

Abstract
Objective
The purpose of the present study was to evaluate the effect of atorvastatin on oxidative stress and angiogenesis in ischemic myocardium in a clinically relevant porcine model of the metabolic syndrome.
Methods
Sixteen Ossabaw pigs were fed either a high-fat diet alone or a high-fat diet supplemented with atorvastatin (1.5 mg/kg daily) for 14 weeks. Chronic myocardial ischemia was induced by ameroid constrictor placement around the circumflex artery. After 6 months of the diet, myocardial perfusion was measured at rest and with demand pacing. The heart was harvested for analysis of perfusion, microvessel relaxation, protein expression, and oxidative stress.
Results
Both groups had similar endothelium-dependent microvessel relaxation to adenosine diphosphate and endothelium-independent relaxation to sodium nitroprusside. Myocardial perfusion in the ischemic territory was also not significantly different either at rest or with demand pacing. Atorvastatin treatment increased total myocardial protein oxidation and serum lipid peroxidation. However, the expression of markers of oxidative stress, including NOX2, RAC1, myeloperoxidase, and superoxide dismutase 1, 2, and 3, were not statistically different. The expression of proangiogenic proteins endothelial nitric oxide synthase, phosphorylated endothelial nitric oxide synthase (Ser 1177), phosphorylated adenosine monophosphate kinase (Thr 172), phosphorylated extracellular signal-regulated kinase (T202, Y204), and vascular endothelial growth factor were all upregulated in the atorvastatin group.
Conclusions
Atorvastatin increased the capillary and arteriolar density and upregulated the proangiogenic proteins endothelial nitric oxide synthase and phosphorylated endothelial nitric oxide synthase, phosphorylated adenosine monophosphate kinase, phosphorylated extracellular signal-regulated kinase, and vascular endothelial growth factor in a swine model of the metabolic syndrome. However, it failed to increase myocardial perfusion. Atorvastatin treatment was associated with increased myocardial and serum oxidative stress, which might contribute to the lack of collateral-dependent perfusion in the setting of angiogenesis.
3-Hydroxy-3-methylglutaryl–co-enzyme A reductase inhibitors, or statins, are a widely prescribed medication for the treatment and prevention of coronary artery disease. In addition to its lipid-lowering effects, statins also protect against ischemia–reperfusion injury, reduce vascular inflammation, and improve endothelial function.1-3 Another important pleiotropic effect is statin’s dose-dependent biphasic effect on angiogenesis.4,5 At low doses, statins induce angiogenesis by promoting endothelial cell migration, maturation, and survival. At high statin doses, the proangiogenic effect is reversed, and statins become anti-angiogenic by inducing endothelial cell apoptosis. In a previous study conducted in our laboratory, high-dose atorvastatin (3 mg/kg) administration in hypercholesterolemic swine resulted in improved endothelial function without improving the angiogenic response in the chronically ischemic myocardium.6,7 Given atorvastatin’s known dose-dependent effect on angiogenesis, we hypothesized that low-dose atorvastatin (1.5 mg/kg) would result in improved angiogenesis in chronically ischemic myocardium in a large animal model of the metabolic syndrome.
METHODS
Animal Model
Sixteen intact male Ossabaw miniswine (Purdue Ossabaw Facility, Indiana University, Indianapolis, Ind) were fed 500 g/day of high-cholesterol chow consisting of 4% cholesterol, 17.2% coconut oil, 2.3% corn oil, 1.5% sodium cholate, and 75% regular chow (Sinclair Research, Columbia, Mo). After 14 weeks of diet initiation, all pigs underwent surgical placement of an ameroid constrictor to induce chronic myocardial ischemia (see “Surgical Interventions”). Postoperatively, the 8 pigs continued to consume an oral hypercaloric/hypercholesterolemic diet (OHC) alone, and the diet of the other 8 pigs was supplemented with oral 1.5 mg/kg atorvastatin daily (OHCS). At 11 weeks after ameroid constrictor placement, all the pigs were weighed and underwent functional cardiac and hemodynamic measurements, were put to death, and the cardiac tissue was harvested. All the pigs were observed to ensure complete consumption of food and supplement, had unlimited access to water, and were housed in a warm, nonstressful environment for the duration of the experiment.
Surgical Interventions
Anesthesia
Anesthesia was induced with an intramuscular injection of telazol (4.4 mg/kg). The pigs were endotracheally intubated and mechanically ventilated at 12 to 20 breaths/min. General anesthesia was maintained with a gas mixture of oxygen at 1.5 to 2 L/min and isoflurane at 0.75% to 3.0% concentration.
