


Abstract
DNA methylation plays a central role in genomic regulation and disease. Sodium bisulfite treatment (SBT) causes unmethylated cytosines to be sequenced as thymine, which allows methylation levels to reflected in the number of ‘C’-‘C’ alignments covering reference cytosines. Di-base color reads produced by lifetech’s SOLiD sequencer provide unreliable results when translated to bases because single sequencing errors effect the downstream sequence. We describe FadE, an algorithm to accurately determine genome-wide methylation rates directly in color or nucleotide space. FadE uses SBT unmethylated and untreated data to determine background error rates and incorporate them into a model which uses Newton–Raphson optimization to estimate the methylation rate and provide a credible interval describing its distribution at every reference cytosine. We sequenced two slides of human fibroblast cell-line bisulfite-converted fragment library with the SOLiD sequencer to investigate genome-wide methylation levels. FadE reported widespread differences in methylation levels across CpG islands and a large number of differentially methylated regions adjacent to genes which compares favorably to the results of an investigation on the same cell-line using nucleotide-space reads at higher coverage levels, suggesting that FadE is an accurate method to estimate genome-wide methylation with color or nucleotide reads. http://code.google.com/p/fade/.
INTRODUCTION
DNA methylation was first proposed to act as a stable and heritable epigenetic modification in 1975 (1) and first observed at cytosine guanine dinuleotides (CpG) in somatic cells (2). Today, we know that DNA methylation plays a vital role in gene regulation such that different levels of methylation can have major ramifications for human health and disease (3). Estimation of the level of methylation at all cytosine nucleotides in an individual (the methylome) has recently become possible with the advent of Next Generation Sequencing (NGS) techniques, specifically sodium bisulfite treated (SBT) sequencing (4,5). In whole genome SBT sequencing, DNA is treated with sodium bisulfite which converts unmethylated cytosine nucleotides to uracil. Because sequencing machines treat uracil the same as thymine, treated reads can be mapped to a reference genome, where the majority of ‘C’-‘C’ alignments will result from methylation. To accurately align each read, the alignment algorithm can first translate all ‘C’ nucleotides to ‘T’ nucleotides on the read and reference sequence. Then the original read sequence can be compared with its aligned location on the translated reference for ‘C’ to ‘T’ mismatches which result from methylation. This method and others which involve translation of the bases on the read have been shown to work successfully (5,6) with reads in nucleotide space.
Unfortunately, these methods are not suitable for color-space as the pre-aligned reads cannot be accurately translated to nucleotide space because single-color errors can change the downstream sequence (7). Post-alignment translation to nucleotides improves accuracy but also introduces errors when the color error rate is high or there exist consecutive or dense polymorphism (i.e. consecutive methylcytosine positions) (8).
Thus, determination of methylation rates is most accurate when SBT color reads are aligned in color-space and methylation is determined directly from the color alignment. Although there exist algorithms to facilitate alignment of SBT color reads (9,10,11), all accomplish estimation of methylation through some type of post-alignment translation from color sequences to called nucleotides which reduces accuracy, especially for consecutive cytosine positions. It is for these reasons that we were motivated to develop an algorithm capable of determining methylation levels directly in color-space. Accurate whole-genome per-base estimation of methylation from color reads requires first that accurate unbiased alignment be acquired, which is itself a non-trivial task. In the Materials and Methods section, we discuss in greater detail how reference bias can be reduced to provide accurate, highly sensitive color-space alignment.
Given such an alignment, an algorithm is tasked with using the colors and quality scores spanning each reference cytosine to estimate the methylation rate in the cell population and determine a statistical level of accuracy for the estimation. For each read covering a particular reference cytosine, one color and quality score encodes the transition from the preceding reference base to the cytosine and another color and quality score encodes the transition from the reference cytosine to the following reference base. This is shown in Figure 1. The quality scores associated with each color are normalized values supplied by the sequencing machine which represent the accuracy for each color sequenced. Rather than representing transitions with one of four colors, the color (x), quality score (q) and read position (i) can be combined to form one of many color-quality tuples (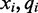 ) which provide more detailed information regarding the underlying nucleotide sequence.
) which provide more detailed information regarding the underlying nucleotide sequence.
Figure 1.
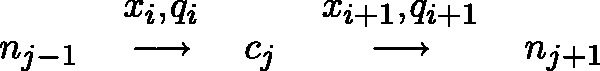
For any alignment spanning a non-consecutive reference cytosine (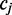 ) at read position i, the transition from the previous nucleotide (
) at read position i, the transition from the previous nucleotide (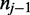 ) and the transition to the following nucleotide (
) and the transition to the following nucleotide (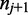 ) each emit a color-quality tuple,
) each emit a color-quality tuple, 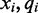 and
and 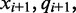 respectively. The rate governing the emission of color-quality tuples for different read positions and adjacent reference nucleotides can be used to estimate the probability that the cytosine sequenced in each read existed in a methylated or unmethylated state.
respectively. The rate governing the emission of color-quality tuples for different read positions and adjacent reference nucleotides can be used to estimate the probability that the cytosine sequenced in each read existed in a methylated or unmethylated state.
