato J, Jin P, Bird C, Short JM, Kimmel BE, Dudley M, Woodnutt G, Hansen G. 2010. Neutralization of Clostridium difficile toxin A using antibody combinations. MAbs. 2:190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hussack G, Arbabi-Ghahroudi M, Van Faassen H, Songer JG, Ng KKS, Mackenzie R, Tanha J. 2011. Neutralisation of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J. Biol. Chem. 286:8961–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowy IMD, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD, Jr, Leney M, Sloan S, Hay CA, Ambrosino DM. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 362:197–205 [DOI] [PubMed] [Google Scholar]
- 39.Lightwood D, O'Dowd V, Carrington B, Veverka V, Carr MD, Tservistas M, Henry AJ, Smith B, Tyson K, Lamour S, Sarkar K, Turner A, Lawson AD, Bourne T, Gozzard N, Palframan R. 2012. The discovery, engineering and characterisation of a highly potent anti-human IL-13 Fab fragment designed for administration by inhalation. J. Mol. Biol. 425:577–593 [DOI] [PubMed] [Google Scholar]
- 40.Heads JT, Adams R, D'Hooghe LE, Page MJT, Humphreys DP, Popplewell AG, Lawson AD, Henry AJ. 2012. Relative stabilities of IgG1 and IgG4 Fab domains: influence of the light-heavy interchain disulfide bond architecture. Protein Sci. 21:1315–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyerly DM, Carrig PE, Wilkins TD. 1989. Nonspecific binding of mouse monoclonal antibodies to Clostridium difficile toxins A and B. Curr. Microbiol. 19:303–306 [Google Scholar]
- 42.Marozsan AJ, Ma D, Nagashima KA, Kennedy BJ, Kang YK, Arrigale RR, Donovan GP, Magargal WW, Maddon PJ, Olson WC. 2012. Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J. Infect. Dis. 206:706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ambrosino D, Babcock GJ, Broering T, Graziano R, Hernandez HJ, Lowy I, Mandell R, Molrine D, Thomas WD, Jr, Zhang HF. December 2009. Antibodies against Clostridium difficile toxins and uses thereof. US patent 7,625,559B2. [Google Scholar]
- 44.Garber E, Demarest SJ. 2007. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem. Biophys. Res. Commun. 355:751–757 [DOI] [PubMed] [Google Scholar]
- 45.Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, Deakin LJ, Pickard DJ, Duncan SH, Flint HJ, Clark TG, Parkhill J, Dougan G. 2012. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 8:e1002995 doi:10.1371/journal.ppat.1002995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. 2012. Profound alteration of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile colitis. Infect. Immun. 80:62–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, Feng H. 2012. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J. Infect. Dis. 205:384–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitchell TJ, Ketley JM, Haslam SC, Stephen J, Burdon DW, Candy DCA, Daniel R. 1986. Effect of toxin A and B of Clostridium difficile on rabbit ileum and colon. Gut 27:78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu AL, Walker WA. 1976. Immunological control mechanism against cholera toxin: interference with toxin binding to intestinal cells. Infect. Immun. 14:1034–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walker WA, Isselbacher KJ, Bloch KJ. 1973. Intestinal uptake of macromolecules. II. Effect of parenteral immunization. J. Immunol. 111:221–226 [PubMed] [Google Scholar]
- 51.Yousaf N, Howard JC, Williams BD. 1986. Studies in cobra venom factor treated rats of antibody coated erythrocyte clearance by the spleen: differential influence of red blood cell antigen number on the inhibitory effects of immune complexes on Fc dependent clearance. Clin. Exp. Immunol. 66:654–660 [PMC free article] [PubMed] [Google Scholar]
- 52.Davies KA, Hird V, Stewart S, Sivolapenko GB, Jose P, Epenetos AA, Walport MJ. 1990. A study of in vivo immune complex formation and clearance in man. J. Immunol. 144:4613–4620 [PubMed] [Google Scholar]
- 53.Mannik M, Arend WP, Hall AP, Gilliland BC. 1971. Studies on antigen-antibody complexes. J. Exp. Med. 133:713–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jacob SS, Sebastian JC, Hiorns D, Jacob S, Mukerjee PK. 2004. Clostridium difficile and acute respiratory distress syndrome. Heart Lung 33:265–268 [DOI] [PubMed] [Google Scholar]
- 55.Steele J, Feng H, Parry N, Tzipori S. 2010. Piglet models of acute or chronic Clostridium difficile illness. J. Infect. Dis. 201:428–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshida M, Kobayashi K, Kuo TT, Bry L, Glickman JN, Claypool SM, Kaser A, Nagaishi T, Higgins DE, Mizoguchi E, Wakatsuki Y, Roopenian DC, Mizoguchi A, Lencer WI, Blumberg RS. 2006. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Invest. 116:2142–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu DZ, Huang KL, Zhao K, Xu LF, Shi N, Yuan ZH, Wen YM. 2005. Vaccination with recombinant HBsAg-HBIG complex in healthy adults. Vaccine 23:2658–2664 [DOI] [PubMed] [Google Scholar]
- 58.Ye L, Zeng R, Bai Y, Roopenian DC, Zhu X. 2011. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat. Biotechnol. 29:158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanna PP, Ramiro-Ibanez F, De Logu D. 2000. Synergistic interactions of antibodies in rate of virus neutralisation. Virology 270:386–396 [DOI] [PubMed] [Google Scholar]
- 60.Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacheri J, Bischof G, Cosentini E, Feil W, Schiessel R, Lamont JT, Wenzl E. 1995. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J. Clin. Invest. 95:2004–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leav BA, Blair B, Leney M, Knauber M, Reilly C, Lowy I, Gerding DN, Kelly CP, Katchar K, Baxter R, Ambrosino D, Molrine D. 2010. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28:965–969 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.