Ameroid constrictor placement
The pigs were given a single dose of intravenous enrofloxacin, 5 mg/kg, for antibiotic prophylaxis, and general anesthesia was induced and maintained. The pigs were prepared and draped in the usual sterile fashion. The heart was exposed through a left minithoracotomy and pericardiotomy. The left atrial appendage was retracted, and the proximal left circumflex artery was dissected immediately distal to the left main coronary artery. The circumflex artery was occluded for 2 minutes, during which, 5 mL of isotope-labeled gold microspheres (BioPhysics Assay Laboratory, Worcester, Mass) was injected into the left atrium to establish shadow labeling of the ischemic myocardium. The ameroid constrictor was placed around the proximal left circumflex artery, just after its branching from the left main coronary artery (Research Instruments SW, Escondito, Calif). The pericardium was loosely reap-proximated, followed by a layered closure of the surgical incision. Postoperative pain was controlled with a single dose of intramuscular buprenorphine (0.03 mg/kg) and a 72-hour fentanyl patch (4 μg/kg). All the pigs received 325 mg of aspirin daily starting 1 day preoperatively and continuing for 5 days for prophylaxis against thromboembolic events. All the pigs continued the perioperative antibiotic (oral enrofloxacin 68 mg/day for 5 days).
Cardiac harvest
Blood samples were drawn from the jugular vein before death and tissue harvest. Blood glucose measurements were taken at baseline (fasting) and 30 and 60 minutes after an intravenous 0.5-mg/kg dextrose infusion. Under general anesthesia, a 5F arterial sheath was placed in the right femoral artery by cutdown, and coronary angiography was performed with an Amplatz R1 catheter (Cordis Corp, Bridgewater, NJ) and advanced to the right and left coronary artery ostia. Contrast was injected into each side to visualize the coronary vessels. The angiograms were read by a cardiologist unaware of the experimental groups and scored according to the Thrombolysis in Myocardial Infarction myocardial perfusion grade (score 0-3). The heart was then exposed by way of a median sternotomy. After microsphere injection (see “Myocardial Perfusion”), the pigs were killed by exsanguination, and chronically ischemic myocardial samples from the left circumflex territory were collected for additional analysis. The cardiac tissue was rapidly frozen in liquid nitrogen for protein expression and histologic analysis and placed in 4°C Krebs solution for microvessel studies or desiccated at 60°C for perfusion analysis.
The Institutional Animal Care and Use Committee of the Rhode Island Hospital approved all experiments. The pigs were cared for in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health publication no. 5377-3, 1996).
Myocardial Perfusion
Myocardial perfusion was measured by injecting gold isotope-labeled microspheres (Biophysics Assay Laboratory) into the left atrium at ameroid placement during a brief left circumflex artery occlusion. At the final operation, Lutetium microspheres were injected into the left atrium at rest, and Europium microspheres were injected during pacing at 160 beats/min. During microsphere injection, the blood was simultaneously withdrawn from a femoral artery catheter. Samples of the ischemic myocardium in the circumflex artery distribution and blood were dried at 60°C for more than 48 hours, and the microsphere density was quantified using a gamma counter after exposure to neutron bean radiation (Biophysics Assay Laboratory). The myocardial blood flow to each sample was calculated using the following equation:
Microvessel Studies
Epicardial arterioles ranging in size from 80 to 120 μm in diameter and 1 to 2 mm in length from the chronically ischemic left circumflex territory were isolated, cannulated, and pressurized (40 mm Hg) with dual micropipettes in a microvessel chamber, as previously described.8 The vessels were preconstricted by 20% to 50% of their baseline diameter with the thromboxane-A2 analog U46619 (0.1-1.0 μM). The microvessels were exposed to an endothelium-dependent vasodilator adenosine-5′-diphosphate (10−9 to 10−4 mol/L), which is a receptor-mediated vasodilator that acts mainly through nitric oxide. The microvessels were also treated with an endothelium-independent vasodilator superoxide dismutase (SOD) (10−9 to 10−4 mol/L), a cyclic guanosine monophosphate-mediated vasodilator (Sigma-Aldrich, St Louis, Mo), as previously described.8 The percentage of relaxation from the initial preconstricted diameter was recorded.
Immunohistochemical Staining for Angiogenesis
The frozen myocardium was cut into 12-μm-thick sections and fixed in 10% formalin for 10 minutes. The sections were blocked with 1% bovine serum albumin in phosphate-buffered saline for 1 hour at room temperature and incubated with antibodies against the porcine endothelial marker CD-31 (R&D Systems, Minneapolis, Minn) and smooth muscle actin (Sigma-Aldrich), followed by the appropriate Alexa-Fluor conjugated antibody (Jackson ImmunoResearch, West Grove, Pa) for 45 minutes. The slides were then mounted with Vectashield with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, Calif). Images were captured at 20× magnification with a Nikon E800 Eclipse microscope (Nikon, Tokyo, Japan) at the same exposure in 3 random fields. Capillaries were defined as structures 5 to 25 μm2 in a cross-sectional area, and arterioles were defined by colocalization of smooth muscle actin and CD31 staining. Arteriolar density and capillary density were measured using Image J software (National Institutes of Health, Bethesda, Md) in a blinded fashion. The percentage of capillary and arteriolar density for each pig was averaged from the 3 randomly selected myocardial tissue sections.