FadE uses the probability of observing each pair of color-quality tuples in the methylated versus unmethylated state to iteratively determine the rate at which the cytosine nucleotide is methylated. While most aligned pairs of color-quality tuples provide convincing evidence as to whether methylation is present, some pairs will not immediately suggest methylation or the lack of it; this is illustrated in Table 1. The probability of observing any such pair of color-quality tuples under methylation depend on the color-quality scores and the machines distribution of color errors. Inferring the error rate distribution or ‘emission rates’ from control data allows FadE to perform methylation estimation that is extremely robust to machine sequencing error as well accurate for consecutive cytosine positions, both of which not possible when reads are translated to nucleotides. The collection of the emission rates and their use in the parameter optimization routine employed by FadE are further described in Materials and Methods section, whereas the Results section demonstrates the accuracy that FadE offers in both real and simulated datasets in color and nucleotide space.
Table 1.
Some pairs of color alignments are difficult to resolve with in a single read
| A |  |
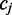 |
 |
G | Likely interpretation | Methylation probability |
|---|---|---|---|---|---|---|
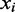 |
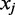 |
– | – | |||
| Green | Red | A-C-G | High | |||
| Red | Green | A-T-G | Low | |||
| Red | Red | A-T-? or ?-C-G | Dep. on error rate | |||
| Green | Green | A-C-? or ?-T-G | Dep. on error rate |
In the example below the colors ‘Green–Red and ‘Red–Green’ lead to the interpretation that the position 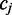 is in the state ‘C’ and ‘T’, respectively. The colors ‘Red–Red’ is likely the result of sequencing error. Whether the sequencing error is assumed to alter the first or second color in the pair will lead to a different interpretation for the position
is in the state ‘C’ and ‘T’, respectively. The colors ‘Red–Red’ is likely the result of sequencing error. Whether the sequencing error is assumed to alter the first or second color in the pair will lead to a different interpretation for the position 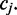
MATERIALS AND METHODS
FadE provides point estimates and credible intervals for the epigenome from an accurate alignment in color or sequence space. While FadE handles both data types the following section explains the development of the algorithm in color-space before changes necessary to facilitate nucleotide alignment are described. In color-space the acquisition of an accurate alignment requires many modifications to the alignment protocol. Thus, it is important to first describe the necessary steps to produce an accurate alignment file and estimate the emission rates before the methylation parameter optimization is described.
Color alignment
The performance of FadE strongly depends on achieving an accurate and complete alignment file. If bisulfite treated reads are aligned to the native human reference, only a small fraction of the reads will find low-mismatch alignments due to the 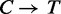 conversion of unmethylated cytosine nucleotides. Alignment to a
conversion of unmethylated cytosine nucleotides. Alignment to a 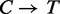 translated reference genome will find produce many more suitable alignments, but also bias the mapping away from methylated cytosines. Hansen et al. (12) describes a method which eliminates bias to CpG positions by creating a custom alignment tool which indexes all combinations of
translated reference genome will find produce many more suitable alignments, but also bias the mapping away from methylated cytosines. Hansen et al. (12) describes a method which eliminates bias to CpG positions by creating a custom alignment tool which indexes all combinations of 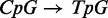 translations for each read-length reference substring and translates all CpH positions (‘H’ is not guanine) to thiamine. If non-CpG cytosine nucleotides are suspected to be methylated or the read length is long, the number of combinations of reference translations may grow prohibitively large for some reference substrings. If an alignment algorithm exists with tolerance to many substitutions, translating the entire reference sequence into multiple sequences will also provide a significantly reduction in bias in comparison to single genome reference alignment without increasing the amount of memory required for alignment. For example, color reads could be mapped to the color sequence corresponding to the following three genomes:
translations for each read-length reference substring and translates all CpH positions (‘H’ is not guanine) to thiamine. If non-CpG cytosine nucleotides are suspected to be methylated or the read length is long, the number of combinations of reference translations may grow prohibitively large for some reference substrings. If an alignment algorithm exists with tolerance to many substitutions, translating the entire reference sequence into multiple sequences will also provide a significantly reduction in bias in comparison to single genome reference alignment without increasing the amount of memory required for alignment. For example, color reads could be mapped to the color sequence corresponding to the following three genomes:
The native reference sequence;
The reference sequence where all
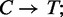 and
andThe reference sequence where all
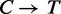 except ‘C’ preceding ‘G’.
except ‘C’ preceding ‘G’.
A highly tolerant alignment algorithm will be able to locate most reads for an alignment to at least one of the reference translations, after which the results can be joined into a single-alignment file. Detailed analysis of this method and others are further discussed in the Supplementary Data. Other ways to increase alignment accuracy include using paired-end reads, using uni-directional (forward strand only) reads, and iteratively translating unmapped reads to sequence space to search for matches (10). Additionally, if untreated reads from the same sample are available, SNP-calling can be performed on the reference sequence and homozygous SNP positions can be altered to facilitate a more accurate alignment.
Emission rate estimation
For any cytosine spanning color alignment, the probability that it was generated from a methylated base is dependent on the observed colors and associated error rate. The supplied quality scores represent the signal intensity and capture only one source of error (13). The SOLiD system in particular has been shown to have a relatively high error rate which increases toward read tails (7). Additionally, it is possible that the bisulfite treatment itself may alter the true error distribution. To most accurately estimate methylation rates, FadE attempts to determine the error distribution for colors adjacent to methylated and unmethyalted cytosines. The error distribution is encapsulated into ‘emission rates’ which describe nucleotide transitions under methylated and unmethylated states.
As shown in Figure 1, for a given cytosine 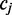 spanning alignment beginning at read position i, emission rates describe the probability that the transition from the preceding reference base
spanning alignment beginning at read position i, emission rates describe the probability that the transition from the preceding reference base 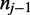 to
to 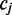 and the transition from
and the transition from 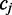 to the following reference base
to the following reference base 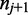 emit the color-quality tuples
emit the color-quality tuples 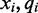 and
and 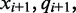 respectively. The emission rates for cytosines in the unmethylated (M = 0) state can be inferred from the alignment of unmethylated bisulfite-treated reads. The phage lambda genome is thought to have little to no methylation, and as such serves as an excellent control to estimate emission rates over unmethylated bisulfite-treated cytosine nucleotides. Using the alignment of bisulfite-treated reads to the phage lambda genome, the emission rate for the unmethylated state (M = 0) for the color-quality tuple
respectively. The emission rates for cytosines in the unmethylated (M = 0) state can be inferred from the alignment of unmethylated bisulfite-treated reads. The phage lambda genome is thought to have little to no methylation, and as such serves as an excellent control to estimate emission rates over unmethylated bisulfite-treated cytosine nucleotides. Using the alignment of bisulfite-treated reads to the phage lambda genome, the emission rate for the unmethylated state (M = 0) for the color-quality tuple 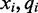 resulting from the transition between reference base
resulting from the transition between reference base 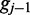 and
and 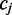 at read position i can be estimated from its frequency (I()) in the read alignment R:
at read position i can be estimated from its frequency (I()) in the read alignment R:
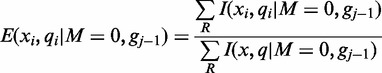 |
(1) |
Emission rates describing the transition away from reference cytosine positions can be calculated similarly, whereas the emission rates for the methylated state (M = 1) should ideally be calculated from experimentally similar reads which have an extremely high rate of methylation. As such data are difficult to acquire, experimentally similar reads which have not been treated with sodium bisulfite (e.g. all cytosine remain unchanged), can serve as an approximation for the distribution expected under methylation. Additionally, if neither of these controls is available, the error distribution can be estimated using the alignment to non-cytosine positions which can serve to provide an approximate estimate of the distribution of observations expected with respect to the methylation state at a reference nucleotide.
Statistical model
FadE uses a hidden data model to describe the methylation level at different cytosine positions across cell populations. At every reference cytosine 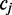 we assume an unknown parameter
we assume an unknown parameter 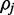 which describes the rate at which the position
which describes the rate at which the position 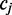 exists in an unmethylated (M = 0) or methylated (M = 1) state. For each read alignment spanning
exists in an unmethylated (M = 0) or methylated (M = 1) state. For each read alignment spanning 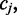 the hidden methylation state produces observable color-quality tuples according to the emission rates for the read position and and adjacent reference nucleotides. This is illustrated in Figure 2. By accurately inferring the emission rates from control data, FadE is able to use each set of reads
the hidden methylation state produces observable color-quality tuples according to the emission rates for the read position and and adjacent reference nucleotides. This is illustrated in Figure 2. By accurately inferring the emission rates from control data, FadE is able to use each set of reads  which align to
which align to  to estimate the posterior probability distribution describing
to estimate the posterior probability distribution describing 
Figure 2.
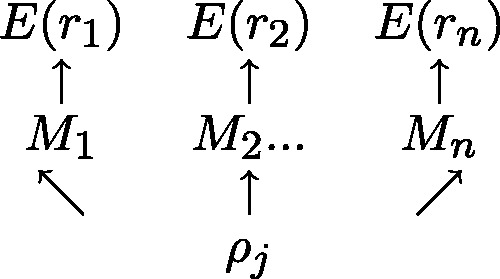
For each of n reads, a hidden binary methylation state (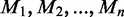 ) is drawn uniformly according to an unknown methylation parameter
) is drawn uniformly according to an unknown methylation parameter 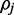 . For each methylation state, observations E(r) in the form of color-quality tuples are produced according to emission rates corresponding to the read position and adjacent reference bases.
. For each methylation state, observations E(r) in the form of color-quality tuples are produced according to emission rates corresponding to the read position and adjacent reference bases.
Parameter optimization
Given an isolated cytosine 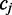 and a single-read alignment r
and a single-read alignment r
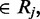 the probability of observing the alignment when
the probability of observing the alignment when 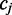 is in the methylated (M = 1) or unmethylated (M = 0) state can be calculated from the product of emission rates:
is in the methylated (M = 1) or unmethylated (M = 0) state can be calculated from the product of emission rates:
| (2) |
where the color-quality tuples 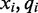 and
and 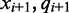 span the transition to and from the reference cytosine
span the transition to and from the reference cytosine 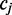 , respectively, as is illustrated in Figure 1. Given the rate of methylation,
, respectively, as is illustrated in Figure 1. Given the rate of methylation, 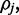 the law of total probability informs us that for any observation r:
the law of total probability informs us that for any observation r:
| (3) |
Assuming independence between read alignments, the probability of observing the set of reads 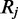 which align to
which align to 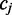 when
when 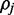 is known is:
is known is:
| (4) |
Given Equation (4), Bayes Theorem can be used to calculate the inverse; the posterior probability for the methylation rate 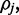 given the set of reads
given the set of reads 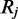 :
:
| (5) |
where 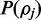 is the prior distribution for
is the prior distribution for 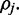 For ease of explanation
For ease of explanation 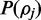 is currently assumed to be the continuous uniform distribution.