Oxidative Stress Assays
Oxidized proteins from chronically ischemic myocardium were detected using a commercial kit (Oxyblot, Millipore, Bellerica, Mass). The carbonyl groups in oxidized protein side chains are derivatized to 2,4-dinitrophenyl (DNP)-hydrazone by incubating lysates with 2,4-dinitrophenylhydrazine. The DNP-derivatized proteins were separated on a polyacrylamide gel electrophoresis and transferred to a polyvinyldine diflouride membrane. The membranes were probed with primary antibody to the DNP moiety of the proteins, followed by incubation with a horseradish peroxidase-linked secondary antibody. Immune complexes were visualized with enhanced chemiluminescense, and the images were captured with a digital camera system (G-Box, Syngene, Cambridge, United Kingdom). Band densitometry was quantified as arbitrary light units using Image-J software (National Institutes of Health).
A commercial enzyme-linked immunosorbent assay kit was used to detect the serum 8-isoprostane levels as a marker of serum lipid peroxidation (Cayman Chemical, Ann Arbor, Mich). 8-Isoprostane is an eicosanoid produced by the oxidation of phospholipids by oxygen free radicals. Serum samples, 8-isoprostane-acetylcholinesterase conjugate, and 8-isoprostane antiserum were added to a microplate coated with mouse monoclonal antibody. Acetylcholinesterase substrate was added, resulting in an enzymatic reaction that produced a distinct yellow that can be detected spectrophotometrically at 412 nm. The signal intensity was inversely proportional to the serum 8-isoprostane levels. 8-Isoprostane levels were quantified using the standards provided by the manufacturer.
Protein Expression
Forty micrograms of the radioimmunoprecipitation assay (Boston Bio-Products, Ashland, Mass) soluble fraction of myocardial lysates were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis 3% to 8% tris-acetate gel (NuPage Novex Mini Gel; Invitrogen, Carlsbad, Calif) for molecular weight targets greater than 100 kD and 4% to 12% Bis-Tris gels for molecular weight targets less than 100 kD (NuPage Novex Mini Gel, Invitrogen). The protein was then transferred to polyvinylidene difluoride membranes (Millipore, Bedford, Mass) and incubated overnight at 4°C with primary antibodies at the dilutions recommended by the manufacturer against phosphorylated Akt (Ser 473), Akt, phosphorylated endothelial nitric oxide synthase (peNOS; Ser 1177), eNOS, RAC1, vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2), extracellular signal-regulated kinase (ERK), phosphorylated ERK (pERK; T202, Y204; all from Cell Signaling, Danvers, Mass), SOD3 and myeloperoxidase (both from Abcam, Cambridge, Mass), angiostatin and VEGF (both from Millipore, Billerica, Mass), SOD1 and SOD2 (both from Stressgen Farmingdale, NY), and NOX2 (Sigma-Aldrich). The membranes were incubated with the appropriate horseradish peroxidase-linked secondary antibody for 1 hour at room temperature (Jackson ImmunoResearch). The immune complexes were visualized with enhanced chemiluminescense, and the images were captured with a digital camera system. Band densitometry was quantified as arbitrary light units using Image-J software (National Institutes of Health). All membranes were probed with glyceral-dehyde-3-phosphate dehydrogenase (Cell Signaling) to correct for loading error.
Histologic Examination
Trichrome and picosirius red staining was performed on frozen tissue sections by the Pathology and Histology Core Facility at Rhode Island Hospital. Images were obtained using Aperio ScanScope technology (Aperio, Vista, Calif) and captured at 5× magnification.
Statistical Analysis
All results are reported as mean ± standard error of the mean. Microvessel responses and postdextrose challenge serum glucose levels were analyzed with 2-way analysis of variance followed by a post-hoc Bonferroni test. Student t test was used to compare the mean values of all other studies using GraphPad Prism, version 5.0 Software (GraphPad Software Inc, San Diego, Calif).
RESULTS
Animal Model
All the pigs included in the analysis survived the entire experiment. One pig in the OHC group and 2 in the OHCS group died 2, 6, and 1 day postoperatively, respectively. Necropsy did not reveal a clear myocardial infarction or other cause of death, and it was assumed they died after acute arrhythmia leading to sudden cardiac death. The pigs that did not survive to completion of the experiment were excluded from the analysis and replaced with new pigs.
In a previously published study, we reported that the OHC group developed the hallmarks of the metabolic syndrome, including obesity, insulin resistance, glucose intolerance, dyslipidemia, and hypertension, compared with the Ossabaw swine fed normal chow.9 In the present study, the body mass index and blood glucose level in response to the dextrose challenge was similar in the OHC and OHCS groups (Table 1). Also, the Thrombolysis in Myocardial Infarction perfusion grade in the collateral dependent zone was not statistically different between the 2 groups.
TABLE 1.