is currently assumed to be the continuous uniform distribution. 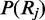 is a normalizing constant which can be calculated by integrating over support of the distribution:
is a normalizing constant which can be calculated by integrating over support of the distribution:
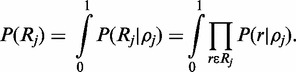 |
(6) |
To produce a point estimate for the methylation rate, we can calculate 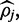 the value for
the value for 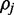 for which
for which 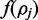 is maximized. To quickly and accurately optimize this function we recall Equation (4) and its expanded form shown in Equation (3). Taking logarithms and differentiating once yields:
is maximized. To quickly and accurately optimize this function we recall Equation (4) and its expanded form shown in Equation (3). Taking logarithms and differentiating once yields:
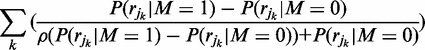 |
(7) |
Differentiating again yields:
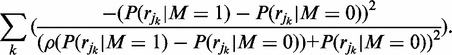 |
(8) |
Which is strictly negative, meaning that Equation (4) is strictly concave on the region (0,1). Thus, the value for the parameter 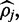 which maximizes the density can be calculated iteratively with Newton’s optimization method, where each estimate of
which maximizes the density can be calculated iteratively with Newton’s optimization method, where each estimate of 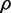 is updated as follows:
is updated as follows:
| (9) |
The value to which this optimization routine converges 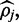 is the maximum value on a concave uni-modal polynomial probability density function. This curve becomes tightly centered around this maximum value provided sufficient read depth, accurate alignment and read quality. Unfortunately, if read quality or read depth is poor the distribution will not be tightly centered and reporting only the maximum value as an estimate for
is the maximum value on a concave uni-modal polynomial probability density function. This curve becomes tightly centered around this maximum value provided sufficient read depth, accurate alignment and read quality. Unfortunately, if read quality or read depth is poor the distribution will not be tightly centered and reporting only the maximum value as an estimate for 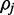 may be misleading, especially if the assumption of a uniform prior is inaccurate (see Implementation section). This can be ameliorated by obtaining the 90% credible interval around
may be misleading, especially if the assumption of a uniform prior is inaccurate (see Implementation section). This can be ameliorated by obtaining the 90% credible interval around 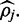 The credible interval can be calculated iteratively by updating the step size
The credible interval can be calculated iteratively by updating the step size 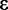 until the following is satisfied:
until the following is satisfied:
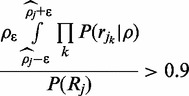 |
(10) |
where 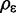 is the prior probability that
is the prior probability that 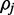 exists on the interval [
exists on the interval [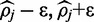 ], which is simply the length of the interval if a uniform prior distribution is assumed. The integrals can be calculated explicitly by expanding the polynomial function
], which is simply the length of the interval if a uniform prior distribution is assumed. The integrals can be calculated explicitly by expanding the polynomial function 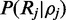 when read depth is moderate or approximated using tools of numerical integration.
when read depth is moderate or approximated using tools of numerical integration.
Implementation
Boundary values for methylation parameter
When read depth and quality are sufficient the Newton–Raphson optimization and credible interval estimation converges quickly and accurately. In practice convergence to within the hundredth decimal point occurs in fewer than 8 iterations in 99% of cases. These conditions also allow a 90% credible interval around 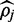 to be quickly located by changing the value of
to be quickly located by changing the value of 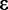 in relation to the distance from 90% achieved in the previous iteration.
in relation to the distance from 90% achieved in the previous iteration.
If read depth is shallow or 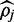 exists near one of the boundary values, convergence is slower and there often is no region which bounds
exists near one of the boundary values, convergence is slower and there often is no region which bounds 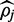 symmetrically to a provide 90% credible interval. In such cases FadE uses Equation (10) to first find the relative support for the symmetric interval which includes one of the boundaries; [
symmetrically to a provide 90% credible interval. In such cases FadE uses Equation (10) to first find the relative support for the symmetric interval which includes one of the boundaries; [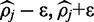 ] where
] where 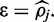 Then the non-boundary edge of the interval is expanded until a non-symmetric 90% credible interval is found.
Then the non-boundary edge of the interval is expanded until a non-symmetric 90% credible interval is found.
Prior methylation probability
As previously described FadE is implemented with a default continuous uniform prior probability. The most accurate prior distribution is highly data dependent. In our tests on the human fibroblast cell-line IMR-90, the methylation rate is highest on isolated CpG sites whereas CpG islands have a bimodal distribution resulting from regions of hypomethylation and otherwise highly methylated sites. In humans CpN dinucleotides (where ‘N’ is not guanine) are relatively unmethylated, but this is not true for all cell types (5). Additionally, the prior methylation probability can be inferred site-by-site from previous studies. If we assume the prior methylation probability is 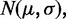 then Equation (8),
then Equation (8), 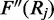 becomes:
becomes:
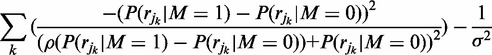 |
(11) |
which is also strictly negative, meaning the Newton–Raphson optimization routine can still be used to calculate 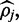 and the numerical methods used to estimate credible intervals can still be employed. FadE is implemented to allow the user to select different normal prior parameters for ‘CpG’ versus ‘CpN’ dinucleotides or supply a file which lists site-specific normal parameters for different reference positions.
and the numerical methods used to estimate credible intervals can still be employed. FadE is implemented to allow the user to select different normal prior parameters for ‘CpG’ versus ‘CpN’ dinucleotides or supply a file which lists site-specific normal parameters for different reference positions.