Thrombolysis in Myocardial Infarction scores, body mass index, and dextrose challenge results
| Variable | OHC | OHCS | P value |
|---|---|---|---|
| TIMI | 1.71 ± 0.36 | 1.50 ± 0.34 | .68 |
| BMI (mg/kg2) | 34.87 ± 0.74 | 37.31 ± 1.14 | .09 |
| Dextrose challenge (min) | |||
| 0 | 59.63 ± 9.56 | 54.13 ± 4.41 | .20* |
| 30 | 214.10 ± 21.06 | 206.00 ± 13.75 | |
| 60 | 130.00 ± 21.90 | 93.00 ± 14.79 |
No difference seen in TIMI, BMI, or response to dextrose challenge in OHC and OHCS groups. OHC, Oral hypercaloric/hypercholesterolemic diet; OHCS, oral hypercaloric/hypercholesterolemic diet plus 1.5 mg/kg atorvastatin daily; TIMI, Thrombolysis in Myocardial Infarction; BMI, body mass index.
Analysis of variance.
Myocardial Perfusion
Myocardial perfusion to the chronically ischemic circumflex artery territory was measured at the final operation, and no difference in flow was found at rest between the OHC and OHCS groups (0.61 ± 0.08 mL/min/g and 0.56 ± 0.07 mL/min/g, respectively; P =.62). Also, no difference was seen in myocardial perfusion during demand pacing at 160 beats/min in the OHC and OHCS groups (0.55 ± 0.03 mL/min/g and 0.38 ± 0.10 mL/min/g, respectively; P =.13; Figure 1).
FIGURE 1.
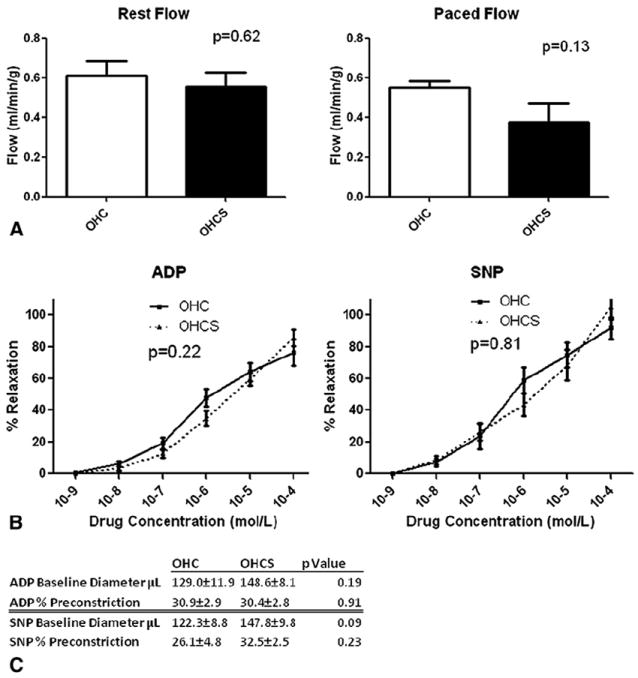
Myocardial perfusion and coronary microvessel reactivity. A, Myocardial perfusion at rest and with demand pacing. No difference was seen in myocardial perfusion at rest or during demand pacing at 160 beats/min. B, Microvessel reactivity. No difference was seen in microvessel reactivity to endothelium-dependent (adenosine diphosphate [ADP]) or endothelium-independent (sodium nitroprusside [SNP]) vasodilators in oral hypercaloric/hypercholesterolemic diet (OHC) and oral hypercaloric/hypercholesterolemic diet plus 1.5 mg/kg atorvastatin daily (OHCS) groups. C, Baseline microvessel diameter and percentage of preconstriction. No difference was seen in baseline diameter or percentage of preconstriction in ADP- or SNP-treated microvessels.
Microvessel Relaxation
Microvessel relaxation in response to adenosine-5′-diphosphate, an endothelium-dependent vasodilator, and sodium nitroprusside (SNP), an endothelium-independent vasodilator, was not significantly different in the OHCS group compared with the OHC group (2-way analysis of variance, P =.22 and P =.81, respectively; Figure 1). The baseline diameter and percentage of preconstriction was also not significantly different in the adenosine-5′-diphosphate and SNP-treated microvessels in each group (Figure 1).
Angiogenesis
The percentage of capillary density/high power field in the OHCS group was significantly greater than in the OHC group (13.9 ± 1.2% vs 9.5 ± 0.4%, P =.02). The percentage of arteriolar density/high power field was also significantly increased in the OHCS group compared with the OHC group (7.71 ± 0.8% vs 3.2 ± 0.9%, P =.003; Figure 2). Significant upregulation was seen of the proangiogenic proteins eNOS (fold change compared with that for OHC, 1.67 ± 0.14; P =.001), peNOS (1.26 ± 0.06, P =.01), phosphorylated adenosine monophosphate kinase (AMPK) (1.62 ± 0.14, P =.03), pERK (3.03 ± 0.47, P =.002), and VEGF (1.29 ± 0.08, P =.04) in the OHCS group and a significant decrease in Akt (0.72 ± 0.07, P =.01) and phosphorylated Akt (0.56 ± 0.08, P =.02). Also, a trend was seen toward decreased anti-angiogenesis protein angiostatin in the OHCS group, but this did not reach statistical significance (0.58 ± 0.08, P =.13; Figure 2).