Adjacent reference cytosines
In the previous section the two reference positions adjacent to 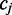 have been assumed to be non-cytosine nucleotides. In such cases, the two-part observation involves color-quality tuples resulting from the transition from the preceding base
have been assumed to be non-cytosine nucleotides. In such cases, the two-part observation involves color-quality tuples resulting from the transition from the preceding base 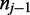 to
to 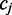 and from
and from 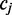 to
to 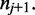 However, (shown in Figure 3) if
However, (shown in Figure 3) if 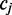 is followed by another cytosine nucleotide, then the emission probability associated with this transition depends on the methylation state at
is followed by another cytosine nucleotide, then the emission probability associated with this transition depends on the methylation state at 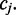 In the general case of m-consecutive reference cytosines, FadE calculates the first and last methylation parameters using only the color-quality tuple spanning the transition involving non-cytosine reference bases. Then, for each of the interior positions the observation probability is conditioned on the adjacent methylation parameter, such that the observation probability for a single-read aligned to the second position in a run of cytosine positions is generalized to:
In the general case of m-consecutive reference cytosines, FadE calculates the first and last methylation parameters using only the color-quality tuple spanning the transition involving non-cytosine reference bases. Then, for each of the interior positions the observation probability is conditioned on the adjacent methylation parameter, such that the observation probability for a single-read aligned to the second position in a run of cytosine positions is generalized to:
| (12) |
Figure 3.
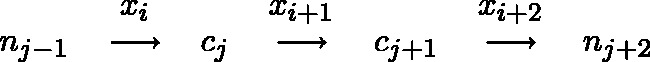
If reference cytosines are adjacent, then the interior emission probabilities will depend on the methylation state of the adjacent cytosine. In this case, the methylation rate at boundary cytosine nucleotides is estimated using the boundary emission probabilities. Then, the rate of interior cytosine nucleotides are calculated conditioned on the identity of boundary positions.
In the Results section, the necessity to remain in color-space to accurately estimate methylation for consecutive cytosine positions is shown. Translating colors to bases over partially methylated cytosine runs results in errors which cannot be recovered once color sequences are discarded.
Implementation in sequence space
FadE can be implemented in nucleotide space by making only a few changes to the algorithm described above. The main difference between sequence and color-space is that emissions in sequence involve only a single nucleotide and quality score rather than a pair of color-quality tuples. Thus, in nucleotide space Equation (2) becomes:
| (13) |
where 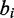 and
and 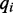 are the base and quality score aligned to a reference cytosine. Estimation of sequence emission rates are calculated without regard to adjacent positions, thus Equation (1), which describes the frequency of observations aligned to a reference cytosine generalizes to:
are the base and quality score aligned to a reference cytosine. Estimation of sequence emission rates are calculated without regard to adjacent positions, thus Equation (1), which describes the frequency of observations aligned to a reference cytosine generalizes to:
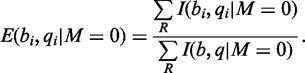 |
(14) |
With these two changes the algorithm can be developed as it is described in color-space, with the exception that methylation estimates at adjacent reference cytosines are carried out independently.
RESULTS
Color-space simulations
To assess FadE’s accuracy and memory requirements for different read depths in color-space, a simulation was performed using Human Chromosome 21. Twenty percent of the over 14 million cytosine positions on either strand (1.7 million of which were members of consecutive blocks) of Chromosome 21 were selected at random and assigned a methylation rate from a 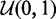 distribution. 100-million 50-bp nucleotide reads were simulated according to a bisulfite conversion rate of 99% and translated to color sequences with uniform color error rates of 1.5%. Alignment was performed with PerM (14) which is capable of providing full sensitivity to up to four substitutions. Only unique alignments with five or fewer substitutions were accepted. 98.8% of the reported alignments were correct and over 96% of the methylated cytosines had at least one unique alignment. In fewer than 3 CPU hours and requiring less than 500 MB of memory, FadE was able to analyse 14-million reference cytosine positions with an average read depth of 83X, which suggests that FadE is feasible for genome-scale use. The size of the credible intervals returned by FadE decreased with respect to coverage; at 10X the simulated parameter was contained within the credible interval
distribution. 100-million 50-bp nucleotide reads were simulated according to a bisulfite conversion rate of 99% and translated to color sequences with uniform color error rates of 1.5%. Alignment was performed with PerM (14) which is capable of providing full sensitivity to up to four substitutions. Only unique alignments with five or fewer substitutions were accepted. 98.8% of the reported alignments were correct and over 96% of the methylated cytosines had at least one unique alignment. In fewer than 3 CPU hours and requiring less than 500 MB of memory, FadE was able to analyse 14-million reference cytosine positions with an average read depth of 83X, which suggests that FadE is feasible for genome-scale use. The size of the credible intervals returned by FadE decreased with respect to coverage; at 10X the simulated parameter was contained within the credible interval 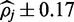 in 91% of cases whereas at 100X coverage the credible interval
in 91% of cases whereas at 100X coverage the credible interval 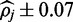 contained the parameter in 91% of cases. This is shown in Figure 4.
contained the parameter in 91% of cases. This is shown in Figure 4.
Figure 4.

At a 1.5% error rate, 20X read depth appears to be sufficient for FadE to produce accurate results using color-space data. Shown here are the average size of the calculated 90% credible interval and the percentage of positions for which the simulated parameter 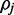 exists in the credible interval. At 20x coverage the average credible interval length is
exists in the credible interval. At 20x coverage the average credible interval length is 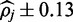 and the interval includes
and the interval includes 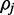 in 92% of cases.
in 92% of cases.
To compare FadE’s performance to traditional pileup methods the aligned reads were converted to Sequence Alignment/Map (SAM) format which translates aligned color sequences into their likely nucleotide sequences using a dynamic programming algorithm (15). After translation methylation can be estimated by simply counting the percentage of ‘C’ nucleotides which cover each reference cytosine. For each of 3 error rates (0, 1.5 and 5%) 20 trials were performed to compare FadE to the base translation strategy. For each test the mean difference between the estimated and true parameter was used as a measure of accuracy. The simulations demonstrated that FadE is capable of moderately outperforming color-translation methods when the color-error rate is low or the cytosine positions are isolated but drastically outperforming color-translation at high color-error rates or on adjacent cytosine nucleotides. As shown in Table 2, in error-free data FadE’s provide more accurate estimation of adjacent cytosine blocks, whereas at high error rates FadE outperforms color-translation by providing a model that is more robust to color-sequencing errors and better at handling alignment errors.
Table 2.
Comparison to color-translation method at 50x average read coverage
| Error rate (%) | Isolated |
Adjacent |
||
|---|---|---|---|---|
| CT-Method | FadE | CT-Method | FadE | |
| 0.0 | 0.066a | 0.064a | 0.281 | 0.070 |
| 1.5 | 0.107 | 0.094 | 0.301 | 0.096 |
| 5.0 | 0.162 | 0.110 | 0.312 | 0.116 |
Shown is the mean absolute difference between the simulated parameter and the value for 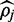 returned by FadE and the color-translation method.
returned by FadE and the color-translation method.
aThe difference is, here is, not less than the standard error in the 20 trials.
To determine if a nucleotide space implementation of FadE increases accuracy compared with estimating methylation by the percentage of aligned cytosines relative to thymine nucleotides, additional simulations were performed on chromosome 21 using nucleotide reads. The methylation distribution and bisulfite conversion rate described in the color simulation were used to for the nucleotide simulation. Sequencing error rates of 0.0, 1.5, 3 and 5% were imposed on the reads and PerM was used to carry out direct nucleotide alignment with parameters identical to those used in the color simulation. However, alignment accuracy in nucleotide space was slightly higher than in color-space; for 1.5% error rate, 99.4% of reported alignments were correct. Computationally, performance was similar to performance in color-space. In nucleotide space FadE reports slightly more accurate estimations for 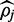 as well as larger credible intervals. The increased accuracy is likely a result of the increase in alignment accuracy that is enjoyed in nucleotide space. The larger credible intervals observed in sequence space (
as well as larger credible intervals. The increased accuracy is likely a result of the increase in alignment accuracy that is enjoyed in nucleotide space. The larger credible intervals observed in sequence space (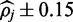 at 25x read depth versus
at 25x read depth versus 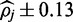 for color alignment) are a result sequence data relying on only a single-aligned base and quality score from which to infer the methylation state responsible for each alignment rather than the two of color-quality tuples which span alignments in color-space. Although valid consecutive color sequencing errors are rare (see Equation (2) versus Equation (13)), the possibility of a single-nucleotide error producing misleading information cannot as easily be discounted, this uncertainty is reflected in larger credible intervals.
for color alignment) are a result sequence data relying on only a single-aligned base and quality score from which to infer the methylation state responsible for each alignment rather than the two of color-quality tuples which span alignments in color-space. Although valid consecutive color sequencing errors are rare (see Equation (2) versus Equation (13)), the possibility of a single-nucleotide error producing misleading information cannot as easily be discounted, this uncertainty is reflected in larger credible intervals.
To compare FadE to an often used method to estimate methylation (5,12), the results obtained by FadE were compared with the result of a native nucleotide-space pileup (where the percentage of ‘C’ relative to ‘T’ alignments was used to infer methylation). In Table 3 the results of the comparison for the three different error rates are shown. When sequencing errors are not present in nucleotide space the Newton–Raphson optimization routine employed by FadE produces the maximum likelihood estimate for 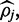 which is the percentage of ‘C’ alignments. While, the advantage is far less than the performance increase seem in color-space, FadE does outperform this method in nucleotide space when the error rate increases. Additionally, by reporting credible intervals as well as methylation estimation in nucleotide space a measure of certainty is supplied which is not obtained by a simple pileup analysis.
which is the percentage of ‘C’ alignments. While, the advantage is far less than the performance increase seem in color-space, FadE does outperform this method in nucleotide space when the error rate increases. Additionally, by reporting credible intervals as well as methylation estimation in nucleotide space a measure of certainty is supplied which is not obtained by a simple pileup analysis.
Table 3.
Comparison to sequence space pileup estimation at 50x average read coverage
| Error rate (%) | Mean absolute difference |
90% Credible interval |
||
|---|---|---|---|---|
| Pileup method | FadE | Size | Percent contained | |
| 0.0 | 0.057a | 0.057a | 0.178 | 0.93 |
| 1.5 | 0.063 a | 0.062a | 0.201 | 0.92 |
| 3.0 | 0.074 | 0.069 | 0.218 | 0.90 |
| 5.0 | 0.079 | 0.072 | 0.234 | 0.89 |
Shown is the mean absolute difference between the simulated parameter and the value for 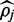 returned by FadE and the percentage of cytosines relative to thymine (pileup) aligned to the location. Additionally, the size of the 90% credible interval returned by FadE and the percentage of locations where the simulated parameter was contained in the interval are shown. Note that in the absence of sequencing error, the algorithm implemented in FadE returns the parameter equal to the rate of cytosines aligned to the location.
returned by FadE and the percentage of cytosines relative to thymine (pileup) aligned to the location. Additionally, the size of the 90% credible interval returned by FadE and the percentage of locations where the simulated parameter was contained in the interval are shown. Note that in the absence of sequencing error, the algorithm implemented in FadE returns the parameter equal to the rate of cytosines aligned to the location.
aThe difference is, here is, not less than the standard error in the 20 trials.
Pileup analysis of sequence data often uses the read depth as a proxy for accuracy, usually filters or minimum coverage levels are set to improve the accuracy of the analysis. Read depth alone does not take into account the read quality or error distribution of the alignment and for this reason does not give as accurate an estimation of the likely discrepancies between the true parameter and the estimated parameter as a credible interval.