FIGURE 2.
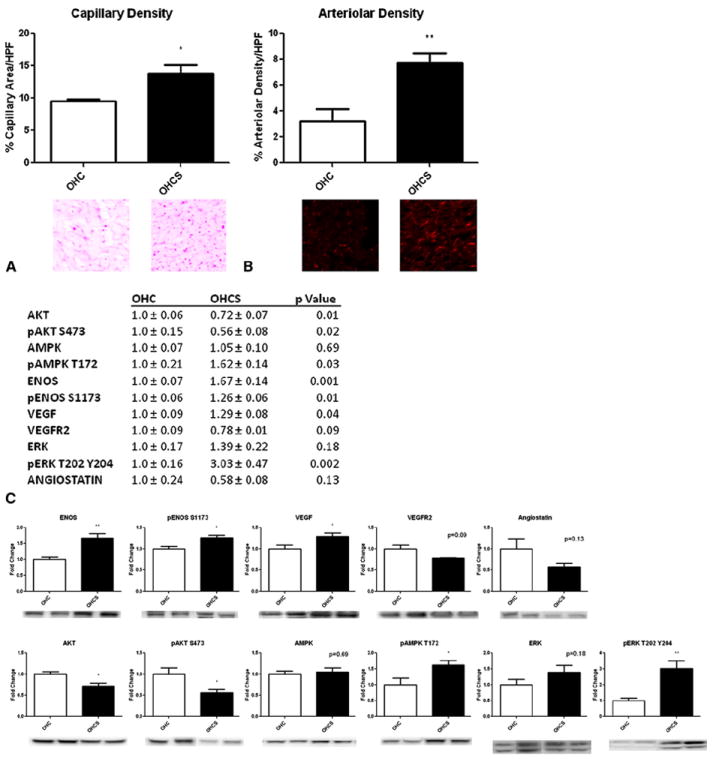
Atorvastatin and angiogenesis. A, Endothelial cell density staining for CD31. Significant increase seen in capillary density in oral hypercaloric/hypercholesterolemic diet plus 1.5 mg/kg atorvastatin daily (OHCS) compared with oral hypercaloric/hypercholesterolemic diet (OHC) groups. B, Arteriolar density staining for smooth muscle actin. No significant increase was seen in arteriolar density in OHCS group compared with OHC. C, Western blot analysis of angiogenesis protein expression showing a significant increase in proangiogenic proteins endothelial nitric oxide synthase (eNOS), phosphorylated eNOS (peNOS), phosphorylated adenosine monophosphate kinase (pAMPK), phosphorylated extracellular signal-regulated kinase (pERK), and vascular endothelial growth factor (VEGF) and a significant decrease in Akt and phosphorylated Akt (pAkt). Also, a trend was seen toward decreased anti-angiogenesis protein angiostatin. *P<.05 and **P<.01. AMPK, Adenosine monophosphate kinase; HPF, high power field; VEGFR2, VEGF receptor 2.
Oxidative Stress
Total myocardial protein oxidation as measured by the Oxyblot assay (Millipore) was significantly increased in the OHCS group (fold change compared with that for OHC, 1.39 ± 0.11; P =.04). Also, significantly increased lipid peroxidation, as measured by the 8-isoprostane levels, was seen in the OHCS group (571.4 ± 72.8 pg/mL) compared with OHC (373.0 ± 29.9 pg/mL, P =.04). Although a significant increase was seen in protein and lipid oxidation, no difference was found in the expression of the proteins involved in oxidative stress, including myeloperoxidase, NOX2, SOD1, SOD2, SOD3, or RAC1 (Figure 3).
FIGURE 3.
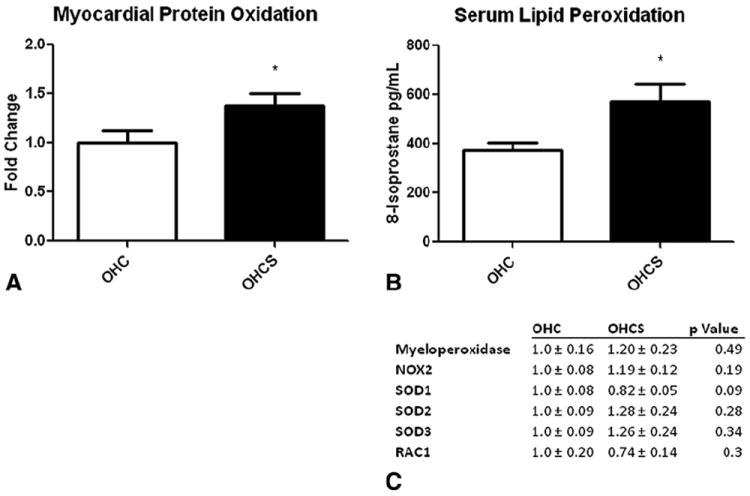
Oxidative stress profile. A, Myocardial protein oxidation showing significantly increased protein oxidation in oral hypercaloric/hypercholesterolemic diet plus 1.5 mg/kg atorvastatin daily (OHCS) compared with oral hypercaloric/hypercholesterolemic diet (OHC) groups. B, Serum lipid peroxidation. Significantly increased lipid peroxidation was seen in OHCS group compared with OHC group. C, Western blot analysis of oxidative stress protein expression showing no difference in expression of proteins involved in oxidative stress. *P<.05. AMPK, Adenosine monophosphate kinase; eNOS, endothelial nitric oxide synthase; pAkt, phosphorylated Akt; pAMPK, phosphorylated AMPK; peNOS, phosphorylated eNOS; VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
Myocardial Collagen and Fibrosis
The chronically ischemic myocardium was stained for collagen (picosirius red stain) and fibrosis (trichrome stain), and no difference was seen in the collagen area fraction or fraction of intramyocardial fibrosis (P =.26 and P =.24, respectively; Figure 4).
FIGURE 4.
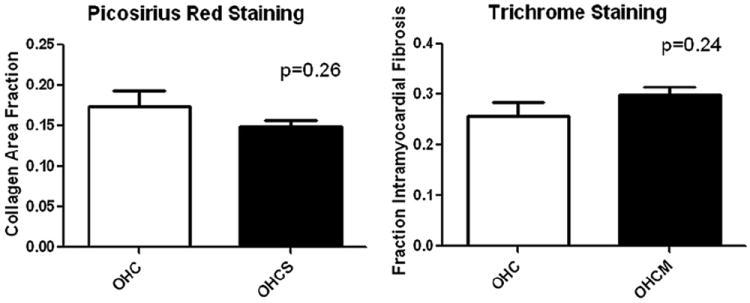
Picosirius red and trichrome staining for collagen and intramyocardial fibrosis showing no difference in collagen area fraction or fraction of intramyocardial fibrosis in oral hypercaloric/hypercholesterolemic diet plus 1.5 mg/kg atorvastatin daily (OHCS) compared with oral hypercaloric/hypercholesterolemic diet (OHC) groups.
DISCUSSION
The results of the present study have demonstrated that high-fat-fed Ossabaw swine with the metabolic syndrome treated with low-dose atorvastatin (1.5 mg/kg) for 14 weeks had an improved angiogenic response in terms of vascular density and increased oxidative stress. However, this failed to lead to increased myocardial perfusion in the collateral-dependent myocardium.
Angiogenesis is a complex process that involves a fine balance between proangiogenesis and anti-angiogenesis mediators. This balance is easily disturbed by coexisting illnesses such as diabetes or hypercholesterolemia or medication. Angiogenesis is activated under conditions such as tissue ischemia and inflammation and is mediated by proliferation, migration, and differentiation of endothelial cells and endothelial progenitor cells.10 In some studies, statins have been shown to activate angiogenesis by promoting migration and proliferation of endothelial progenitor cells derived from bone marrow and tissue in the foci of ischemia and neovascularization.11 Although not entirely well understood, Akt signaling is central in statin-mediated angiogenesis by promoting endothelial cell survival by inhibiting apoptosis, activating eNOS, and increasing nitric oxide bioavailability.12 However, in the present study, Akt and phosphorylated Akt was down-regulated. In contrast, significant upregulation of phosphorylated AMPK and pERK was seen in the OHCS group, consistent with previous studies of statin treatment.13 AMPK has been shown to activate and phosphorylate eNOS, and ERK has been shown to promote cell survival.13,14 Thus, atorvastatin seems to promote angiogenesis in an Akt-independent manner, perhaps through AMPK and ERK upregulation.