Color-space methylome estimation
Data acquisition and alignment
To demonstrate FadE’s scalability to large datasets as well as to compare color-space results to those achieved using nucleotide-space data, two full slides of IMR-90 (fibroblast cell-line) bisulfite-converted fragment libraries were sequenced in color-space. The IMR-90 cell-line has been previously used to study the methylome (5). In total, 1.1 billion unidirectional, paired-end (50 and 25 colors, in the reverse and forward directions) reads were generated using the SOLiD 4 system. The DNA was spiked with phage lambda DNA that had been digested with the restriction endonuclease ALu-I to allow the emission rates under complete bisulfite conversion (no methylation) to be estimated. The 50 color reads and 25 color reads were aligned with PerM, using the same three-reference protocol described in the Materials and Methods section. PerM provided full sensitivity to 3 color mismatches in the 50 color sequences and full sensitivity to two-adjacent color mismatches in the 25 color sequences. To increase alignment accuracy, resulted were combined and read pairs which did not find a 50 color forward strand alignment and 25 color reverse strand alignment within the 1200 bp fragment window were discarded. After this filter an alignment file was produced in which the average horizontal coverage was ∼3.8 reads per genome position.
Fibroblast methylation analysis
For each chromosome FadE was ran on every reference CpG site with at least one read covering it, which resulted in the analysis of ∼85% of the over 28 million CpG sites. The average coverage to the analysed CpG sites was ∼5.14 reads.
Globally the mean estimated methylation rate returned by FadE was ∼63%. Methylation rates across CpG sites were not distributed uniformly. FadE identified 13 841 CpG islands which had more than 10 CpG sites with >20X coverage using the definition described by Takai and Jones (16). Figure 5 shows that the distribution among of methylation rates along CpG islands differed wildly from that observed in an isolated CpG context. Over 20% of CpG islands had an average methylation rate more than 5 times less than the global average across CpG sites and 435 sites had an estimated average methylation rate more than 10 times less than the global average. More than half of these islands (218 of the 435) were within 1000 bp of a gene. In comparison only 23% of all CpG islands analysed were within 1000 bp of a gene, which matches the results of previous studies which aim to show that hypomethylated CpG islands are involved in gene regulation (4,16). The methylation rates around CpG island promoter regions are further discussed in the Supplementary Data.
Figure 5.

Analysis of experimental SOLiD color-space data taken from the stem cell-line IMR90 show a different in methylation distribution in CpG islands when compared with isolated CG positions. Across Hg19 the global distribution of ‘CG’ dinucleotides is 1%. Here, a CpG island is defined as a region of at least 500 bp and >5% ‘CG’ dinucleotides.
Comparison to previous nucleotide-space analysis
To demonstrate the feasibility of color-space epigenome analysis our results were compared with a study carried out using nucleotide-space reads which were also sequenced from the IMR-90 fibroblast cell-line (5). Lister et al. used 91.0 gigabases of SBT Illumina reads (14.8x avg read depth per strand, 29.6x total average depth) to study the methylation patterns in different cell types, including the fibroblast IMR-90. Despite differences in coverage, both FadE and the protocol used by Lister et al. found very similar average CpG methylation rates (62.7% and 63.5%, respectively) and also found a very similar global distribution for methylation levels across CpG sites. In total there were over 23 million CpG sites which were analysed in both nucleotide and color-space. In over 89% of shared sites the rate estimated by Lister et al. was contained in the credible interval returned by FadE. Across the shared sites fewer than 10% of the positions analysed by both protocols had reported methylation rates which differed by more than 30%. These statistics are summarized in Table 4.
Table 4.
The methylation rate at 23 152 801 IMR90 CpG positions analyse by FadE and Lister et al.
| FadE | Lister et al. | Validation ratea (%) | |
|---|---|---|---|
| Global rate (%) | 62.7 | 63.5 | |
| Total shared sites | 23 152 801 | 23 152 801 | 89.1 |
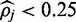 |
5 646 187 | 4 106 700 | 84.8 |
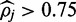 |
10 943 429 | 11 160 396 | 91.1 |
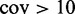 |
1 030 308 | 92.9 |
aThe validation rate refers to the percentage of positions where the methylation rate calculated by Lister et al. fell within the credible interval returned by FadE.
The work by Lister et al. also described biologically relevant partially methylated domains (PMDs), which can be identified by iteratively adding 10 kb windows to a region until a window contains fewer than 10 well covered ( 5x coverage) CpG sites or has >70% average methylation. In Hg19 coordinates they identified 8030 such regions made up of 125 601 sliding 10 kb windows (125 601 000 total bases). In the 110 748 10 kb sliding windows in which FadE contained contained sufficient coverage, 96% had <70% average methylation over 99% had <75% average methylation. In comparison, the global distribution of methylation over sufficiently covered 10 kb windows included over 30% and 22% which had average methylation rates of 70% and 75%. That the results are so similar when viewed over 10 kb windows suggest that sources of noise may be largely responsible for the site-by-site differences in methylation estimation between FadE and the nucleotide-space analysis performed by Lister et al.
5x coverage) CpG sites or has >70% average methylation. In Hg19 coordinates they identified 8030 such regions made up of 125 601 sliding 10 kb windows (125 601 000 total bases). In the 110 748 10 kb sliding windows in which FadE contained contained sufficient coverage, 96% had <70% average methylation over 99% had <75% average methylation. In comparison, the global distribution of methylation over sufficiently covered 10 kb windows included over 30% and 22% which had average methylation rates of 70% and 75%. That the results are so similar when viewed over 10 kb windows suggest that sources of noise may be largely responsible for the site-by-site differences in methylation estimation between FadE and the nucleotide-space analysis performed by Lister et al.