The observed histologic increase in capillary and arteriolar density in the ischemic myocardium was supported by the protein expression analysis, in which atorvastatin increased expression of the proangiogenic proteins eNOS, peNOS, and VEGF. Previous studies have shown that statin treatment stabilizes eNOS mRNA, resulting in increased eNOS protein, which might account for the increased eNOS protein expression in the ischemic myocardium.5,15 Increased nitric oxide bioavailability promotes angiogenesis by increasing VEGF expression.16-18 VEGF binds VEGFR2, which promotes cell survival and proliferation by activating ERK and inhibiting apoptosis.19 The decreased VEGFR2 protein expression might result from a negative-feedback loop in which VEGFR2 activation leads to receptor internalization.20
The significant increase in angiogenesis in a swine model of the metabolic syndrome supplemented with low-dose atorvastatin is in contrast to a previous study conducted in our laboratory in which we showed a decreased angiogenic response to high-dose atorvastatin.6,7 These results confirm previous animal and cell culture studies demonstrating that statins have a biphasic, dose-dependent effect on angiogenesis.4,5 At high statin doses, toxic mevalonate-derived intermediates are increased as a result of statin-mediated inhibition of biosynthetic pathways that rely on mevalonic acid as a precursor. The mevalonate intermediates lead to endothelial progenitor cell apoptosis and angiostasis.5 At lower statin doses, endothelial progenitor cell survival is preserved and angiogenesis promoted.4
An interesting result was the significant increase in myocardial protein oxidation and serum lipid peroxidation. These results were similar to our previous study of high-dose atorvastatin treatment.21 Statins have been reported to have systemic antioxidant properties based on an assessment of oxidative stress markers in blood and urine.22,23 No difference was seen in the expression of proteins involved in oxidative stress, such as NOX2, RAC1, myeloperoxidase, SOD1, SOD2, or SOD3. Thus, the mechanism for the increased oxidative stress has yet to be elucidated. Perhaps the increased tissue and peripheral oxidative stress results from statin-mediated inhibition of the reducing agent Q10, resulting in redox imbalance toward oxidation.24 The clinical significance of the increased oxidative stress is unclear; however, it is possible that the oxidative stress contributed to the increased angiogenic response in the ischemic myocardium.25
Although angiogenesis was significantly increased, this did not result in the expected increase in myocardial perfusion. Three reasons could explain this result. First, atorvastatin increased oxidative stress in the serum and ischemic myocardium, as previously reported.21 Increased oxidative stress, in turn, could result in vasoconstriction owing to a reduction of bioavailable nitric oxide. This could account for the lack of improved perfusion.26 In contrast to previous studies, the microvessel reactivity was not different between the 2 groups. With increased oxidative stress, one would predict a reduction in endothelium-dependent relaxation as a result of diminished bioavailable nitric oxide. This could be due to the in vitro examination of the microvessels, in which they are removed from the oxidative environment before the vascular experiments. Second, quantification of CD31-positive endothelial cells identifies all vessels, including the immature, leaky, unstable capillaries that might not contribute to myocardial perfusion. Third, myocardial perfusion is more reflective of the larger arteriole vessel blood supply and not dependent on capillary-mediated perfusion.
Study Limitations
Although providing important functional and molecular data on the role of low-dose atrovastatin in chronic myocardial ischemia, the present study had several limitations. First, although atorvastatin is 1 of the most commonly prescribed statins, these results are not necessarily generalizable to all statins currently prescribed. For example, cerivastatin has been shown to have overall angiostatic effects by inhibiting endothelial cell migration and increased cell cycle arrest.27 Second, because all cardiac tissue was harvested at a single point, 11 weeks after ameroid constrictor placement, it is likely that some of the acute and longer term changes in angiogenesis and vessel maturation were not captured in this model. Third, morphometric analysis of new vessel formation by quantification of CD31-positive endothelial cells identifies all vessels, including the newly formed, immature, unstable capillaries that might not contribute to myocardial perfusion. This could also account for the increased angiogenesis, without the expected increase in perfusion.
Acknowledgments
Funding for this research was provided by the National Heart, Lung, and Blood Institute (grants R01HL46716, R01HL69024, and R01HL85647, Dr Sellke), National Institutes of Health Training grant 5T32-HL076134 (Dr Lassaletta), and National Institutes of Health Training grant 5T32-HL094300-03 (Drs Chu and Elmadhun).
Abbreviations and Acronyms
- AMPK
adenosine monophosphate kinase
- DNP
2,4-dinitrophenyl
- eNOS
endothelial nitric oxide synthase
- ERK
extracellular signal-regulated kinase
- OHC
oral hypercaloric/hypercholesterolemic diet
- OHCS
oral hypercaloric/hypercholesterolemic diet plus 1.5 mg/kg atorvastatin daily
- pERK
phosphorylated ERK
- SOD
superoxide dismutase
- SNP
sodium nitroprusside
- TIMI
Thrombolysis in Myocardial Infarction
- VEGF
vascular endothelial growth factor
- VEGFR2
VEGF receptor 2
Footnotes
Disclosures: Authors have nothing to disclose with regard to commercial support.
Read at the 38th Annual Meeting of The Western Thoracic Surgical Association, Maui, Hawaii, June 27-30, 2012.