To determine if noise is indeed responsible for these differences FadE was used to perform analysis in nucleotide space using the dataset supplied by Lister et al. As described in detail in the Supplementary Data, the nucleotide alignment was used to directly estimate emission rates and FadE was run on each human autosome. In the Supplementary Data we show that the site-by-site concordance between the two programs to be much higher when the same dataset is used, which provides evidence that differences in the color-space study likely result from biological variation, differences between the sequencing platforms, or other forms of noise rather than differences or errors in implementation.
DISCUSSION
FadE’s most novel concept is it’s capability of working directly in color-space or nucleotide space and calculating not just an estimation for the level of methylation at a site but also the credible interval to provide information about the distribution of the parameter. Working directly in color-space allows FadE to provide a higher level of accuracy than algorithms which rely on translation of color reads, especially near consecutive cytosine positions. Recent scientific studies have shown not only that the specific levels of methylation in a cell population can have massive biological implications (17,18) but also that non-CpG cytosine positions, which are often found in consecutive blocks are heavily methylated in some cell types (5). In light of these discoveries, there is a need for an algorithm which uses a statistical model to estimate methylation directly in color or nucleotide space and provide accurate results and a statistical framework from which to interpret them. FadE is able to provide similar results regarding methylated regions with low-coverage color-space data to a high-coverage nucleotide-space experiment. This is evidence which supports what is displayed in the simulated (Figure 4) datasets; very high coverage and alignment quality is needed to determine the specific methylation level in cell population at a particular site but moderate read depth is sufficient to determine differentially methylated regions.
CONCLUSION
FadE is a novel tool which implements a Bayesian statistical model to estimate methylation levels in color and nucleotide space. FadE is both fast and memory efficient and uses the natural parallelism of the 24 human chromosomes to quickly determine the methylation rate at every reference covered reference cytosine. FadE supplies the scripts and tools to allow it to pair well with any mapping program capable of outputting color or nucleotide alignment in SAM, bed, mapping or a user-defined column separated format. FadE also increases accuracy by allowing the user to supply prior probabilities if the tissue in question has been previously studied as well as including the emission rates calculated from the Phage-Lambda genome to increase accuracy if controls are not available. FadE’s result’s on simulation data show the utility of using a statistical model to determine methylation rather than a simple color-conversion or sequence pileup method whereas the results on the human fibroblast dataset provide some insight into the distribution of methylation across the human genome and also corroborate with a study conducted using higher coverage nucleotide data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3 and Supplementary Figures 1–4.
FUNDING
Funding for open access charge: National Science Foundation [NSF/OCE 1136818, NSF/DMS ATD 7031026]; National Institutes of Health [NIH/NIMH 1 RC1 MH090047-01]; Center of Excellence of Genome Sciences [NIH/HG 2 P50 HG002790-06] as well as funding from the Life Technology Collaboration Grant.
Conflict of interest statement. Z.Z. is employed by, and shareholder of Life Technologies.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Fengzhu Sun and Reza Ardekani for helpful suggestions regarding statistics and also to Zach Frazier and Yangho Chen for their programming suggestions. Additionally, this work would have been possible if not for the work of Gavin Meredith and the staff scientists at Life Technologies who prepared the dataset.
REFERENCES
- 1.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. [PubMed] [Google Scholar]
- 2.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 4.Li, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lister, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ondov BD, Varadarajan A, Passalacqua KD, Bergman NH. Efficient mapping of Applied Biosystems SOLiD sequence data to a reference genome for functional genomic applications. Bioinformatics. 2008;24:2776–2777. doi: 10.1093/bioinformatics/btn512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Souaiaia T, Frazier Z, Chen T. ComB: SNP calling and mapping analysis for color and nucleotide space platforms. J. Comput. Biol. 2011;18:795–807. doi: 10.1089/cmb.2011.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pedersen B, Hsieh TF, Ibarra C, Fischer RL. MethylCoder: software pipeline for bisulfite-treated sequences. Bioinformatics. 2011;27:2435–2436. doi: 10.1093/bioinformatics/btr394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ondov BD, Cochran C, Landers M, Meredith GD, Dudas M, Bergman NH. An alignment algorithm for bisulfite sequencing using the Applied Biosystems SOLiD System. Bioinformatics. 2010;26:1901–1902. doi: 10.1093/bioinformatics/btq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bormann Chung CA, Boyd VL, McKernan KJ, Fu Y, Monighetti C, Peckham HE, Barker M. Whole methylome analysis by ultra-deep sequencing using two-base encoding. PLoS One. 2010;5:e9320. doi: 10.1371/journal.pone.0009320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansen, et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Nordborg M, Li LM. Adjust quality scores from alignment and improve sequencing accuracy. Nucleic Acids Res. 2004;32:5183–5191. doi: 10.1093/nar/gkh850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Souaiaia T, Chen T. PerM: efficient mapping of short sequencing reads with periodic full sensitive spaced seeds. Bioinformatics. 2009;25:2514–2521. doi: 10.1093/bioinformatics/btp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. SHRiMP: accurate mapping of short color-space reads. PLoS Comput.-Biol. 2009;5:e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl Acad. Sci. USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 18.Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, Rehli M. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009;7:1165–1174. doi: 10.1101/gr.091470.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.