References
- 1.Dupuis J, Tardif JC, Cernacek P, Theroux P. Cholesterol reduction rapidly improves endothelial function after acute coronary syndromes: the RECIFE (Reduction of Cholesterol in Ischemia and Function of the Endothelium) trial. Circulation. 1999;99:3227–33. doi: 10.1161/01.cir.99.25.3227. [DOI] [PubMed] [Google Scholar]
- 2.Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation. 1999;100:178–84. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- 3.Pruefer D, Scalia R, Lefer AM. Simvastatin inhibits leukocyte-endothelial cell interactions and protects against inflammatory processes in normocholesterolemic rats. Arterioscler Thromb Vasc Biol. 1999;19:2894–900. doi: 10.1161/01.atv.19.12.2894. [DOI] [PubMed] [Google Scholar]
- 4.Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–45. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 5.Urbich C, Dernbach E, Zeiher AM, Dimmeler S. Double-edged role of statins in angiogenesis signaling. Circ Res. 2002;90:737–44. doi: 10.1161/01.res.0000014081.30867.f8. [DOI] [PubMed] [Google Scholar]
- 6.Boodhwani M, Mieno S, Voisine P, Feng J, Sodha N, Li J, et al. High-dose atorvastatin is associated with impaired myocardial angiogenesis in response to vascular endothelial growth factor in hypercholesterolemic swine. J Thorac Cardiovasc Surg. 2006;132:1299–306. doi: 10.1016/j.jtcvs.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 7.Boodhwani M, Nakai Y, Voisine P, Feng J, Li J, Mieno S, et al. High-dose atorvastatin improves hypercholesterolemic coronary endothelial dysfunction without improving the angiogenic response. Circulation. 2006;114:I402–8. doi: 10.1161/CIRCULATIONAHA.105.000356. [DOI] [PubMed] [Google Scholar]
- 8.Tofukuji M, Metais C, Li J, Franklin A, Simons M, Sellke FW. Myocardial VEGF expression after cardiopulmonary bypass and cardioplegia. Circulation. 1998;98:II242–8. [PubMed] [Google Scholar]
- 9.Lassaletta AD, Chu LM, Robich MP, Elmadhun NY, Feng J, Burgess TA, et al. Overfed Ossabaw swine with early stage metabolic syndrome have normal coronary collateral development in response to chronic ischemia. Basic Res Cardiol. 2012;107:243. doi: 10.1007/s00395-012-0243-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang C, Zhang ZH, Li ZJ, Yang RC, Qian GQ, Han ZC. Enhancement of neovascularization with cord blood CD133+ cell-derived endothelial progenitor cell transplantation. Thromb Haemost. 2004;91:1202–12. doi: 10.1160/TH03-06-0378. [DOI] [PubMed] [Google Scholar]
- 11.Ma FX, Han ZC. Statins, nitric oxide and neovascularization. Cardiovasc Drug Rev. 2005;23:281–92. doi: 10.1111/j.1527-3466.2005.tb00173.x. [DOI] [PubMed] [Google Scholar]
- 12.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655–62. doi: 10.1161/CIRCULATIONAHA.106.630194. [DOI] [PubMed] [Google Scholar]
- 14.Merla R, Ye Y, Lin Y, Manickavasagam S, Huang MH, Perez-Polo RJ, et al. The central role of adenosine in statin-induced Erk1/2, Akt, and eNOS phosphorylation. Am J Physiol Heart Circ Physiol. 2007;293:H1918–28. doi: 10.1152/ajpheart.00416.2007. [DOI] [PubMed] [Google Scholar]
- 15.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG-CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 16.Dulak J, Jozkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, et al. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659–66. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- 17.Murohara T, Witzenbichler B, Spyridopoulos I, Asahara T, Ding B, Sullivan A, et al. Role of endothelial nitric oxide synthase in endothelial cell migration. Arterioscler Thromb Vasc Biol. 1999;19:1156–61. doi: 10.1161/01.atv.19.5.1156. [DOI] [PubMed] [Google Scholar]
- 18.Noiri E, Lee E, Testa J, Quigley J, Colflesh D, Keese CR, et al. Podokinesis in endothelial cell migration: role of nitric oxide. Am J Physiol. 1998;274:C236–44. doi: 10.1152/ajpcell.1998.274.1.C236. [DOI] [PubMed] [Google Scholar]
- 19.Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–12. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 20.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 21.Sodha NR, Boodhwani M, Ramlawi B, Clements RT, Mieno S, Feng J, et al. Atorvastatin increases myocardial indices of oxidative stress in a porcine model of hypercholesterolemia and chronic ischemia. J Card Surg. 2008;23:312–20. doi: 10.1111/j.1540-8191.2008.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marketou ME, Zacharis EA, Nikitovic D, Ganotakis ES, Parthenakis FI, Maliaraki N, et al. Early effects of simvastatin versus atorvastatin on oxidative stress and proinflammatory cytokines in hyperlipidemic subjects. Angiology. 2006;57:211–8. doi: 10.1177/000331970605700212. [DOI] [PubMed] [Google Scholar]
- 23.Sugiyama M, Ohashi M, Takase H, Sato K, Ueda R, Dohi Y. Effects of atorvastatin on inflammation and oxidative stress. Heart Vessels. 2005;20:133–6. doi: 10.1007/s00380-005-0833-9. [DOI] [PubMed] [Google Scholar]
- 24.Hargreaves IP, Duncan AJ, Heales SJ, Land JM. The effect of HMG-CoA reductase inhibitors on coenzyme q10: possible biochemical/clinical implications. Drug Saf. 2005;28:659–76. doi: 10.2165/00002018-200528080-00002. [DOI] [PubMed] [Google Scholar]
- 25.West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–6. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cox DA, Cohen ML. Effects of oxidized low-density lipoprotein on vascular contraction and relaxation: clinical and pharmacological implications in atherosclerosis. Pharmacol Rev. 1996;48:3–19. [PubMed] [Google Scholar]
- 27.Skaletz-Rorowski A, Walsh K. Statin therapy and angiogenesis. Curr Opin Lipidol. 2003;14:599–603. doi: 10.1097/00041433-200312000-00008. [DOI] [PubMed] [Google Scholar]